

Abstract
The growth arrest and DNA damage-inducible 45β (GADD45β) gene product has been implicated in the stress response, cell cycle arrest, and apoptosis. Here we demonstrated the unexpected expression of GADD45β in the embryonic growth plate and uncovered its novel role as an essential mediator of matrix metalloproteinase-13 (MMP-13) expression during terminal chondrocyte differentiation. We identified GADD45β as a prominent early response gene induced by bone morphogenetic protein-2 (BMP-2) through a Smad1/Runx2-dependent pathway. Because this pathway is involved in skeletal development, we examined mouse embryonic growth plates, and we observed expression of Gadd45β mRNA coincident with Runx2 protein in pre-hypertrophic chondrocytes, whereas GADD45β protein was localized prominently in the nucleus in late stage hypertrophic chondrocytes where Mmp-13 mRNA was expressed. In Gadd45β−/− mouse embryos, defective mineralization and decreased bone growth accompanied deficient Mmp-13 and Col10a1 gene expression in the hypertrophic zone. Transduction of small interfering RNA-GADD45β in epiphyseal chondrocytes in vitro blocked terminal differentiation and the associated expression of Mmp-13 and Col10a1 mRNA in vitro. Finally, GADD45β stimulated MMP-13 promoter activity in chondrocytes through the JNK-mediated phosphorylation of JunD, partnered with Fra2, in synergy with Runx2. These observations indicated that GADD45β plays an essential role during chondrocyte terminal differentiation.
Growth arrest and DNA damage-inducible (GADD)4 45β is a member of the GADD45 family of small (18 kDa) proteins, also including GADD45α and GADD45γ. The GADD45 family is known to be associated with cell growth control, apoptotic cell death, and the cellular response to DNA damage (1, 2). Initially, GADD45β, encoded by MyD118, was identified as a myeloid differentiation primary response gene activated by IL-6 in murine myeloid leukemia cells upon induction of terminal differentiation (1, 3). More recently, GADD45β, which is induced by TGF-β in a SMAD-dependent manner, has been identified as a positive regulator of TGF-β-induced apoptosis (4). Although GADD45α has been identified on DNA microarrays as prominently expressed genes in chondrocytes from adult articular cartilage and in chondrosarcoma or immortalized chondrocyte cell lines (5, 6), a role for GADD45 family members, including GADD45β, during cartilage development has not been reported previously.
Formation of the vertebrate skeleton through endochondrial ossification, involving progressive differentiation of proliferating chondrocytes to growth-arrested hypertrophic cells, is one of the most complex processes in biology. In the embryonic or postnatal growth plate, terminal chondrocyte differentiation occurs during conversion of cartilage to a vascularized tissue that supports matrix remodeling, cartilage calcification, and recruitment of osteogenic precursors. Cascades of growth and differentiation factors act through positive and negative signaling kinases and transcription factors to tightly control this process. Bone morphogenetic proteins (BMPs), which were identified originally as molecules that induce ectopic endochondral ossification (7), set the stage for bone morphogenesis by initiating chondrogenesis and by regulating chondrocyte maturation and terminal differentiation to the hypertrophic phenotype (8–11). The actions of the BMPs are determined by spatial and temporal differences in the distribution of BMP-binding proteins, such as noggin and chordin, BMP receptors, and the associated signal-transducing acceptor proteins, Smads 1 and 5, and inhibitory Smads 6 and 7.
BMP-2, -4, -6, and -7 are known to stimulate the expression of markers of the hypertrophic phenotype, including type X collagen and alkaline phosphatase (11, 12), and these responses to BMPs in the growth plate are restricted to pre-hypertrophic chondrocytes derived from regions where endochondral bone will form (13). Previous studies have demonstrated that BMP-induced Smad1 and interactions with the Runt domain transcription factor, Runx2, or Cbfa1, are important for chondrocyte hypertrophy (14–16). Runx2 is required for bone formation (17, 18), serving as a positive regulatory factor in chondrocyte maturation to the hypertrophic phenotype (15). Matrix metalloproteinase (MMP)-13, a downstream target of Runx2, is expressed by terminal hypertrophic chondrocytes during endochondral ossification (19–22). Recent evidence indicates that MMP-13 deficiency results in significant interstitial collagen accumulation leading to the delay of endochondral ossification in the growth plate (23, 24). Although the induction of MMP-13 expression by cytokines in chondrocytes (25) and by parathyroid hormone-related protein in osteoblasts (26, 27) is known to involve cooperation between AP-1 and Runx2 transcription factors, the exact molecular mechanism of MMP-13 induction in hypertrophic chondrocytes remains unclear. Runx2 is expressed mainly in prehypertrophic and less in late hypertrophic chondrocytes (28, 29), whereas MMP-13 is expressed only in the late hypertrophic zone (30, 31). This spatial discrepancy suggests that unknown intermediate molecules may synchronize with Runx2 to regulate Mmp-13 gene expression.
During the course of a study to identify the BMP-2-induced early genes that might be involved in signaling and transcriptional regulation in human chondrocytes, we identified GADD45β as one of the most highly induced genes. We further showed specific induction of GADD45β, but not GADD45α and GADD45γ, by BMP-2, but not by EGF, FGF-2, or IGF-I, by a mechanism involving Smad1-dependent signaling and synergism with Runx2. Because BMP-2 is an important regulator of skeletal development, we examined mouse embryos and demonstrated for the first time that Gadd45β mRNA is expressed by pre-hypertrophic chondrocytes coincident with the Runx2 protein, whereas GADD45β protein accumulates in hypertrophic chondrocytes. Analysis of Gadd45β−/− mouse embryos showed defective mineralization and decreased bone growth accompanied by decreased levels of Mmp-13 and Col10a1 mRNA in hypertrophic chondrocytes. In addition, lentiviral expression of siRNA-GADD45β blocked Mmp-13 gene expression during hypertrophic differentiation of epiphyseal chondrocytes in vitro. Furthermore, we show that GADD45β induces AP-1 transcriptional activity through JNK-mediated phosphorylation of JunD partnered with Fra2 and stimulates Mmp-13 promoter activity in synergism with Runx2. These results indicate that GADD45β has a critical role in mediating matrix remodeling during the final stages of chondrocyte terminal differentiation.
MATERIALS AND METHODS
Cell Culture
The immortalized human chondrocyte cell line, C-28/I2, was cultured in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 (1/1, v/v; Invitrogen) containing 10% fetal calf serum (FCS) (BioWhittaker), as described previously (32, 33). For experiments with growth factors, subconfluent cultures were changed to medium containing 1% Nutridoma-SP (Roche Applied Science) for 24 h prior to incubation in the presence of growth factors. Primary chondrocytes were isolated from human articular cartilage, obtained from intact regions of femoral condyles at the time of total knee replacement surgery, and cultured for 7–10 days in DMEM/Ham’s F-12 containing 10% FCS. ATDC5 cells were grown in DMEM/Ham’s F-12 containing 5% FCS, 10 μg/ml human insulin, 10 μg/ml human transferrin, and 10 μg/ml selenious acid, as described (34). After incubation in the presence of growth factors for 1 h, cells were harvested for RNA isolation. Three-dimensional pellet cultures of murine rib growth plate chondrocytes were prepared by the method of Ballock and Reddi (35). Chondrocytes from the ventral parts of rib cartilage of 2-day-old C57BL/6 mice were cultured in monolayer for 1 day followed by infection with lentiviral vector and culture for 2 days. Using trypsin/EDTA, the cells were resuspended at 1.6 × 105 cells/ml, pelleted at 1 ml per tube in 15-ml conical polypropylene tubes by centrifugation at 1,000 rpm for 5 min at 4 °C, and cultured at 37 °C for periods of up to 21 days.
RNA Isolation and Microarray Analysis
Total RNA was isolated using RNase-free DNase and RNeasy mini kit (Qiagen). Transcriptional profiling was performed at the Genomics Center of the Beth Israel Deaconess Medical Center on HU133A Affymetrix GeneChips containing 22,283 genes, according to protocols supplied by Affymetrix, with 8 μg of total RNA per sample. For each sample, 20 μg of fragmented cDNA was hybridized with a pre-equilibrated HU133A Affymetrix chip, washed, stained, and scanned in the HP ChipScanner (Affymetrix Inc., Santa Clara, CA), as described (36).
Microarray data pre-processing was carried out by assigning each sample a quality measure based on criteria such as 3′–5′ratios, strength of hybridization, and image analysis. The samples that passed these a priori quality control criteria were analyzed using dChip software (37), by which smoothing spline normalization was applied prior to obtaining model-based gene expression indices or signal values. When comparing two groups of samples to identify the genes enriched in a given phenotype, we used the lower confidence bound of the fold change between the experiment and the base line. If the 90% lower confidence bound of the fold change between the experiment and the base line was above 1.2, the corresponding gene was considered to be differentially expressed (38).
Real Time PCR
For each sample, cDNA was generated as described previously (33, 39). Amplifications were carried out using SYBR Green I-based real time PCR on the MJ Research DNA Engine OpticonTM Continuous Fluorescence Detection System (MJ Research Inc., Waltham, MA), as described previously (40).
