

Abstract
Objective:
To evaluate clinical features among patients with neuromyelitis optica spectrum disorders (NMOSD) who have myelin oligodendrocyte glycoprotein (MOG) antibodies, aquaporin-4 (AQP4) antibodies, or seronegativity for both antibodies.
Methods:
Sera from patients diagnosed with NMOSD in 1 of 3 centers (2 sites in Brazil and 1 site in Japan) were tested for MOG and AQP4 antibodies using cell-based assays with live transfected cells.
Results:
Among the 215 patients with NMOSD, 7.4% (16/215) were positive for MOG antibodies and 64.7% (139/215) were positive for AQP4 antibodies. No patients were positive for both antibodies. Patients with MOG antibodies represented 21.1% (16/76) of the patients negative for AQP4 antibodies. Compared with patients with AQP4 antibodies or patients who were seronegative, patients with MOG antibodies were more frequently male, had a more restricted phenotype (optic nerve more than spinal cord), more frequently had bilateral simultaneous optic neuritis, more often had a single attack, had spinal cord lesions distributed in the lower portion of the spinal cord, and usually demonstrated better functional recovery after an attack.
Conclusions:
Patients with NMOSD with MOG antibodies have distinct clinical features, fewer attacks, and better recovery than patients with AQP4 antibodies or patients seronegative for both antibodies.
Neuromyelitis optica (NMO) is characterized by severe attacks of optic neuritis (ON) and longitudinally extensive transverse myelitis (LETM) with 3 or more vertebral segment spinal cord lesions observed on MRI.1 Limited forms of the disease are known as NMO spectrum disorders (NMOSD). NMOSD currently include patients with either ON or LETM (single or recurrent events of LETM or recurrent or simultaneous bilateral ON).2 Approximately 90% of the patients with NMO and more than half of the patients with NMOSD are positive for autoantibodies against aquaporin-4 (AQP4).3,4 Therefore, a proportion of patients with NMO or NMOSD remains AQP4 antibody-negative despite the use of the best assays available on serum samples collected during an acute attack before any treatment.
Recently, autoantibodies against myelin oligodendrocyte glycoprotein (MOG) were reported in 4 patients who were clinically diagnosed with NMOSD and negative for AQP4 antibodies.5 High-titer MOG antibodies are predominantly of the immunoglobulin G (IgG) 1 subtype and efficiently mediate complement-dependent cytotoxicity in vitro.6 However, none of these previous studies investigated comprehensively the features that may distinguish patients with AQP4 antibodies from those with high-titer MOG antibodies or those who are negative for both antibodies, even though such information is useful for clinical practice. In this study, we compared the clinical, MRI, and laboratory characteristics of patients with high-titer MOG antibodies with those of patients with AQP4 antibodies and seronegative patients.
METHODS
Patients and serum samples.
We enrolled a total of 215 patients from 3 tertiary centers for this study: 1) Hospital das Clínicas, Faculty of Medicine, University of Sao Paulo, Brazil; 2) Center for the Investigation of MS at Federal University of Minas Gerais, Belo Horizonte, Brazil; and 3) Tohoku University Hospital, Sendai, Japan. We included pediatric and adult patients who had received a clinical diagnosis of definitive NMO or NMOSD, which currently includes patients with one attack or recurrent LETM and those with bilateral simultaneous or recurrent ON. For simplicity, we use the term NMOSD to encompass both NMO and NMOSD. We only included consecutive patients followed up in one of the 3 centers for whom information regarding the clinical attacks, brain and spinal cord MRIs, and serum for antibody testing were available; 7 patients were excluded because of a lack of information (5 patients with AQP4 antibodies and 2 seronegative patients). All patients seronegative for both AQP4 and MOG antibodies were fully investigated, and alternative diagnoses were ruled out. The serum samples from the Brazilian centers were stored at −80 °C after centrifugation in each center, shipped on dry ice to Sendai, Japan, and stored again at −80 °C until analysis.
AQP4 and MOG antibody assays.
