

Abstract
The freshwater cyanotoxins, microcystins (MCs), pose a global public health threat as potent hepatotoxins in cyanobacterial blooms; their persistence in drinking and recreational water has been associated with potential chronic effects in addition to acute intoxications. Rapid and accurate detection of the over 80 structural congeners is challenged by the rigorous and time consuming clean up required to overcome interference found in raw water samples. MALDI-MS has shown promise for rapid quantification of individual congeners in raw water samples, with very low operative cost, but so far limited sensitivity and lack of available and versatile internal standards (ISs) has limited its use. Two easily synthesized S-hydroxyethyl–Cys(7)-MC-LR and –RR ISs were used to generate linear standard curves in a reflectron MALDI instrument, reproducible across several orders of magnitude for MC –LR, - RR and –YR. Minimum quantification limits in direct water samples with no clean up or concentration step involved were consistently below 7 μg/L, with recoveries from spiked samples between 80 and 119%. This method improves sensitivity by 30 fold over previous reports of quantitative MALDI-TOF applications to MCs and provides a salient option for rapid throughput analysis for multiple MC congeners in untreated raw surface water blooms as a means to identify source public health threats and target intervention strategies within a watershed. As demonstrated by analysis of a set of samples from Uruguay, utilizing the reaction of different MC congeners with alternate sulfhydryl compounds, the m/z of the IS can be customized to avoid overlap with interfering compounds in local surface water samples.
Keywords: Microcystins, cyanotoxins, cyanobacterial blooms, quantitative MALDI-TOF, internal standards
1. Introduction
Contamination of natural surface waters by toxic cyanobacteria is a growing problem worldwide, resulting in serious animal and human health hazards. Microcystins (MCs) are the most common hepatotoxins and tumour promoters produced by freshwater cyanobacteria (de Figueiredo et al., 2004). They encompass a large family of cyclic heptapeptide toxins, composed of uncommon amino acids, including several D-amino acids and the characteristic Adda group (3-amino-9-methoxy-2,6,8-trimethyl-10-phenyldeca-4(E),6(E)-dienoic acid) (figure 1). Structural variation is observed in all amino acids, but major MC analogues occur due to variations in the L-amino acids “X” and “Z” (Rinehart et al., 1994). More than 80 MC congeners have been described to date, ranging in size between 909–1115 Da, and are produced by a broad range of cyanobacteria species in fresh and brackish surface water blooms (Codd, 2000; Donohue et al., 2008). Potent inhibitors of highly conserved protein phosphatase 1 and 2A (Runnegar et al., 1995), microcystin toxicity impacts a broad range of plant and animal species, including humans. In spite of similar phosphatase inhibitory capacity, MCs differ greatly in their acute toxicity from poorly toxic, LD50 >1200 mg/kg, to highly toxic variants such as MC-LR (LD50 = 50 mg/kg) (Rinehart et al., 1994). These differences appear to be related to their toxicokinetics, since the mechanism of cellular uptake occurs via specific organic anion transporting polypeptides that mediate selective congener transport (Fischer et al., 2010).
Figure 1. Structure and synthesis of thiol-modified microcystins.
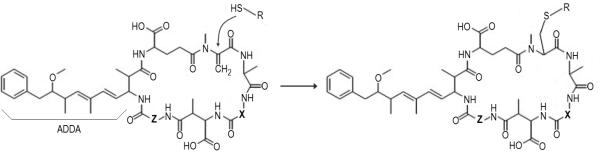
The general structure of microcystins is displayed at the left, denoting the characteristic Adda β-amino acid, and “X” and “Z” representing variable L-amino acids. Internal standards were prepared by the well-known reaction of sulfhydryl compounds with the methylene group of the N methyldehydroalanine (Mdha) residue of MCs. The reaction occurred almost instantaneously under nitrogen gas.
Cyanobacterial blooms result from heavy nutrient loading, are enhanced by environmental perturbations, increasing global water temperatures, and their increase occurrence has emerged as worldwide concern (Codd, 2000; Pearson et al., 2010). While many cyanobacterial species are known producers of microcystins, reasons for toxin production remain unknown and the presence of a bloom does not guarantee the presence of toxic compounds (Pearson et al., 2010). Moreover, the production of individual MC congeners varies among genera, species and strains, and toxin production can vary considerably within hours (Wood et al., 2011). This variability presents a serious challenge to public health officials and water treatment facility managers requiring to optimize sampling, minimize public health risk and target interventions in the management of recreational or source water bodies (Dodds et al., 2009). The WHO has established a provisional guideline value of 1 μg/L for microcystin LR in drinking water, but recent evidence in animal intoxications suggests the prevalence and consequences of potentially overlooked congeners (Dietrich and Hoeger, 2005; Donohue et al., 2008). Similarly, a reference health alert value of 20 μg/L has been suggested to minimize the toxicity risk by accidental ingestion of recreational waters (Chorus et al., 2000), and many countries have adopted similar limits for bloom risk assessment ranging from 15-25 μg/L (Chorus, 2012).