Immunoprecipitation and Western Blotting Analysis
After incubation without or with BMP-2 at 100 ng/ml for 1 h, the C-28/I2 cells were collected by scraping, and total protein was extracted with 50 mM Tris-HCl buffer (pH 7.4) containing 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor mixture (Roche Applied Science). The cell lysates were analyzed on Western blots using antibody against total Smad1 or phospho-Smad1/5/8 (Cell Signaling). For immunoprecipitations, the capture protein for the Runx2 antibody (PEBP2aA, Santa Cruz Biotechnology) was coated at 200 μl/well in a 96-well flat bottom, high binding microplate (Costar Corp., Cambridge, MA) at a final protein concentration of 2.4 μg/ml in 50 mM carbonate/bicarbonate buffer (pH 9.6) and incubated overnight at 4 °C. The plate was washed with phosphate-buffered saline (pH 7.4) containing 0.05% Tween 20 (PBST), blocked by applying 200 μl of 1% bovine serum albumin in PBST to each well, and incubated at room temperature for 1 h. Plates were washed three times with PBST. Cell lysate (1.1 mg of protein) was applied to each well, incubated at room temperature for 4 h, and washed three times with PBST. The bound protein was eluted with 1×SDS sample buffer and carefully loaded on a Tris-glycine SDS-10% polyacrylamide gel followed by Western blotting with rabbit anti-phosphoserine antibody (Zymed Laboratories Inc.) or the total Smad1 antibody (Cell Signaling).
Immunohistochemistry (IHC) and in Situ Hybridization
Hind limbs were collected from C57BL/6 mouse embryos fixed for 24 h in 4% paraformaldehyde. Limbs were embedded in paraffin, and 5-μm sections were obtained. Tissue sections were deparaffinized in xylene and rehydrated through an ethanol series for use in either IHC or in situ hybridization. For IHC, tissue sections were subjected to microwave antigen retrieval in 10 mM EDTA (pH 7.5) at 93 °C for 7 min and allowed to cool for at least 2 h. Sections were blocked with normal horse serum (for GADD45β IHC) or normal swine serum (for Runx2 IHC). IHC for GADD45β was performed as described previously (41), using goat polyclonal anti-GADD45β (sc-8776, Santa Cruz Biotechnology; 1:400 dilution, 0.5 μg/ml final concentration), rabbit biotinylated anti-goat IgG (Sigma), and the Vectastain Elite ABC kit (Vector Laboratories, Inc.). Anti-Runx2 antibody (PEBP2αA, sc-10758, Santa Cruz Biotechnology; 1:25 dilution, 8 μg/ml final concentration), swine biotinylated anti-rabbit F(ab)2, and streptavidin/horseradish peroxidase-conjugated (DAKO, Denmark) were used to detect Runx2 by IHC. Sections were counterstained with hematoxylin. For negative controls, normal goat IgG (sc-2028) and normal rabbit IgG (sc-2027) were used in place of the primary antibodies against GADD45β and Runx2, respectively. We optimized the staining methods for detection of cytoplasmic and nuclear staining with each antibody by testing both microwave and enzyme retrieval of each antigen.
For in situ hybridization, a 391-bp fragment of human GADD45β cDNA (sense, 5′-CTGGTTGTTGCCCCGGCTTTCTTC-3′; anti-sense, 5′-CGCGGTGGAGGAGCTTTTGGTG-3′) and a 1006-bp fragment of mouse MMP-13 cDNA (sense, 5′CATCCACATGGTTGGGAAGTTCTG-3′;antisense,5′-CATTCAGCTATCCTGGCCACCTTC-3′) were obtained by reverse transcription-PCR and subcloned in the pCR II vector (Invitrogen). Plasmids were linearized using either HindIII or NotI for GADD45β and either KpnI or XhoI for MMP-13, respectively. The linearized plasmids served as templates for riboprobe synthesis using either T7 or SP6 RNA polymerases and digoxigenin-UTP RNA labeling nucleotide mix (Roche Applied Science) according to the manufacturer’s instructions. The cDNA encoding type X collagen was a kind gift of Dr. Bjorn Olsen, Harvard Medical School, Boston. Hybridization was performed according to the method described previously (42).
Analysis of Gadd45β−/− Mice
Gadd45β−/− mice were generated on the C57BL/6 background, as described previously (43). Briefly, the targeting vector was constructed to replace exons 3 and 4, encoding the region crucial for interaction with MTK-1/MEKK4 (44), with the neomycin resistance cassette. Gadd45β−/− embryos and wild type littermates at 14.5, 15.5, 16.5, and 17.5 days post-conception (dpc), generated with the approval of the University of Pittsburgh Institutional Animal Care and Use Committee, were fixed in 4% paraformaldehyde/phosphate-buffered saline overnight at 4 °C and stored in 70% ethanol at room temperature. Whole embryos were stained with Alcian Blue and Alizarin Red. Long bones were sectioned and stained with toluidine blue or von Kossa or analyzed by in situ hybridization as described above.
Plasmids
The human GADD45β promoter fragment spanning −1604 to +141 was prepared by PCR using human genomic DNA (Clontech) as template. The PCRs were carried out using the Pfu Turbo DNA polymerase (Stratagene), and the products were subcloned in the pGL2-basic vector (Promega, Madison WI) and verified by DNA sequencing. Smad1, Smad4, and alk6 expression vectors were gifts from Dr. Xu Cao (University of Alabama). The Runx2 and ΔRunx2 expression vectors were described previously (45). The pcDNA3-GADD45β-FLAG plasmid was generated as described previously (40). The pAP-1 Luc plasmid was obtained from Stratagene. The MMP-13 promoter sequence spanning −1007/+26 bp was prepared by PCR using a pCAT-MMP-13 promoter construct5 as a template, and cloned in the pGL2-B vector in the XmaI and XhoI sites. Expression vectors for JunD, JunB, Fra1, and Fra2 were kindly provided by Dr. Paul R. Dobner (University of Massachusetts Medical Center, Worcester, MA). The dominant negative JunD was described previously (46).
Transient Transfections and Luciferase Assay
Transient transfection experiments were performed in C-28/I2 cells and ATDC5 cells using Lipofectamine Plus (Invitrogen), as described (33, 40), but in 12-well plates. Cells were plated at 100,000 cells per well and transfected with 300 ng of pGL2-promoter plasmid and 10–100 ng of expression plasmid. Luciferase activity was measured 24 h later using the dual-luciferase reporter assay system (Promega). Cotransfection with pRL-TK Renilla luciferase control vector was used for controlling transfection efficiency. Cotransfections of the promoter-less pGL2-B vector with expression plasmids did not significantly increase activity compared with promoter-containing vectors. This is in contrast to a study showing that 1 μg of Runx2 expression vector increased luciferase activity when cotransfected with 1 μg of promoter-less pGL2 or pGL3 vector in cells plated in 6-well plates (47). Transfections were performed in triplicate, and experiments were repeated three times with different plasmid preparations with similar results.
Knockdown of GADD45β Expression by siRNA
Two approaches were used. First, chemically synthesized complementary RNA oligonucleotides (siRNA-GADD45β sense-strand sequence, 5′-AAGTTGATGAATGTGGACCCA) were annealed, deprotected, and desalted as recommended by the manufacturer (Qiagen). A total of 50 μM RNA duplexes was transfected into cells using TKO transfection reagent (Mirus, Madison, WI) and tested for specificity and efficiency, as described previously (40). Second, a lentiviral vector containing siRNA-GADD45β was constructed using single 83-mer oligonucleotides containing an XbaI site at the 5′ end, an intermediate short spacer, and a partial sequence of the H1-RNA promoter at the 3′ end, as described previously (40). Standard PCR procedures (Advantage 2 PCR kit, Clontech) were performed using specific siRNA oligonucleotides and T3 primer plus pSuper-like plasmid as template to provide H1-mediated siRNA cassettes with an additional XbaI site at the 3′ end (48). PCR products were purified (Qiagen), digested with XbaI, and cloned into the 3′-long terminal repeat NheI site of a CMV-GFP lentiviral vector as described (48). The LV-siRNAGFP construct (control) was kindly donated by Dr. Oded Singer (Salk Institute). The following siRNA oligonucleotide for GADD45β was used, 5′-CTGTCTAGACAAAAAGTTGATGAATGTGGACCCAtctcttgaaTGGGTCCACATTCATCAACGGGGATCTGTGGTCTCATACA-3′. Vesicular stomatitis virus G envelope protein-pseudotyped lentiviruses were prepared and purified as described previously (48–50). Vector concentrations were analyzed by immunocapture p24 gag enzyme-linked immunosorbent assay (Alliance, PerkinElmer Life Sciences) (49). The C-28/I2, ATDC5, or murine epiphyseal chondrocytes were plated in monolayer at 25,000 cells/cm2 for 1 or 2 days, followed by infection with LV-siRNA-GFP or LV-siRNA-GADD45β at a multiplicity of infection of 10. In tests for specificity and efficiency, cells were transfected with GFP-FLAG and GADD45β-FLAG. Cells infected with LV-siRNA-GFP showed no staining for GFP, whereas GFP staining was not affected in cells infected with LV-siRNA-GADD45β. On Western blots, transfected GADD45β-FLAG was detected on Western blots using anti-FLAG antibody in cells infected with LV-siRNA-GFP, but not in cells infected with LV-siRNA-GADD45β. (see supplemental Fig. S1).
EMSA and Supershift Assay
Whole cell extracts were made from uninfected, LV-siRNA-GFP-infected (GFP KD), or LV-siRNA-GADD45β-infected (GADD45β KD) C-28/I2 cells, and DNA binding reactions and EMSAs were performed using a 32P-labeled oligonucleotide containing the AP-1 consensus (Santa Cruz Biotechnology), as described (33, 46). Extracts from DU145 cells were used as a positive control for activated AP-1 (data not shown) (46). For supershift analysis, the cell extracts were preincubated with specific antibodies against different members of the AP-1 family (Santa Cruz Biotechnology) for 20 min before the incubation with labeled probe for an additional 20 min (46).