All serum samples were analyzed at Tohoku University to detect AQP4 and MOG antibodies. The cell-based assay (CBA) for AQP4 antibody detection in living cells has been described7 using HEK-293 cells stably transfected with the M23 isoform of AQP4. Two investigators (D.K.S. and T.T.) scored the assays. These samples were also analyzed for the presence of MOG antibodies using a CBA with live HEK-293 cells transiently transfected with a plasmid containing full-length human MOG cDNA (pIRES2-Dsred2 vector, BD Biosciences, San Jose, CA; provided by P.J.W.) using the FUGENE6 transfection agent (Promega Corp., Madison, WI). Goat anti-human IgG labeled with Alexa488 (Invitrogen, Carlsbad, CA) was used as a secondary antibody after the transfected cells were exposed to the patients' diluted serum. The samples were tested for MOG antibodies at least twice at dilution of 1:128, and only patients whose samples were judged to be positive by 2 observers (D.K.S. and T.T.) were considered positive with high titers. See details of the patients tested during the validation of MOG antibody assay in the e-Methods on the Neurology® Web site at www.neurology.org. Later, positivity for the MOG antibodies was also confirmed by P.J.W. at Oxford University in a blinded fashion. The titers from both assays were calculated semiquantitatively using consecutive twofold dilutions.
Statistical analysis.
Nonparametric tests (Wilcoxon tests) were used to compare the clinical data and CBA titers among the groups of patients. The categorical data from the study were analyzed using Fisher exact test, and p < 0.05 was considered to be significant.
Standard protocol approvals, registration, and patient consent.
This study was approved by the ethics committee of each center and conducted in accordance with internationally recognized ethical standards. All study participants provided written informed consent.
RESULTS
In total, 215 patients with NMOSD were included: 152 from Brazil (Sao Paulo and Belo Horizonte) and 63 from Japan (Sendai). Among these patients, 7.4% (16/215) were positive for MOG antibodies, 64.7% (139/215) were positive for AQP4 antibodies, and the remaining 27.9% (60/215) were seronegative. No patients were positive for both antibodies. Patients with MOG antibodies represented 21.1% (16/76) of AQP4-antibody seronegative cases.
The clinical characteristics of each patient with MOG antibodies are summarized in table 1. None of the patients had encephalopathy or seizures. In the acute phase, all patients with MOG antibodies received IV high-dose methylprednisolone (1 g/d for 3–5 days) as the first-line treatment for acute attacks, and 87.5% (14/16) of them experienced good recovery. Only one patient with a severe LETM attack, with tetraparesis, urinary retention, and a sensory level of Th10 and below, was further treated with 5 sessions of plasmapheresis. However, this patient had only a partial recovery of motor strength in the trunk and upper limbs, remaining paraplegic and without bowel/bladder control.
Table 1.
Clinical characteristics of 16 patients with NMOSD with MOG antibodies
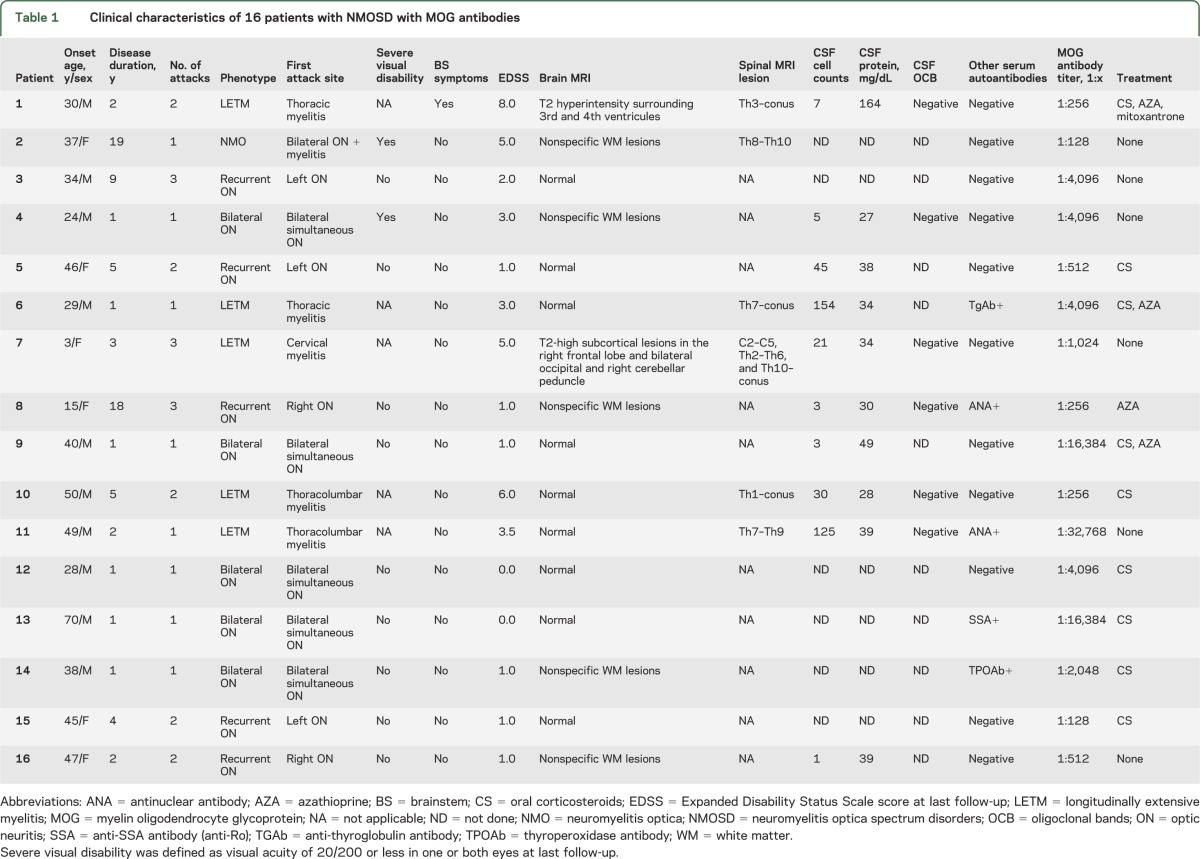
The median (range) MOG antibody titers were higher in patients with a single attack than in recurrent patients (4,096 [128–32,768] vs 384 [128–4,096], respectively, p = 0.0193). Later, we were able to confirm persistent positivity for MOG antibodies in consecutive samples from 7 patients after a median (range) of 246 (34–400) days.
Comparison of demographic and main clinical syndrome features according to antibody status.
Among the patients, 168 were female and 47 were male. The female:male ratio was 7.2:1.0 (122/17) in the AQP4 antibody group and 2.0:1.0 (40/20) in the seronegative group. By contrast, males predominated (ratio of 0.6:1.0 [6/10]) in the MOG antibody group. The median (range) onset age was 37.5 (3–70) years for patients with MOG antibodies, 37 (4–78) years for patients with AQP4 antibodies, and 32.5 (10–69) for patients who were seronegative (p = 0.0915).
In the group of patients who met the criteria for definite NMO (n = 101), only 1 patient had MOG antibodies, whereas the majority had AQP4 antibodies (84.2% [85/101]) and 14.8% (15/101) were seronegative. We found 12 patients with a single attack who presented initially with ON and myelitis, thus fulfilling the definitive NMO criteria in the first attack. Among them, 1 patient had MOG antibodies, 8 had AQP4 antibodies, and 3 were seronegative.
In the group of patients with only LETM, 6.4% (5/78) had MOG antibodies, 55.1% (43/78) had AQP4 antibodies, and 38.5% (30/78) were seronegative. Among the patients with bilateral simultaneous or recurrent ON, 27.8% (10/36) had MOG antibodies, 30.5% (11/36) had AQP4 antibodies, and 41.7% (15/36) were seronegative.
The comparison of the demographic and clinical characteristics among patients with MOG antibodies, patients with AQP4 antibodies, and seronegative patients is summarized in table 2 and table e-1.
Table 2.
Comparison of clinical features between patients with NMOSD with MOG antibodies, AQP4 antibodies, and seronegative patients

Comparison of detailed clinical features among patients with MOG and AQP4 antibodies and seronegative patients.