Analysis of Protein Kinases
To examine protein kinase activities, the C-28/I2 cells without or with siRNA-GADD45β were transfected with pcDNA3 expression vectors encoding control-FLAG or GADD45β-FLAG. After transfection, the cells were cultured for 12 h with DMEM/F-12, 1% Nutridoma, followed by addition of BMP-2 (100 ng/ml) and further incubation for 12 h before collection of total cell lysates. Western blots were performed using antibodies against JunD (Santa Cruz Biotechnology) and phospho-c-Jun (Ser-73) (Cell Signaling) according to the manufacturers’ protocols. PD98059 (10 μM), SB203580 (10 μM), or SP600125 (100 μM) was added to cultures 1 h before BMP-2. JNK, ERK and p38 kinase activities were measured by using the SAPK/JNK, p44/42, and p38 MAPK assay kit (Cell Signaling) according to the manufacturer’s protocol.
RESULTS
Specific Up-regulation of GADD45β by BMP-2 in Human Chondrocytes
To characterize the genes involved in the early response to BMP-2 in chondrocytes, we performed oligonucleotide microarray-based gene expression profiling using RNA isolated from the immortalized human chondrocyte cell line, C-28/I2, after treatment with BMP-2 for 1 h. BMP-2 induced distinct patterns of coordinated changes in a wide range of transcripts, which encode cell cycle-related and anti-apoptotic molecules and transcription factors that are known to regulate growth and survival cascades. Identification of highly expressed genes induced by BMP-2 (more than 2 times >control) yielded 249 transcript variants. Actin-related protein 2/3 complex subunit 4 was the most highly expressed gene stimulated by BMP-2 (18.4 times). Analysis of gene ontology molecular function revealed 31 transcription factors, including ZNF238 (7.5 times), JunD (5.3 times), JunB (4 times), ID3 (4 times), and DLX2 (3.7 times) and 11 kinases, such as phosphatidylinositol 3-kinase regulatory subunit 3 (7.5 times), AKT2 (6.5 times), TK 2 (4 times), and AXL (3 times), which were induced by BMP-2 after 1 h. A complete set of the microarray data is provided as supplemental material. GADD45β, which was among the most highly up-regulated BMP-2-induced genes, was detected by all three representative probe sets present on the Affymetrix GeneChip Human Genome U133A, whereas GADD45α and GADD45γ transcripts were not altered by the treatment with BMP-2 (TABLE ONE). Real time PCR corroborated the microarray data showing that BMP-2 at concentrations of 50 and 100 ng increased GADD45β mRNA (Fig. 1A). Other growth and differentiation factors, including epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), and insulin-like growth factor (IGF)-I, had no effect (Fig. 1A), indicating the specificity of the BMP-2 response. Real time PCR also confirmed that BMP-2 had no effect on GADD45α or GADD45γ mRNA (Fig. 1, B and C). Furthermore, the peak of induction was 1 h after the addition of BMP-2 with a gradual decline thereafter, such that there was no difference between treated and untreated at 24 h (Fig. 1D). Finally, the BMP-2-induced increase in GADD45β mRNA was confirmed in primary human articular chondrocytes (Fig. 1E).
TABLE ONE.
Microarray analysis of GADD45 family genes in chondrocytes treated with BMP-2
Subconfluent cultures of the immortalized human chondrocyte cell line, C-28/I2, were incubated for 1 h in the absence (control) or presence of 100 ng/ml of BMP-2. Total RNA was applied to the HU133A Affymetrix GeneChip.
| Probe id | Gene | Control | BMP-2 | FC | LCB | Rank |
|---|---|---|---|---|---|---|
| 207574_s_at | GADD45β | 285.86 | 832.17 | 2.91 | 2.71 | 13 |
| 209304_x_at | GADD45β | 420.60 | 1051.55 | 2.5 | 2.26 | 31 |
| 209305_s_at | GADD45β | 266.40 | 668.53 | 2.51 | 2.2 | 37 |
| 203725_at | GADD45α | 529.55 | 512.18 | −1.03 | −1 | NA |
| 204121_at | GADD45γ | 221.52 | 251.23 | 1.13 | 1.04 | NA |
Probe identification (id), Affymetrix identifier for separate probes on the microarray; FC, fold change between control and BMP-2-treated sample; LCB, lower confidence bound; NA, not applicable.
FIGURE 1. Specific up-regulation of GADD45β mRNA by BMP-2 in chondrocytes.

Monolayer cultures of the human chondrocyte cell line C-28/I2 were treated as indicated, and total RNA extracts were analyzed by real time PCR. Each value was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the same sample and shown as the mean ± S.D. of triplicate cultures. A, GADD45β mRNA levels were increased in cultures of the human chondrocyte cell line, C-28/I2, after treatment with BMP-2 for 1 h but not with EGF, bFGF, or IGF-I. B and C, incubation of C-28/I2 cells with BMP-2 for 1 h did not increase the levels of GADD45α or GADD45γ mRNA. D, time course of BMP-2-induced GADD45β mRNA in C-28/I2 cells showed transient induction with a peak at 1 h. E, treatment of primary human articular chondrocytes with BMP-2 for 1 h increased the levels of GADD45β mRNA by 2-fold.
Induction of GADD45β Promoter Activity by BMP-2 in Chondrocytes Requires Signaling by Smad1 Interacting with Runx2
Because BMP-2 is known to regulate gene expression by Runx2-mediated recruitment of Smad 1 to subnuclear sites (51, 52), we examined GADD45β promoter activity in the C-28/I2 cells after BMP-2 treatment to determine the consequence of the direct cooperation between Runx2 and Smad1 in this model. After transfection of a luciferase reporter vector driven by the GADD45β promoter sequence spanning −1604 to +141 bp, BMP-2 increased promoter activity by around 2-fold (Fig. 2A). Overexpression of alk6, the BMP type IB receptor, activated GADD45β promoter activity to a similar extent as BMP-2. Cotransfection with Smad1 or Runx2 alone further enhanced the BMP-2-induced activation of the GADD45β promoter. In contrast, Smad6, which is known to repress transcriptional activity by competing with Smad4 for binding to Smad1, and ΔRunx2 inhibited GADD45β promoter activation by Smad1 and Runx2, respectively (Fig. 2A). To confirm the Runx2-Smad1 interaction in this model, we examined Smad1 and Runx2 phosphorylation in C-28/I2 chondrocytes treated with BMP-2. As shown in Fig. 2, B and C, both Runx2 and Smad1 were phosphorylated after treatment with BMP-2 for 1 h. The Runx2 interaction with Smad1 was further investigated by coimmunoprecipitation of total cell lysates with anti-Runx2 and Western blotting with anti-Smad1. As shown in Fig. 2C, Western blotting with anti-Smad1 detected a single BMP-2-induced band after immunoprecipitation with anti-Runx2. Together, these data indicate that the induction of GADD45β by BMP-2 in chondrocytes is mediated by Smad1-dependent signaling and that Runx2 contributes to the full induction.
FIGURE 2. The induction of GADD45β promoter activity by BMP-2 requires signaling by Smad1 interacting with Runx2.
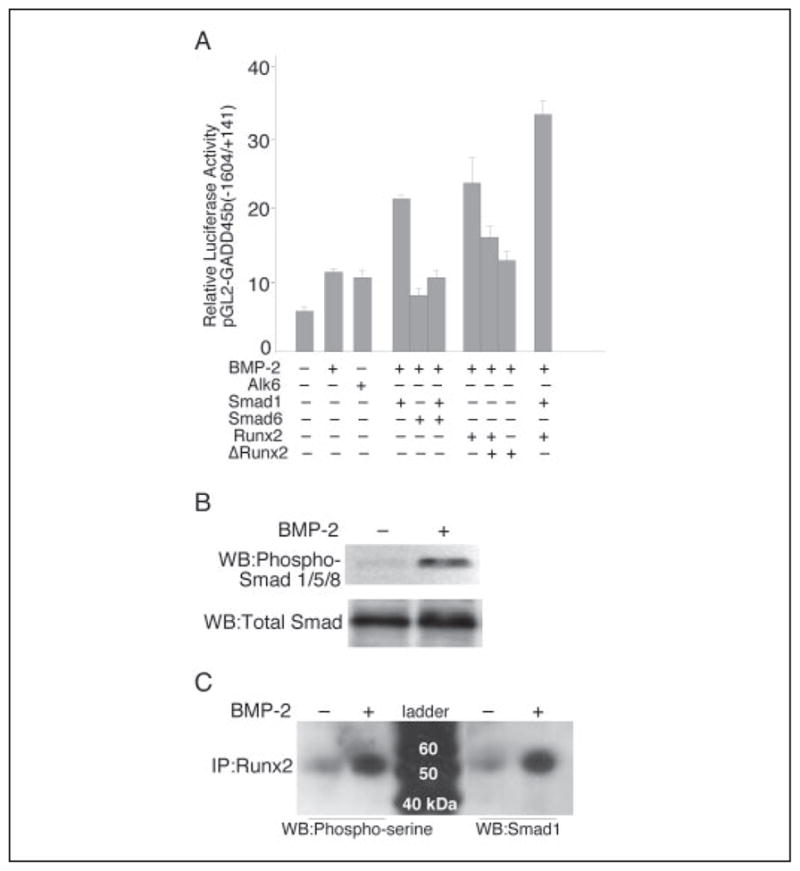
A, the pGL2-GADD45β promoter (−1604/+141 bp) was cotransfected with expression vectors encoding the BMP type IB receptor (alk6), Smad1, or Smad6 alone or in combination, or Runx2 or ΔRunx2 alone or in combination. The results show the means ± S.D. of determinations of samples from three or more separate wells. B, the C-28/I2 cells were incubated without (−) or with (+) BMP-2 for 1 h, and total lysates were analyzed on Western blots (WB) using antibodies against phospho-Smad1/5/8 and total Smad1. C, total cell lysates were subjected to immunoprecipitation (IP) with anti-Runx2 and analyzed on Western blots using anti-phosphoserine or Smad1 antibody.