In total, 155 patients were positive for either MOG or AQP4 antibodies. Brainstem symptoms commonly reported in AQP4 antibody-positive patients, such as persisting nausea/vomiting episodes for longer than 48 hours, were found in 40.6% (56/139) of patients with AQP4 antibodies, whereas only a single patient with MOG antibodies and 11.7% (7/60) of the seronegative patients reported similar symptoms during attacks (p < 0.0001). Intractable hiccups were only reported in patients with AQP4 antibodies and one seronegative patient (0 [0/16] vs 23.7% [33/139] vs 1.7% [1/60] for patients with MOG antibodies, patients with AQP4 antibodies, and seronegative patients, respectively; p < 0.0001). Painful tonic spasms were present in 6.3% (1/16) of the patients with MOG antibodies, 42.5% (59/139) of the patients with AQP4 antibodies, and 31.7% (19/60) of seronegative patients (p = 0.0073).
Among patients with ON attacks, we found that 18.8% (3/11) of the MOG antibody group, 72.9% (72/96) of the AQP4 antibody group, and 46.7% (14/30) of the seronegative group had a visual acuity <20/200 (p = 0.0010). One patient (9.1%) positive for MOG antibodies, 30.2% (29/96) of the patients positive for AQP4 antibodies, and 13.3% (4/30) of the seronegative patients had no light perception after an ON attack. Bilateral, simultaneous ON attacks were observed in all groups, but such attacks were more common in patients with MOG antibodies (see illustrative imaging scan in figure 1) than in those with AQP4 antibodies or those who were seronegative (72.7% [8/11] vs 24.0% [23/96] vs 36.7% [11/30], respectively; p = 0.0035).
Figure 1. Bilateral optic neuritis in a patient with MOG antibodies.

Axial short TI inversion recovery (STIR) and T1-weighted with gadolinium MRI of the optic nerves from a 28-year-old man with bilateral simultaneous optic neuritis and myelin oligodendrocyte glycoprotein (MOG) antibodies (titer 1:4,096) shows bilateral STIR hyperintense lesions with contrast enhancement.
Patients with a single attack comprised 50.0% (8/16) of patients with MOG antibodies and 16.6% (23/139) of patients with AQP4 antibodies (p = 0.0044). In these patients, the median (range) follow-up was not different among patients with MOG antibodies, those with AQP4 antibodies, and those who were seronegative (1 [1–19] vs 2 [1–27] vs 1 [0–11] years, respectively; p = 0.3046). The median disability at the last visit after a single attack measured by the Expanded Disability Status Scale (EDSS) was 2 (range 0–5) in the MOG antibody-positive group, 6 (2–8.5) in the AQP4 antibody-positive group, and 4.5 (0–7) in the seronegative group (p = 0.0173).
Recurrent patients with MOG antibodies (n = 8) had a median follow-up of 4.5 years (range 2–18), in contrast to 8 years (range 0–45) in the patients with AQP4 antibodies and 3 years (range 0–32) in seronegative patients (p = 0.0046), but there were no differences between patients with MOG and AQP4 antibodies (p = 0.1493) or between patients with MOG antibodies and those who were seronegative (p = 0.5399). Recurrent patients who were positive for MOG antibodies had a lower median number of attacks than patients positive for AQP4 antibodies and those who were seronegative (2 [2–3] vs 4 [2–33] vs 3 [2–18], p = 0.0011), most likely because of the higher median (range) number of myelitis attacks in the patients with AQP4 antibodies and in those who were seronegative (0 [0–2] vs 2 [0–30] vs 2 [0–15], respectively; p = 0.0026). However, the annual relapse rate was not different among recurrent disease patients with MOG antibodies (0.5 [0.1–1]), those with AQP4 antibodies (0.7 [0.1–4]), and those who were seronegative (0.9 [0.1–4], p = 0.1453). The median (range) EDSS at the last visit of the patients with recurrent disease was 1.5 (1–8) in patients with MOG antibodies, 5.5 (1–8.5) in patients with AQP4 antibodies, and 4 (1–7) in seronegative patients (p = 0.0125). Patients with MOG antibodies clearly appear to have a better outcome regardless of the course of the disease.
Comparison of MRI findings among patients with MOG antibodies, patients with AQP4 antibodies, and seronegative patients.