Expression of GADD45β in the Mouse Embryonic Growth Plate
Because Runx2 is expressed in the growth plate as a major transcriptional regulator of chondrocyte terminal differentiation, we asked whether GADD45β is also expressed during skeletal development. Therefore, we performed in situ hybridization and immunohistochemical assays in the mouse embryonic limb. We detected expression of Gadd45β mRNA at 14.5 (data not shown) and 15.5 dpc (Fig. 3, A and B) primarily in prehypertrophic chondrocytes, where Runx2 protein was localized most prominently in the nucleus (Fig. 3C). On the other hand, immunoreactive GADD45β protein was detected most strongly in the nuclei of intact hypertrophic chondrocytes (Fig. 3, D, F, and J). Runx2 persisted through this region into the late hypertrophic zone of the femur (Fig. 3C), where Mmp-13 mRNA was also detected (Fig. 3, H and K). As shown in Fig. 3 (H and K), Mmp-13 mRNA colocalized with nuclear GADD45β protein in the late hypertrophic zone of the tibia. These data demonstrate that Gadd45β mRNA is induced in prehypertrophic chondrocytes coincident with prominent nuclear Runx2 protein, whereas GADD45β protein accumulates in the nuclei of hypertrophic chondrocytes in the region destined to undergo ossification, where Mmp-13 mRNA is also expressed.
FIGURE 3. Expression of Gadd45β mRNA together with Runx2 protein in prehypertrophic chondrocytes and intracellular GADD45β protein in the hypertrophic chondrocytes that express Mmp-13 mRNA.

Adjacent longitudinal sections from mouse embryonic femurs or tibiae (15.5 dpc) were analyzed by immunohistochemistry and in situ hybridization to illustrate the expression of GADD45β in relation to Runx2 protein and MMP-13 mRNA in proliferating (pro), prehypertrophic (pre-hy), and hypertrophic (hyp) zones indicated in A. Antisense Gadd45β (A and B) and anti-GADD45β antibody (D) were used to detect Gadd45β mRNA, and protein and anti-Runx2 antibody (C) detected Runx2 protein in mouse femurs. Sense Gadd45β (insets, A and B), normal goat IgG (inset, C), and normal rabbit IgG (inset, D) were applied as negative controls. Tibiae at 15.5 dpc were stained with toluidine blue (T.Blue, E). GADD45β protein was detected in late stage hypertrophic chondrocytes (F and J), in which Mmp-13 mRNA was also detected using antisense MMP-13 (H and K). Normal goat IgG (G) and sense Mmp-13 (I) were applied as negative controls. Brown staining indicates immunodetectable protein, and purple staining indicates a positive signal using antisense probe. Original magnification: A, E–I, ×40; B–D, F, and G, ×200.
Defective Mineralization and Decreased Bone Growth Accompany Decreased Mmp-13 mRNA Expression in Hypertrophic Chondrocytes of the Gadd45β−/− Mouse Embryo
To determine whether GADD45β has a crucial function in hypertrophic chondrocytes during endochondral ossification, we performed histological analysis of embryonic limbs from Gadd45β−/− mice. At 15.5 dpc, the tibiae showed the first signs of the formation of a primary ossification center in wild type (WT) mice (Fig. 4A). Mineralization of the extra-cellular matrix (ECM) in WT mice was clearly identified by von Kossa staining (Fig. 4B). Mmp-13 mRNA-expressing hypertrophic chondrocytes were distributed throughout this zone (Fig. 4, C and G). On the other hand in Gadd45β−/− mice, the primary ossification center was decreased in size (Fig. 4D). Mineralization of the ECM was defective, as shown by the decreased von Kossa staining (Fig. 4E), but persisted in the surrounding bone collar, and the number and distribution of Mmp-13 mRNA-expressing hypertrophic chondrocytes were markedly diminished (Fig. 4, F and H).
FIGURE 4. Defective mineralization accompanies decreased Mmp-13 mRNA expression in hypertrophic chondrocytes of Gadd45β−/− mouse embryos at 15 dpc.
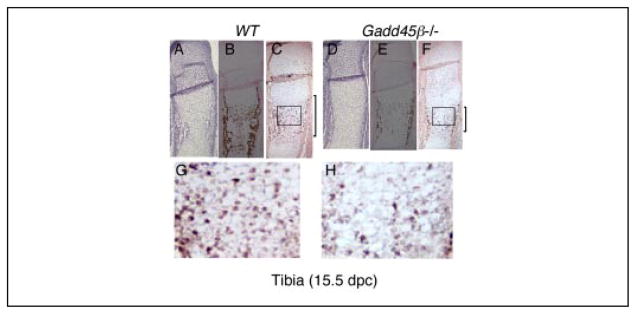
Tibiae from WT and Gadd45β−/− mouse embryos at 15.5 dpc were examined by toluidine blue staining (A and D), von Kossa staining (B and E), and in situ hybridization for Mmp-13 mRNA (C, F, G, and H). Original magnifications: A–F, ×40; G and H, ×200.
The defective mineralization and deficient Mmp-13 mRNA expression at the site of the primary ossification center at 15 dpc preceded the decreased bone growth observed in Gadd45β−/− mouse embryos at 16.5 dpc (Fig. 5). A delay in mineralization, indicated by Alizarin Red staining was noted in the ribs of Gadd45β−/− embryos (arrows, Fig. 5A) and in both fore and hind limbs (Fig. 5, B and C). The hypertrophic zone of the tibia of the Gadd45β−/− embryo was decreased in length compared with WT (Fig. 5D). Decreased expression of type X collagen (Col10a1) mRNA, a specific marker of the hypertrophic phenotype (14), throughout the hypertrophic zone accompanied the markedly decreased Mmp-13 mRNA expression in the late hypertrophic zone (Fig. 5E). These data suggest that GADD45β is involved in terminal differentiation of hypertrophic chondrocytes and may contribute to Mmp-13 gene regulation.
FIGURE 5. Gadd45β−/− mouse embryos at 16. 5 dpc exhibit decreased bone growth associated with decreased levels of Mmp-13 and Col10a1 mRNA in hypertrophic chondrocytes.

Whole mounted embryos at 16.5 dpc were stained with Alcian Blue and Alizarin Red to show decreased cartilage matrix and mineralization in ribs (A, arrowheads), forearms (B), and hind limbs (C) of Gadd45β−/− compared with WT mice. Adjacent longitudinal sections from the tibia at 16.5 dpc showed a smaller hypertrophic zone by toluidine blue (T.Blue) staining (D) accompanied by markedly decreased levels of Mmp-13 mRNA (E, arrowhead) and Col10a1 mRNA in Gadd45β−/− mouse embryos compared with WT. Original magnifications: D, ×40; E, ×200.
Requirement of GADD45β for Expression of Mmp-13 mRNA during Hypertrophic Chondrocyte Differentiation in Vitro
To confirm the results in Fig. 5 and to determine the extent of dependence of Mmp-13 gene expression in hypertrophic chondrocytes upon GADD45β, we used murine rib growth plate chondrocytes in three-dimensional pellet culture, an established model in which terminal chondrocyte differentiation occurs spontaneously (35). In control GFP knockdown (KD) cultures infected with a lentiviral vector encoding siRNA-GFP, chondrocytes with hypertrophic morphology were present at days 14 and 21 and were surrounded by matrix that stained metachromatically with toluidine blue (pink staining in matrix in Fig. 6A), evidence of the early proteoglycan deposition that occurs in this model. Strong expression of Mmp-13 and Col10a1 mRNA was confirmed by in situ hybridization in the GFP KD cultures at day 21 (Fig. 6B). In contrast, GADD45β KD cells, which were infected with a lentivirus encoding siRNA-GADD45β, did not develop hypertrophic morphology or increase metachromatic staining with toluidine blue even after 21 days of culture (Fig. 6A). Furthermore, in situ hybridization analysis showed that the levels of Mmp-13 mRNA, as well as Col10a1 mRNA, specific marker of the hypertrophic phenotype, were decreased markedly in the GADD45β KD cells compared with the GFP KD cells (Fig. 6B). These results support the in vivo data, where GADD45β expression is restricted in a temporal-spatial manner, and suggest that GADD45β is an essential regulator of chondrocyte terminal differentiation that may influence, directly or indirectly, Mmp-13 gene expression during hypertrophy.
FIGURE 6. GADD45β is required for hypertrophic chondrocyte differentiation in vitro and associated expression of Mmp-13 and Col10a1 mRNA.

A, murine rib growth plate chondrocytes were infected with lentivirus encoding siRNA-GFP to generate GFP KD cells or with siRNA-GADD45β to generate GADD45β KD cells and cultured as three-dimensional pellets for 3, 14, and 21 days. Toluidine blue staining showed the progressive hypertrophy in cultures of GFP KD chondrocytes but not in the cultures of GADD45β KD chondrocytes. Magnification is ×40. B, after 21 days of culture, toluidine blue (T.Blue) staining and the levels of Col10a1 and Mmp-13 mRNA, detected by in situ hybridization using antisense probes, were markedly decreased in GADD45β KD compared with GFP KD cells. Magnification is ×200.