An abnormal brain MRI was present in 37.5% (6/16) of patients with MOG antibodies, 56.8% (79/139) of patients with AQP4 antibodies, and 56.7% (34/60) of seronegative patients. Medullary lesions commonly associated with persisting nausea/vomiting or hiccups episodes in NMOSD were observed in a single patient with MOG antibodies, in 12.9% (18/139) of patients with AQP4 antibodies, and in 3.3% (2/60) of seronegative patients.
Spinal cord lesions on MRI were present in 37.5% (6/16), 92.1% (128/139), and 71.7% (43/60) of patients with MOG antibodies, patients with AQP4 antibodies, and seronegative patients, respectively (table 3, table e-2). Lesions in patients with MOG antibodies were distributed more frequently in the thoracolumbar region (figure 2). By contrast, patients with AQP4 antibodies and seronegative patients had more lesions distributed in the cervicothoracic region. All patients with MOG antibodies (6/6), all patients who were seronegative (43/43), and 89.1% (114/128) of patients with AQP4 antibodies had lesions covering 3 or more vertebral segments in the sagittal spinal cord MRI. The median (range) number of vertebral segments of the longest spinal cord lesion was 6.5 (3–14) in patients with MOG antibodies, 6 (1–19) in patients with AQP4 antibodies, and 4.5 (3–19) in seronegative patients (p = 0.3658). The involvement of the central portion of the spinal cord in the axial spinal cord MRI was found in 66.7% (4/6) of patients with MOG antibodies, 89.8% (115/128) of patients with AQP4 antibodies, and 79.1% (34/43) of the seronegative patients (p = 0.0721).
Table 3.
MRI and laboratory findings in patients with NMOSD with MOG antibodies, patients with NMOSD with AQP4 antibodies, and seronegative patients

Figure 2. Recurrent longitudinally extensive transverse myelitis in a patient with MOG antibodies.

Sagittal T2-weighted MRI of the spinal cord from a 6-year-old girl with recurrent longitudinally extensive transverse myelitis and myelin oligodendrocyte glycoprotein (MOG) antibodies (titer 1:1,024) shows 2 longitudinally extensive lesions. On T1-weighted sagittal sequence, the lesion in the thoracolumbar transition shows a ring contrast enhancement.
Comparison of laboratory findings among patients with MOG antibodies, patients with AQP4 antibodies, and seronegative patients.
Analysis of the CSF was available in 73.4% (158/215) of the patients, but there were no differences on the results among the 3 groups. Antinuclear autoantibodies (ANA) in the serum were found less frequently in patients with MOG antibodies than in the other groups. The frequency of other antibodies was not different among the 3 groups (table 3).
Comparison of long-term treatments used in patients with MOG antibodies, patients with AQP4 antibodies, and seronegative patients.
Among the patients with MOG antibodies, 56.3% (9/16) were on oral corticosteroids for relapse prevention, whereas oral corticosteroids were used by 80.6% (112/139) of patients with AQP4 antibodies and 56.7% (34/60) of patients who were seronegative (p = 0.0007). Azathioprine was used in 25.0% (4/16) of patients with MOG antibodies, 54.7% (76/139) of patients with AQP4 antibodies, and 38.3% (23/60) of patients who were seronegative (p = 0.0166). A single patient with MOG antibodies, 8.6% (12/139) of patients with AQP4 antibodies, and 3.3% (2/60) of seronegative patients used mitoxantrone. None of the patients with MOG antibodies, 8.6% (12/139) of the patients with AQP4 antibodies, and only 1 seronegative patient used rituximab.
DISCUSSION
We compared patients with AQP4 and MOG antibodies who received a clinical diagnosis of NMOSD. We also compared some of the clinical features of these patients with seronegative patients. Patients with MOG antibodies tended to have a single or a lower number of attacks, whereas patients with AQP4 antibodies were more likely to be prototypical NMO patients, with relapsing disease characterized by severe ON and LETM attacks, as previously described.1 The degree of recovery, measured by EDSS and visual acuity, also indicates that patients with MOG antibodies tended to have a better recovery after an attack. Nevertheless, it is important to emphasize some patients with MOG antibodies experience severe disability after ON or LETM attacks, and thus patients in this group may not recover well.