Regulation of AP-1 Activity by GADD45β in Chondrocytes
Because members of the AP-1 family of transcription factors are known to be involved in MMP-13 gene regulation (25), we examined whether GADD45β could regulate AP-1 activity in chondrocytes. We used the C-28/I2 cell line to generate sufficient numbers of lentivirally transduced cells for reproducible transient expression assays and for nuclear extracts for EMSA. Cotransfection GADD45β-FLAG together with the cis-reporter plasmid containing AP-1-responsive elements resulted in concentration-dependent transactivation (2–3-fold increase) of the AP-1-dependent reporter (Fig. 7A). Transduction of siRNA-GADD45β decreased constitutive AP-1 activity by around 25% and blocked the GADD45β-FLAG-dependent activation to the control level (Fig. 7B). EMSAs were also performed using a radiolabeled probe containing the AP-1 consensus binding site and whole cell extracts from cells infected with lentiviral siRNA-GADD45β and transfected with GADD45β-FLAG. As shown in Fig. 7C, the low level of AP-1 binding activity was increased by overexpression of GADD45β. Overexpression of GADD45β further increased AP-1 binding activity in GFP KD cells, but not in GADD45β KD cells, which showed decreased AP-1 binding activity compared with GFP KD cells. The AP-1 binding activity due to endogenous GADD45β was also decreased in GADD45β KD cells in the absence of GADD45β-FLAG. These results correspond directly to the GADD45β-dependent AP-1 promoter activity shown in Fig. 7, A and B.
FIGURE 7. GADD45β positively regulates AP-1 activity and JunD/Fra2 binding in chondrocytes.

The AP-1-driven luciferase reporter vector (pAP-1-Luc) was cotransfected in C-28/I2 cells with the GADD45β expression vector (GADD45β-FLAG) at 50 and 100 ng/ml (A) or with GADD45β-FLAG alone or together with siRNA-GADD45β (B). C, extracts were prepared from uninfected C-28/I2 cells (1st and 2nd lanes), cells infected with lentiviral siRNA-GFP (GFP KD; 3rd and 5th lanes), or siRNA-GADD45β (GADD45β KD; 4th and 6th lanes). Cells were then transfected with GFP-FLAG (control) or GADD45β-FLAG, as indicated. AP-1 binding activity was examined by EMSA using the double-stranded AP-1 consensus oligonucleotide as labeled probe. The cells extracted for the 5th and 6th lanes (lower exposure of gel shift using probe labeled at different time) were not transfected to show that decreasing endogenous GADD45β also decreases AP-1 binding activity. D, supershift analysis of binding activities in GADD45β-expressing C-28/I2 cells was performed using antibodies against different AP-1 family members. Note that the Fra1, JunB, and JunD antibodies produce supershifts, whereas the Fra2 antibody produces a block shift. E, the C-28/I2 cells were cotransfected with pAP-1-Luc together with expression vectors encoding Fra1, Fra2, JunD, and JunB alone or in combination in the absence or presence of the GADD45β-FLAG. Luciferase activities are shown as means ± S.D. of replicates from representative transfections. *, p < 0.05; **, p < 0.01; by analysis of variance with subsequent Dunnett test in multiple comparisons. Comparisons in E are with respective controls containing single expression vectors.
To characterize further the AP-1 binding activity in extracts from GADD45β-expressing chondrocytes, we performed supershift analyses using antibodies that recognize various members of the Jun and Fos families. JunD, JunB, Fra1, and Fra2 were identified as the predominant proteins present in the AP-1-binding complex (Fig. 7D). Because Jun proteins are known to bind to AP-1 sites as heterodimers with Fos proteins such as Fra1 or Fra2, we assessed the AP-1 reporter activity after cotransfection with JunD, JunB, Fra1, and Fra2 expression vectors alone and in various combinations (Fig. 7E). Each expression vector alone was ineffective as an activator, whereas JunD/Fra1 and JunD/Fra2 increased AP-1 reporter activity by around 5-fold, and JunB/Fra1 and JunB/Fra2 increased activity by 2–2.5-fold. Most interestingly, cotransfection with GADD45β in the presence of JunD, which can form homodimers, increased AP-1 reporter activity by more than 4-fold, whereas the activity in the presence of each of the other factors alone or in combination increased 2-fold above that in control-FLAG cells (Fig. 7E). These results suggest that JunD is a major component of the AP-1 activity induced by GADD45β in chondrocytes.
JNK Activity Is Necessary for JunD Activation by GADD45β
To unravel the mechanism of JunD activation by GADD45β, we evaluated the phosphorylation status of JunD in GADD45β-expressing and GADD45β KD chondrocytes. We analyzed cell extracts from C-28/I2 cells for kinase activation using antibodies against phospho-c-Jun, phospho-Elk-1, and phospho-ATF-2, specific substrates for JNK, ERK, and p38, respectively. As shown in Fig. 8A, transduction of GADD45β induced JunD phosphorylation at serine 100, which is reported as a direct JNK target residue (53). The phosphorylation was completely blocked in the presence of the JNK inhibitor, SP600125, but not by the ERK inhibitor, PD98059, or by the p38 inhibitor, SB203580. Transduction of siRNA-GADD45β blocked the GADD45β-dependent JunD phosphorylation (Fig. 8A). JNK activity, detected as phospho-c-Jun, was clearly increased in GADD45β-expressing cells and siRNA-GADD45β blocked its activity, whereas ERK was activated only slightly by GADD45β, and p38 activity was not altered (Fig. 8B). These results are consistent with previous studies reporting that GADD45 family members are upstream activators of the JNK pathway (2, 54–56).
FIGURE 8. JNK activity is necessary for JunD activation by GADD45β.

A, extracts from C-28/I2 cells expressing GADD45β-FLAG or siRNA-GADD45β were analyzed by Western blotting using antibodies against phosphorylated (phospho) or total JunD. GADD45β-expressing cells were also treated with inhibitors of ERK (PD98059), p38 (SB203580), and JNK (SP600125) to examine the roles of the different kinases in JunD phosphorylation by GADD45β. B, kinase activities were analyzed on Western blots to show increased JNK (phospho-c-Jun) and ERK (phospho-Elk) activities in GADD45β-FLAG cells and decreased activities in cells expressing siRNA-GADD45β. Note that p38 activity (phospho-ATF-2) did not change.
JunD-dependent MMP-13 Promoter Activation by GADD45β in ATDC5 Cells and Synergism with Runx2
To determine the functional consequences of GADD45β-induced AP-1 transcriptional activity in hypertrophic chondrocytes, we investigated the response of the MMP-13 promoter using ATDC5 cells, a well characterized chondrogenic cell line that undergoes terminal differentiation to the hypertrophic phenotype in vitro (34). Because MMP-13 gene transcription is known to involve Runx2 and AP-1 (25–27), we performed cotransfection experiments using FLAG-GADD45β, Runx2, and JunD expression vectors in various combinations with the pGL2-MMP-13 reporter vector containing promoter sequences spanning −1007 to +26 bp. As shown in Fig. 9A, overexpression of either GADD45β or Runx2 up-regulated MMP-13 promoter activity by 2.5–3-fold. Cotransfection with both Runx2 and GADD45β expression vectors enhanced promoter activity by around 4-fold (Fig. 9A). Furthermore, transduction of siRNA-GADD45β reduced MMP-13 promoter activity to 25% of the constitutive activity in ATDC5 cells (Fig. 9B).
FIGURE 9. JunD-dependent MMP-13 promoter activation by GADD45β in cooperation with Runx2 in the ATDC5 chondrogenic cell line.

The ATDC5 cells were cotransfected with the pGL2-MMP-13 promoter (−1007 bp/+26 bp) construct and expression vectors encoding GADD45β and Runx2 (A), siRNA-GADD45β (B), Runx2 and JunD (C), or GADD45β and dominant negative (DN) JunD (D). The results show the means ± S.D. of determinations in triplicate wells from representative experiments. Empty expression vector controls or promoter-less pGL2-B did not induce or express any significant luciferase activity (not shown).
To evaluate further the contribution of JunD in the GADD45β- and Runx2-dependent MMP-13 promoter activation, we performed cotransfections with expression vectors encoding JunD or dominant negative JunD. Most strikingly, JunD mimicked the GADD45β-dependent up-regulation of the MMP-13 promoter, enhancing the basal or Runx2-dependent activity by 2-fold (Fig. 9C), and dominant negative-JunD down-regulated the GADD45β-dependent activity (Fig. 9D). Similar results were obtained using the C-28/I2 cells, which express higher basal MMP-13 activity but with similar responses to transfected expression vectors. These results indicate that Mmp-13 gene expression in terminally differentiating chondrocytes is predominantly a result of JunD activation by GADD45β in cooperation with Runx2.
DISCUSSION
In this study, we have highlighted a new function of GADD45β, a member of a group of stress-response proteins that are associated with growth arrest and cell survival. We initially identified GADD45β as a candidate target of BMP-2 using microarray analysis. Because BMP-2 is known to be involved in hypertrophic differentiation of chondrocytes, we asked whether GADD45β could play a role during endochondral ossification in the mouse embryo. Thus, we made the novel observation that Gadd45β mRNA is expressed by pre-hypertrophic chondrocytes in the mouse embryonic growth plate, where Runx2 is also detected, whereas GADD45β protein accumulates in the nuclei of chondrocytes of the hypertrophic zone, where Runx2 protein also persists, prominently in late hypertrophic chondrocytes expressing Mmp13 mRNA prior to mineralization. In further studies, we found that GaddD45β−/− mice embryos display defective mineralization accompanied with a decreased number of Mmp-13- and Col10a1-expressing hypertrophic chondrocytes with the consequence of decreased bone growth. By using models of chondrocyte hypertrophic differentiation in vitro and siRNA technology, we provide clear evidence to support an essential role for GADD45β in the regulation of Mmp-13 gene expression. Finally, we reveal that GADD45β-mediated up-regulation of MMP-13 promoter activity is because of an increase in AP-1 activity through the JNK/JunD pathway and that Runx2 cooperates with GADD45β and JunD to enhance this response.
The BMPs along with other growth and differentiation factors are responsible for initiating and directing cartilage morphogenesis in the embryonic limb and generally set the stage for bone morphogenesis (31, 57–60). We initiated this study by examining directly the global responses of chondrocytes to BMP-2 to profile on microarrays the early induced genes that are likely to play regulatory roles. Many of these gene products are already known as BMP-induced transcription factors and signaling kinases, including JunB, JunD, ID3, and DLX2 (61–64). A recent report indicates that phosphatidylinositol 3-kinase-AKT signaling is involved Runx2-induced chondrocyte and osteoblast differentiation (65). The markedly enhanced expression of GADD45β on these arrays and our recent findings on the differential roles of GADD45 family members in apoptosis and tumor growth (40) led us to ask whether GADD45β may play a role in chondrocyte differentiation. Thus, we report for the first time that Gadd45β mRNA is expressed by pre-hypertrophic chondrocytes and that the protein accumulates in hypertrophic chondrocytes in the mouse embryonic growth plate.
Runx2, or Cbfa1, belongs to the family of Runt domain genes, which play fundamental roles in organ development and cell differentiation (17), and together with SMAD proteins is involved in the regulation of phenotypic gene expression during osteogenesis (51). We demonstrate in our study that GADD45β promoter activity is regulated by Smad1-dependent signaling in cooperation with Runx2. Previous studies have focused on regulation by NF-κB of GADD45β gene expression, which is induced by interleukin (IL)-6, IL-1, TGF-β, TNF-α, IL-18, and genotoxic and oxidative stress in various cell types (44, 66). Three κB elements at positions −447/−438 bp, −426/−417 bp, and −377/−368 bp are responsive to the NF-κB subunit RelA (66). Yoo et al. (4) suggested the existence of a TGF-β-responsive sequence within the GADD45β −220/−1-bp promoter region, and potential SMAD enhancesomes have been identified in both the murine and human promoters (67). Recent studies suggest that both SMAD-dependent and -independent mechanisms could be involved in BMP-2-induced GADD45β expression (68, 69). Thus, analysis of the GADD45β promoter will require a more extensive study in our chondrocyte models.
Regarding the specific function of Runx2 during endochondral ossification, it serves as a positive regulatory factor in chondrocyte maturation to the hypertrophic phenotype (15, 19, 20) and is expressed in the adjacent perichondrium and in prehypertrophic chondrocytes but less in late hypertrophic chondrocytes (28, 29), overlapping with Col10a1 (70, 71). Our findings indicate that Runx2 plays a synergistic role in both the induction and activity of GADD45β by enhancing BMP-2-induced Smad1 signaling and AP-1-dependent MMP-13 gene expression. The distinct spatial localization of Gadd45β mRNA compared with the accumulated intracellular protein in the late hypertrophic zone suggests that it acts in a temporal-spatial manner to regulate the duration of chondrocyte terminal differentiation.
In studies using Mmp-13−/− mice, MMP-13 deficiency results in significant interstitial collagen accumulation leading to the delay of endochondral ossification in the growth plate and increased length of the hypertrophic zone (23, 24). In Gadd45β−/− mice, we observe defective mineralization at 15.5 and 16.5 dpc persisting at least until 18.5 dpc (data not shown), but decreased length of the hypertrophic zone. This discrepancy may be due to regulation by GADD45β of other activities in hypertrophic chondrocytes, but also the selective loss of MMP-13 in the hypertrophic zone of the Gadd45β−/− growth plate appears to affect the late stage of terminal differentiation only. Furthermore, GADD45β is not the only determinant of the expression of MMP-13, which is expressed also in osteoblasts, osteoclasts, and periosteal cells below the inner periosteal region of ossification (72). Consistent with the persistent mineralization in the bone collar surrounding the Gadd45β−/− growth plate, abundant tartrate-resistant acid phosphatase (TRAP)-positive osteoclast-like cells were present in the diaphyseal collar in Gadd45β−/− mice.6
In contrast to Mmp-13 knock-out mice but similar to the Gadd45β knock-out mice, both Col10a1 knock-out mice and transgenic mice with a dominant interference Col10a1 mutation have subtle growth plate phenotypes with compressed proliferative and hypertrophic zones and altered mineral deposition (73, 74). The decreased Col10a1 expression because of GADD45β deficiency suggests that GADD45β plays a role in matrix remodeling at the late hypertrophic stage The chondrodysplasia (cho/cho) mouse, which carries a loss of function mutation in the Col11a1 gene, has wider long bones and shorter length possibly because type XI collagen influences the formation of type II collagen fibrils at early stages of chondrogenesis (75). Other ECM proteins, including osteocalcin and osteopontin, are known to play functional roles in cell-matrix adhesion during endochondral ossification. Thus, the tight regulation of matrix composition at different stages is required for normal development of long bones.
The dwarfism observed in human chondrodysplasias with COL10A1 mutations involves skeletal elements that are under great mechanical stress because of disruption in the pericellular matrix in the hypertrophic zone, although a role for defective vascularization has been proposed (73). Several previous studies have focused on vascularization as a critical step in endochondral ossification. Removal of angiogenic stimuli by ablating vascular endothelial growth factor (VEGF) (76) or VEGF receptors (77) resulted in increased length of the hypertrophic zone. A similar phenotype was observed in Mmp-9−/− mice (78). These and other studies indicate that ECM remodeling is the dominant rate-limiting process for chondrocyte hypertrophy, angiogenesis, and osteoblast recruitment during endochondral ossification (24, 30). Whether GADD45β plays a role in coupling ECM remodeling and vascularization by regulating the expression of MMP-9 or VEGF, in addition to MMP-13, in hypertrophic chondrocytes will require further study.
Although our analyses of Gadd45β−/− mice did not demonstrate complete suppression of hypertrophic differentiation itself, our finding that GADD45β silencing decreases the expression of Mmp-13 as well as Col10a1 during terminal differentiation in vitro supports a role of GADD45β as a crucial regulator in this process. Col10a1 is also a Runx2-responsive gene in hypertrophic chondrocytes (14). Thus, GADD45β may be involved, directly or indirectly, in the regulation of other key processes in hypertrophic differentiation through cooperation with Runx2. GADD45β is associated with cell cycle G2-M arrest (79), suggesting that it may act as a switch to inhibit chondrocyte proliferation during the transition to hypertrophy. The remodeling of matrix proteins induces an alteration in the environmental stress experienced by hypertrophic chondrocytes (12, 30). Our data suggest that GADD45β, which is highly expressed in response to environmental stresses (44), is an essential stress-response protein during chondrocyte hypertrophy.
The process of chondrocyte death subsequent to terminal differentiation to hypertrophy is also one of the most complicated steps during endochondral ossification, but the mechanism remains unknown. The GADD45 family members are indispensably involved in the mechanism of cell survival and apoptosis in cancer cells (54). Recently, we reported that NF-κB-mediated repression of GADD45α and GADD45γ is essential for the survival of cancer cells, but we found no relevance for GADD45β in cell survival and apoptosis (40). In fibroblasts, GADD45β is essential for JNK-mediated blockade of TNF-α-induced apoptosis (80). Although our present study does not address whether GADD45β is an anti-apoptotic or pro-apoptotic protein in chondrocytes, its intracellular localization in intact hypertrophic chondrocytes suggests a role as a survival factor that maintains expression of critical genes prior to apoptosis. The decreased length of the hypertrophic zone in Gadd45β−/− mice suggests that GADD45β may prolong survival of hypertrophic chondrocytes at later stages.
AP-1 transcription factors, including c-Jun, JunB, and JunD, are substrates of the JNK signaling pathway (81), and GADD45β is known to activate the JNK pathway in response to environmental stress (44). This interaction activates MEKK4 kinase leading to MKK4 and, subsequently, JNK activation. However, GADD45β also mediates the NF-κB suppression of JNK signaling in the apoptotic response to TNF-α (54, 80). This controversy surrounding GADD45β and JNK has left open the question of how GADD45β contributes to cell differentiation. We applied a mass spectrometric assay (MALDI-TOF MS) to identify protein-protein interaction partners of GADD45β proteins in vivo resulting in identification of MEKK4, the upstream kinase of JNK, as an in vivo interactor of GADD45β,7 as reported previously (44). Several mitogen-activated protein kinase cascades are involved in BMP-induced signaling during skeletal development. The p38 pathway contributes to the initiation of chondrogenic cellular condensation (82), and ERK1/2 activation cross-interacts with BMP-2-induced signaling to regulate chondrogenesis in a positive manner (83). Our results indicate that chondrocytes also use the JNK/MEKK4 pathway during terminal differentiation.
Several genetic studies have provided insight into the role of AP-1 family members, including c-Fos, Fra-1, c-Fos, FosB, JunB, and ATF-2 in skeletal development in vivo (84–89). MMP-13 is also a target of c-Maf, which can form heterodimers with AP-1 family members and regulates the differentiation of hypertrophic chondrocytes (90). Our results show that GADD45β-induced AP-1/JunD activity is essential for MMP-13 expression in hypertrophic chondrocytes and are consistent with previous findings of functional interaction between AP-1 and Runx2 during parathyroid hormone-related protein-dependent MMP-13 expression in osteoblasts (26, 27). The nuclear localization of GADD45β in hypertrophic chondrocytes also suggests a direct mechanism by which AP-1 family members could regulate terminal differentiation.