Our study results suggest that patients with MOG antibodies fulfilling the definitive NMO criteria as recently reported5 may actually be rare. Some NMOSD cases with MOG antibodies reported previously (table e-3) and the single NMO case in our study had a monophasic presentation with both ON and myelitis occurring during the same attack, similarly to the original description by Eugene Devic in 1894. In fact, some of these patients with MOG antibodies and single attack in the previous and current studies may have a spatially limited form of acute demyelinating encephalomyelitis (ADEM) but without encephalopathy and typical brain lesions, which could mimic NMOSD. However, we also found recurrent patients with persistent MOG antibodies, which is not usually observed in ADEM and precludes the generalization of this hypothesis to all patients.
The strong female predominance in patients with AQP4 antibodies was not found in the MOG antibody group, and a much lower female:male ratio was found in seronegative patients. A similar difference in the female:male ratio between the diseases (with MOG antibodies and with AQP4 antibodies) was observed in previous smaller studies5,6 and this finding seems to parallel the serum ANA positivity. By contrast, and similarly to MOG antibody-mediated disease, ADEM does not appear to have a sex bias.8
We found a difference in the spatial distribution of spinal cord lesions on MRI between patients with MOG antibodies and those with AQP4 antibodies. The patients with AQP4 antibodies frequently had cervical lesions, including some extending to the medullary region, whereas patients with MOG antibodies more frequently had long lesions extending to the lumbar spinal cord. Brainstem symptoms, such as persistent nausea/vomiting and hiccups, as previously reported in NMOSD,9,10 were more commonly found in the AQP4 antibody group than in the MOG antibody or seronegative groups. Typical pathologic NMO lesions in the area postrema have been found in postmortem specimens of AQP4 antibody-positive patients,11 and, although no pathologic studies have been performed on patients with MOG antibodies, no lesions in area postrema would be expected in this group.
Although MOG antibodies have been shown to be potentially pathogenic and to efficiently activate complement in vitro, these antibodies have been reported in a variety of demyelinating diseases such as ADEM and multiple sclerosis (MS),12 especially in pediatric populations,13,14 and also in NMOSD.6 It is possible that differences in antibody assays or cohort ascertainments have a role to play in the apparent nondisease specificity of the MOG antibodies. One could argue that the presence of MOG antibodies in a number of distinct diseases may currently limit the use of the MOG antibody assay as a specific diagnostic biomarker, and it is possible that other undiscovered autoantibodies may exist in the seronegative patients. However, it is important to consider that excluding the patients with NMOSD with AQP4 antibodies, all of these diagnoses are currently based on clinical and MRI features. Because of the study design, patients with MOG antibodies who had received a diagnosis of ADEM and MS were not included in this study, so our study findings are currently limited to patients who received a diagnosis of NMOSD. MOG antibody-positive patients, regardless of whether they had a single attack or recurrent disease, had some distinct clinical presenting features, usually fewer attacks, and a better recovery after an attack than patients with AQP4 antibodies or seronegative patients. Further long-term prospective studies are required to investigate whether patients with a single attack and patients with recurrent MOG antibodies—with clinical diagnosis of ADEM, MS, or NMOSD—have a singular disease, but with some differences in the clinical phenotype, disease course, and treatment response.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Paula Zago, Dr. Jose Antonio Livramento, Dr. Elizabeth Regina Comini Frota, Sayuko Kasahara, and Karina Suera Bosso for technical assistance.
GLOSSARY
- ADEM
acute demyelinating encephalomyelitis
- ANA
antinuclear autoantibodies
- AQP4
aquaporin-4
- CBA
cell-based assay
- EDSS
Expanded Disability Status Scale
- IgG
immunoglobulin G
- LETM
longitudinally extensive transverse myelitis
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- NMOSD
neuromyelitis optica spectrum disorders
- ON
optic neuritis
Footnotes
Editorial, page 466
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Study design and conceptualization: D.K. Sato, I. Nakashima, A.M.M. Lino, D. Callegaro, and K. Fujihara. Drafting of manuscript: D.K. Sato, T. Takahashi, I. Nakashima, P.J. Waters, M.I. Leite, and K. Fujihara. Acquisition, analysis, and interpretation of data: D.K. Sato, F.M.d.H. Jorge, S.L. Apostolos-Pereira, N. Talim, T. Takahashi, P.J. Waters, I. Nakashima, D. Callegaro, and M.A. Lana-Peixoto. Statistical analysis: D.K. Sato, T. Takahashi, and I. Nakashima. Critical revision of the manuscript: D.K. Sato, P.J. Waters, M.I. Leite, I. Nakashima, T. Misu, T. Takahashi, R.F. Simm, A.M.M. Lino, K. Fujihara, and M. Aoki.