In conclusion, our data demonstrate a novel role in chondrocyte hypertrophy for GADD45β, which has been implicated in the stress response and cell survival during terminal differentiation of other cell types. It appears to be a major BMP-2-responsive gene induced in chondrocytes by Smad1-dependent signaling in cooperation with Runx2, which immunolocalizes with GADD45β mRNA in the prehypertrophic zone of the embryonic growth plate. The intracellular accumulation of Gadd45β protein prominently in the nucleus in intact hypertrophic chondrocytes in the mouse embryonic growth plate and the consequences of GADD45β deficiency in vivo and in vitro indicate that Gadd45β is required for MMP-13 expression in the late hypertrophic zone where Runx2 is also expressed. Our observations that GADD45β can stimulate MMP-13 gene transcription via the JNK/JunD pathway in synergy with Runx2 provides a new concept regarding a temporal and spatial link between BMP-2-induced pathways upstream and the expression of genes required at terminal stages. Together, our findings support a previously undiscovered role for GADD45β as a critical mediator of chondrocyte hypertrophy during endochondral ossification.
Acknowledgments
We are grateful to Dr. Inder M. Verma for help with the generation of LV-siRNA-GADD45β, to Dr. Elisabeth Morris (Wyeth, Cambridge, MA) for providing recombinant human BMP-2, and to Dr. Benjamin Bierbaum (New England Baptist Hospital) for providing human articular cartilage. We also thank Dr. Bob Choy for technical advice.
Footnotes
This work was supported in part by National Institutes of Health Grants R01-AR45378 and R01-AG22021 (to M. B. G.) and R01-AI49527 (to T. A. L.) and a Biomedical Science Grant from the Arthritis Foundation (to M. B. G.).
The on-line version of this article (available at http://www.jbc.org) contains Fig. S1.
The abbreviations used are: GADD45β, growth arrest and DNA damage-inducible 45β; BMP, bone morphogenetic protein; MMP, matrix metalloproteinase; EMSA, electrophoretic mobility shift assays; JNK, c-Jun N-terminal kinase; siRNA, small interfering RNA; EGF, epidermal growth factor; VEGF, vascular endothelial growth factor; IGF, insulin-like growth factor; bFGF, basic fibroblast growth factor; IL, interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor; DMEM, Dulbecco’s modified Eagle’s medium; FCS, fetal calf serum; IHC, immunohistochemistry; GFP, green fluorescent protein; ECM, extracellular matrix; WT, wild type; dpc, days post-conception; KD, knockdown; ERK, extracellular signal-regulated kinase.
M. Rahman, unpublished data.
K. Ijiri, N. Walsh, and M. B. Goldring, unpublished data.
L. F. Zerbini, K. Ijiri, M. B. Goldring, and T. A. Libermann, unpublished data.
References
- 1.Abdollahi A, Lord KA, Hoffman-Liebermann B, Liebermann DA. Oncogene. 1991;6:165–167. [PubMed] [Google Scholar]
- 2.Amanullah A, Azam N, Balliet A, Hollander C, Hoffman B, Fornace A, Liebermann D. Nature. 2003;424:741–742. doi: 10.1038/424741b. [DOI] [PubMed] [Google Scholar]
- 3.Selvakumaran M, Lin HK, Sjin RT, Reed JC, Liebermann DA, Hoffman B. Mol Cell Biol. 1994;14:2352–2360. doi: 10.1128/mcb.14.4.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, Jr, Liebermann DA, Bottinger EP, Roberts AB. J Biol Chem. 2003;278:43001–43007. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 5.Aigner T, Zien A, Gehrsitz A, Gebhard PM, McKenna L. Arthritis Rheum. 2001;44:2777–2789. doi: 10.1002/1529-0131(200112)44:12<2777::aid-art465>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 6.Sironen RK, Karjalainen HM, Elo MA, Kaarniranta K, Torronen K, Takigawa M, Helminen HJ, Lammi MJ. Biochim Biophys Acta. 2002;1591:45–54. doi: 10.1016/s0167-4889(02)00247-1. [DOI] [PubMed] [Google Scholar]
- 7.Urist MR. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- 8.Hogan BL. Genes Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- 9.Enomoto-Iwamoto M, Iwamoto M, Mukudai Y, Kawakami Y, Nohno T, Higuchi Y, Takemoto S, Ohuchi H, Noji S, Kurisu K. J Cell Biol. 1998;140:409–418. doi: 10.1083/jcb.140.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valcourt U, Gouttenoire J, Moustakas A, Herbage D, Mallein-Gerin F. J Biol Chem. 2002;277:33545–33558. doi: 10.1074/jbc.M202086200. [DOI] [PubMed] [Google Scholar]
- 11.Ballock RT, O’Keefe RJ. J Bone Jt Surg Am. 2003;85:715–726. [PubMed] [Google Scholar]
- 12.Colnot CI, Helms JA. Mech Dev. 2001;100:245–250. doi: 10.1016/s0925-4773(00)00532-3. [DOI] [PubMed] [Google Scholar]
- 13.Volk SW, Luvalle P, Leask T, Leboy PS. J Bone Miner Res. 1998;13:1521–1529. doi: 10.1359/jbmr.1998.13.10.1521. [DOI] [PubMed] [Google Scholar]
- 14.Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. J Cell Biol. 2003;162:833–842. doi: 10.1083/jcb.200211089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y, Kishimoto T, Komori T. J Biol Chem. 2000;275:8695–8702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- 16.Leboy P, Grasso-Knight G, D’Angelo M, Volk SW, Lian JV, Drissi H, Stein GS, Adams SL. J Bone Jt Surg Am. 2001;83(Suppl 1):15–22. [PubMed] [Google Scholar]
- 17.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 18.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 19.Jimenez MJ, Balbin M, Lopez JM, Alvarez J, Komori T, Lopez-Otin C. Mol Cell Biol. 1999;19:4431–4442. doi: 10.1128/mcb.19.6.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T, Komori T. Dev Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 21.Porte D, Tuckermann J, Becker M, Baumann B, Teurich S, Higgins T, Owen MJ, Schorpp-Kistner M, Angel P. Oncogene. 1999;18:667–678. doi: 10.1038/sj.onc.1202333. [DOI] [PubMed] [Google Scholar]
- 22.Wu CW, Tchetina EV, Mwale F, Hasty K, Pidoux I, Reiner A, Chen J, Van Wart HE, Poole AR. J Bone Miner Res. 2002;17:639–651. doi: 10.1359/jbmr.2002.17.4.639. [DOI] [PubMed] [Google Scholar]
- 23.Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, Krane SM. Proc Natl Acad Sci U S A. 2004;101:17192–17197. doi: 10.1073/pnas.0407788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z. Development (Camb) 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mengshol JA, Vincenti MP, Brinckerhoff CE. Nucleic Acids Res. 2001;29:4361–4372. doi: 10.1093/nar/29.21.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess J, Porte D, Munz C, Angel P. J Biol Chem. 2001;276:20029–20038. doi: 10.1074/jbc.M010601200. [DOI] [PubMed] [Google Scholar]
- 27.D’Alonzo RC, Selvamurugan N, Karsenty G, Partridge NC. J Biol Chem. 2002;277:816–822. doi: 10.1074/jbc.M107082200. [DOI] [PubMed] [Google Scholar]
- 28.Kim IS, Otto F, Zabel B, Mundlos S. Mech Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- 29.Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ortega N, Behonick DJ, Werb Z. Trends Cell Biol. 2004;14:86–93. doi: 10.1016/j.tcb.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colnot C, Lu C, Hu D, Helms JA. Dev Biol. 2004;269:55–69. doi: 10.1016/j.ydbio.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 32.Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S, Glowacki J, Arbiser JL, Apperley JF. J Clin Investig. 1994;94:2307–2316. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan L, Peng H, Osaki M, Choy BK, Auron PE, Sandell LJ, Goldring MB. J Biol Chem. 2003;278:17688–17700. doi: 10.1074/jbc.M301676200. [DOI] [PubMed] [Google Scholar]
- 34.Shukunami C, Ohta Y, Sakuda M, Hiraki Y. Exp Cell Res. 1998;241:1–11. doi: 10.1006/excr.1998.4045. [DOI] [PubMed] [Google Scholar]
- 35.Ballock RT, Reddi AH. J Cell Biol. 1994;126:1311–1318. doi: 10.1083/jcb.126.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spentzos D, Levine DA, Ramoni MF, Joseph M, Gu X, Boyd J, Libermann TA, Cannistra SA. J Clin Oncol. 2004;22:4648–4658. doi: 10.1200/JCO.2004.04.070. [DOI] [PubMed] [Google Scholar]