STUDY FUNDING
Supported by KAKENHI (22229008) of The Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and by the Health and Labour Sciences Research Grant on Intractable Diseases (Neuroimmunological Diseases) from the Ministry of Health, Labour and Welfare of Japan.
DISCLOSURE
D. Sato receives scholarship from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and has received research support from Ichiro Kanehara Foundation. D. Callegaro and M. Lana-Peixoto report no disclosures. P. Waters is a named inventor and has received royalties for antibody assays, and has received a speaker honorarium from Biogen Idec Japan. F. Jorge and T. Takahashi report no disclosures. I. Nakashima has received funding for travel and received speaker honoraria from Bayer Schering Pharma and Biogen Idec and has received research funding from Mitsubishi Chemical Medience Corporation and the Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology of Japan. S. Apostolos-Pereira, N. Talim, R. Simm, and A. Lino report no disclosures. T. Misu has received speaker honoraria from Bayer Schering Pharma, Biogen Idec Japan, Mitsubishi Tanabe Pharma Corporation, Asahi Kasei Medical Co., and Astellas Pharma Inc., and has received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Kuraray Medical Co., The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corporation, Teijin Pharma, and Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology, and the Ministry of Health, Labor and Welfare of Japan. M. Leite is supported by NHS National Specialised Commissioning Group for Neuromyelitis Optica, UK, and by NIHR Oxford Biomedical Research Centre, and has received speaking honoraria from Biogen Idec and travel grants from Novartis. M. Aoki reports no disclosures. K. Fujihara serves on scientific advisory boards for Bayer Schering Pharma, Biogen Idec, Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Merck Serono, Alexion Pharmaceuticals, Medimmune and Medical Review; has received funding for travel and speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai Inc., Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Astellas Pharma Inc., Takeda Pharmaceutical Company Limited, Asahi Kasei Medical Co., Daiichi Sankyo, and Nihon Pharmaceutical; serves as an editorial board member of Clinical and Experimental Neuroimmunology (2009–present) and an advisory board member of Sri Lanka Journal of Neurology; has received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, and Genzyme Japan; and is funded as the secondary investigator (22229008, 2010–2015) by the Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology of Japan and as the secondary investigator by the Grants-in-Aid for Scientific Research from the Ministry of Health, Welfare and Labor of Japan (2010–present). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114 [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815 [DOI] [PubMed] [Google Scholar]
- 3.Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007;130:1235–1243 [DOI] [PubMed] [Google Scholar]
- 4.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012;78:665–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273–1277 [DOI] [PubMed] [Google Scholar]
- 6.Mader S, Gredler V, Schanda K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 2011;8:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato DK, Nakashima I, Takahashi T, et al. Aquaporin-4 antibody-positive cases beyond current diagnostic criteria for NMO spectrum disorders. Neurology 2013;80:2210–2216 [DOI] [PubMed] [Google Scholar]
- 8.de Seze J, Debouverie M, Zephir H, et al. Acute fulminant demyelinating disease: a descriptive study of 60 patients. Arch Neurol 2007;64:1426–1432 [DOI] [PubMed] [Google Scholar]
- 9.Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y. Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis optica. Neurology 2005;65:1479–1482 [DOI] [PubMed] [Google Scholar]
- 10.Sato D, Fujihara K. Atypical presentations of neuromyelitis optica. Arq Neuropsiquiatr 2011;69:824–828 [DOI] [PubMed] [Google Scholar]
- 11.Popescu BF, Lennon VA, Parisi JE, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 2011;76:1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Connor KC, McLaughlin KA, De Jager PL, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med 2007;13:211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Probstel AK, Dornmair K, Bittner R, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 2011;77:580–588 [DOI] [PubMed] [Google Scholar]
- 14.Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 2013;9:455–461 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.