- 37.Li C, Wong WH. Proc Natl Acad Sci U S A. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 39.Xu L, Peng H, Wu D, Hu K, Goldring MB, Olsen BR, Li Y. J Biol Chem. 2005;280:548–555. doi: 10.1074/jbc.M411036200. [DOI] [PubMed] [Google Scholar]
- 40.Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, Wei W, Joseph M, Gu X, Grall F, Goldring MB, Zhou JR, Libermann TA. Proc Natl Acad Sci U S A. 2004;101:13618–13623. doi: 10.1073/pnas.0402069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu W, David D, Zhou B, Chu PG, Zhang B, Wu M, Xiao J, Han T, Zhu Z, Wang T, Liu X, Lopez R, Frankel P, Jong A, Yen Y. Am J Pathol. 2003;162:1961–1974. doi: 10.1016/s0002-9440(10)64329-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kartsogiannis V, Moseley J, McKelvie B, Chou ST, Hards DK, Ng KW, Martin TJ, Zhou H. Bone (NY) 1997;21:385–392. doi: 10.1016/s8756-3282(97)00180-4. [DOI] [PubMed] [Google Scholar]
- 43.Lu B, Ferrandino AF, Flavell RA. Nat Immun. 2004;5:38–44. doi: 10.1038/ni1020. [DOI] [PubMed] [Google Scholar]
- 44.Takekawa M, Saito H. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 45.Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, Karsenty G. Genes Dev. 1999;13:1025–1036. doi: 10.1101/gad.13.8.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zerbini LF, Wang Y, Cho JY, Libermann TA. Cancer Res. 2003;63:2206–2215. [PubMed] [Google Scholar]
- 47.Thirunavukkarasu K, Miles RR, Halladay DL, Onyia JE. BioTechniques. 2000;28:506–510. doi: 10.2144/00283st09. [DOI] [PubMed] [Google Scholar]
- 48.Tiscornia G, Singer O, Ikawa M, Verma IM. Proc Natl Acad Sci U S A. 2003;100:1844–1848. doi: 10.1073/pnas.0437912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 50.Pfeifer A, Verma IM. Annu Rev Genomics Hum Genet. 2001;2:177–211. doi: 10.1146/annurev.genom.2.1.177. [DOI] [PubMed] [Google Scholar]
- 51.Zaidi SK, Sullivan AJ, van Wijnen AJ, Stein JL, Stein GS, Lian JB. Proc Natl Acad Sci U S A. 2002;99:8048–8053. doi: 10.1073/pnas.112664499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Afzal F, Pratap J, Ito K, Ito Y, Stein JL, van Wijnen AJ, Stein GS, Lian JB, Javed A. J Cell Physiol. 2005;204:63–72. doi: 10.1002/jcp.20258. [DOI] [PubMed] [Google Scholar]
- 53.Yazgan O, Pfarr CM. J Biol Chem. 2002;277:29710–29718. doi: 10.1074/jbc.M204552200. [DOI] [PubMed] [Google Scholar]
- 54.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 55.Lu B, Yu H, Chow C, Li B, Zheng W, Davis RJ, Flavell RA. Immunity. 2001;14:583–590. doi: 10.1016/s1074-7613(01)00141-8. [DOI] [PubMed] [Google Scholar]
- 56.Mita H, Tsutsui J, Takekawa M, Witten EA, Saito H. Mol Cell Biol. 2002;22:4544–4555. doi: 10.1128/MCB.22.13.4544-4555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoon BS, Lyons KM. J Cell Biochem. 2004;93:93–103. doi: 10.1002/jcb.20211. [DOI] [PubMed] [Google Scholar]
- 58.Wan M, Cao X. Biochem Biophys Res Commun. 2005;328:651–657. doi: 10.1016/j.bbrc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 59.Goldring MB. In: Kelley’s Textbook of Rheumatology. 7. Harris ED, Ruddy S, Sledge CB, Sergent JS, Budd RC, editors. W. B. Saunders Co; Philadelphia: 2004. pp. 203–234. [Google Scholar]
- 60.Provot S, Schipani E. Biochem Biophys Res Commun. 2005;328:658–665. doi: 10.1016/j.bbrc.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 61.Locklin RM, Riggs BL, Hicok KC, Horton HF, Byrne MC, Khosla S. J Bone Miner Res. 2001;16:2192–2204. doi: 10.1359/jbmr.2001.16.12.2192. [DOI] [PubMed] [Google Scholar]
- 62.Xu SC, Harris MA, Rubenstein JLR, Mundy GR, Harris SE. DNA Cell Biol. 2001;20:359–365. doi: 10.1089/10445490152122479. [DOI] [PubMed] [Google Scholar]
- 63.Lai CF, Cheng SL. J Biol Chem. 2002;277:15514–15522. doi: 10.1074/jbc.M200794200. [DOI] [PubMed] [Google Scholar]
- 64.Maeda Y, Tsuji K, Nifuji A, Noda M. J Cell Biochem. 2004;93:337–344. doi: 10.1002/jcb.20154. [DOI] [PubMed] [Google Scholar]
- 65.Fujita T, Azuma Y, Fukuyama R, Hattori Y, Yoshida C, Koida M, Ogita K, Komori T. J Cell Biol. 2004;166:85–95. doi: 10.1083/jcb.200401138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin R, De Smaele E, Zazzeroni F, Nguyen DU, Papa S, Jones J, Cox C, Gelinas C, Franzoso G. DNA Cell Biol. 2002;21:491–503. doi: 10.1089/104454902320219059. [DOI] [PubMed] [Google Scholar]
- 67.Balliet AG, Hollander MC, Fornace AJ, Jr, Hoffman B, Liebermann DA. DNA Cell Biol. 2003;22:457–468. doi: 10.1089/104454903322247334. [DOI] [PubMed] [Google Scholar]
- 68.Ungefroren H, Groth S, Ruhnke M, Kalthoff H, Fandrich F. J Biol Chem. 2005;280:2644–2652. doi: 10.1074/jbc.M411925200. [DOI] [PubMed] [Google Scholar]
- 69.Qiao B, Padilla SR, Benya PD. J Biol Chem. 2005;280:17562–17571. doi: 10.1074/jbc.M500646200. [DOI] [PubMed] [Google Scholar]
- 70.Ferguson CM, Miclau T, Hu D, Alpern E, Helms JA. Ann N Y Acad Sci. 1998;857:33–42. doi: 10.1111/j.1749-6632.1998.tb10105.x. [DOI] [PubMed] [Google Scholar]
- 71.Colnot C. J Cell Biochem. 2005;95:688–697. doi: 10.1002/jcb.20449. [DOI] [PubMed] [Google Scholar]
- 72.Johansson N, Saarialho-Kere U, Airola K, Herva R, Nissinen L, Westermarck J, Vuorio E, Heino J, Kahari VM. Dev Dyn. 1997;208:387–397. doi: 10.1002/(SICI)1097-0177(199703)208:3<387::AID-AJA9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 73.Gress CJ, Jacenko O. J Cell Biol. 2000;149:983–993. doi: 10.1083/jcb.149.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacenko O, Chan D, Franklin A, Ito S, Underhill CB, Bateman JF, Campbell MR. Am J Pathol. 2001;159:2257–2269. doi: 10.1016/S0002-9440(10)63076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li Y, Lacerda DA, Warman ML, Beier DR, Yoshioka H, Ninomiya Y, Oxford JT, Morris NP, Andrikopoulos K, Ramirez F. Cell. 1995;80:423–430. doi: 10.1016/0092-8674(95)90492-1. [DOI] [PubMed] [Google Scholar]
- 76.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 77.Maes C, Carmeliet P, Moermans K, Stockmans I, Smets N, Collen D, Bouillon R, Carmeliet G. Mech Dev. 2002;111:61–73. doi: 10.1016/s0925-4773(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 78.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vairapandi M, Balliet AG, Hoffman B, Liebermann DA. J Cell Physiol. 2002;192:327–338. doi: 10.1002/jcp.10140. [DOI] [PubMed] [Google Scholar]
- 80.Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, De Smaele E, Tang WJ, D’Adamio L, Franzoso G. Nat Cell Biol. 2004;6:146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 81.Davis RJ. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 82.Nakamura K, Shirai T, Morishita S, Uchida S, Saeki-Miura K, Makishima F. Exp Cell Res. 1999;250:351–363. doi: 10.1006/excr.1999.4535. [DOI] [PubMed] [Google Scholar]
- 83.Seghatoleslami MR, Roman-Blas JA, Rainville AM, Modaressi R, Danielson KG, Tuan RS. J Cell Biochem. 2003;88:1129–1144. doi: 10.1002/jcb.10458. [DOI] [PubMed] [Google Scholar]
- 84.Grigoriadis AE, Schellander K, Wang ZQ, Wagner EF. J Cell Biol. 1993;122:685–701. doi: 10.1083/jcb.122.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jochum W, David JP, Elliott C, Wutz A, Plenk H, Jr, Matsuo K, Wagner EF. Nat Med. 2000;6:980–984. doi: 10.1038/79676. [DOI] [PubMed] [Google Scholar]
- 86.Reimold AM, Grusby MJ, Kosaras B, Fries JW, Mori R, Maniwa S, Clauss IM, Collins T, Sidman RL, Glimcher MJ, Glimcher LH. Nature. 1996;379:262–265. doi: 10.1038/379262a0. [DOI] [PubMed] [Google Scholar]
- 87.Jochum W, Passegue E, Wagner EF. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- 88.Karreth F, Hoebertz A, Scheuch H, Eferl R, Wagner EF. Development (Camb) 2004;131:5717–5725. doi: 10.1242/dev.01414. [DOI] [PubMed] [Google Scholar]
- 89.Hess J, Hartenstein B, Teurich S, Schmidt D, Schorpp-Kistner M, Angel P. J Cell Sci. 2003;116:4587–4596. doi: 10.1242/jcs.00772. [DOI] [PubMed] [Google Scholar]
- 90.MacLean HE, Kim JI, Glimcher MJ, Wang J, Kronenberg HM, Glimcher LH. Dev Biol. 2003;262:51–63. doi: 10.1016/s0012-1606(03)00324-5. [DOI] [PubMed] [Google Scholar]