The most widely used analytical methods for the detection and quantification of MCs rely on chromatographic separation techniques such as high pressure liquid chromatography (HPLC) coupled to UV or MS detection, and a reference standard HPLC/UV method (ISO 20179:2005) has been issued by the International Organization for Standardization. In normal practice and for regulatory purposes, the analysis of the MC content in drinking water is restricted to the use of these reference methods. To fulfill the sensitivity required by the WHO advisory level of <1 μg/L of MC-LR, these chromatographic methods often require large sample volumes, concentration and clean steps, as well as the sequential analysis of sample (Meriluoto et al., 2000)s. This processing makes the methods comparatively less attractive for screening purposes, particularly when large numbers of samples need to be analyzed on a regular basis within a timely fashion. To this end, other techniques, particularly immunochemical methods, are currently used to study the ecology of cyanobacterial communities or monitor the evolution of these communities in source or recreational waters (Pirez et al., 2013; Qin et al., 2010; Znachor et al., 2006). ELISA is a convenient option because it allows for the parallel analysis of large sample loads in a fast and cost efficient manner. However, commercially available ELISAs, detecting the side chain ADDA common to all microcystins, show variable cross reactivity and only detect total microcystins, missing the information on individual congeners with variable degree of toxicity (Msagati et al., 2006). In addition, most ELISAs perform within a narrow linear range, about one order, while environmental water and scum samples may contain MC concentrations ranging from few μg/L to hundreds of thousands. With an extended linear range, short time of analysis (< 30 seconds), high-throughput capacity and minimal requirements for sample preparation, Matrix Assisted Laser Desorption Ionization (MALDI) coupled with time of flight (TOF) mass spectrometry (MS) offers a salient option for comprehensive analysis of environmental water samples (Pan et al., 2007; Pusch and Kostrzewa, 2005).
To date, MALDI-TOF MS mostly has been utilized qualitatively to identify MC variants in cyanobacterial colonies and surface water blooms (Erhard et al., 1997; Ferranti et al., 2011; Welker et al., 2002), and its use for MC quantification is rather recent (Howard and Boyer, 2007; Puddick et al., 2011). Quantitative detection by MALDI-TOF based on peak response has poor reproducibility, mainly due to spot-to-spot variability in crystal formation (Duncan et al., 2008; Szajli et al., 2008). However, this can be overcome using an internal standard (IS), and the best results are obtained when this compound mimics the physicochemical properties of the analyte throughout all steps of the analytical process (Szajli et al., 2008). The use of a stable isotope-labeled form of the analyte (isotopomer) is an obvious way to accomplish that goal, but requires special chemicals and can be difficult to prepare and purify. In the initial report of a quantitative MALDI-TOF application for MCs (Howard and Boyer, 2007), three potential internal standards were assayed, and the best method detection limit (MDL) for MC-LR was obtained with nodularin (a pentapeptide cyanotoxin with similar basic ring structure to microcystins), which outperformed a [15N]10-isotopomer of microcystin-YR and the peptide hormone angiotensin I. More recently, Puddick et al, in order to reduce the cost of the analysis, used angiotensin I as internal standard in combination with different sample preparation techniques, but being a linear peptide, angiotensin as IS did not attain the MDL values previously obtained with nodularin (Puddick et al., 2011). This highlights the importance of using internal standards that resemble the physicochemical properties of the target compound.
With this in mind, we explored the use of derivatives of MCs as internal standards for quantitative detection of MC by MALDI-TOF MS. To this end, we utilized a well-known and simple synthesis to prepare ISs by reaction of MCs with thiol-containing compounds readily available in most laboratories. To further improve the detection limit of the method introduced by Howard and Boyer, we utilized reflectron MALDI-TOF equipment that provides improved resolution and better sensitivity. We demonstrate that these ISs can be used for sensitive detection of MCs by direct analysis of unprocessed surface water samples, with an extended linear range and high recoveries. The ability to identify and quantify common microcystins with minimal processing of the raw water sample will dramatically reduce time for screening of a broad number of samples.
2. Materials and Methods
2.1 Materials
Microcystin-LR, and microcystin-RR were obtained from GreenWater Laboratories (Palatka, FL); microcystin-YR, β-mercaptoethanol (99.9%), and trifluoroacetic acid (TFA) were obtained from Sigma-Aldrich (St. Louis, MO). Alpha-cyano-4-hydroxycinnamic acid (CHCA) and a standard peptide calibration solution were obtained from Bruker Daltonics (Denmark). HPLC grade acetonitrile and methanol were obtained from Baker (Phillipsburg, NJ), along with sodium borate and sodium chloride for the preparation of the borate buffer.
2.2 Preparation of Internal Standards
Two internal standards (IS-1 and IS-2) were prepared from microcystin-LR (MC-LR) and microcystin-RR (MC-RR), respectively, by reaction of the N methyldehydroalanine (Mdha) group with sulfhydryl compounds, figure 1 (Smith and Boyer, 2009). For each 0.1 μM of MC dissolved in 50% methanol/50% 100 μM borate buffer, pH 10.5; 10 μl of β-mercaptoethylamine or β-mercaptoethanol were added and allowed to react under nitrogen gas for several hours at room temperature. One μl of the reaction mix was taken initially every 15 minutes and then hourly, and after dilution in 100 μl of MilliQ water was analyzed by MALDI-TOF for relative appearance and disappearance of peaks. After completion, the reaction mixture was diluted with 10% methanol, acidified to pH 3.0 with 5% glacial acetic acid and purified using solid phase extraction (SPE C18, Phenomenex, Torrance, CA). The methanol eluted (S-aminoethyl-/S-hydroxyethyl-Cys(7) microcystin) adduct was dried under nitrogen gas to eliminate any residual sulfhydryl compound, reconstituted in 200 μl of methanol, quantified using HPLC, aliquoted into 50 μl and stored at -20°C until further use.
2.3 MALDI-TOF-MS
CHCA, which has been reported to deliver relatively homogenous samples in crystal formation (Szajli et al., 2008), was prepared as a saturated solution (>21 mg/ml) in 50% (v/v) acetonitrile and 50% (v/v) 0.1% TFA MilliQ water. The matrix was mixed with internal standard and sample and permitted to air dry. A Microflex LRF MALDI-TOF (Bruker Daltonics, Billerica, MS, USA) with a 337 nm nitrogen laser was operated in positive ion reflectron mode with delayed extraction and optimized in the m/z range of 500 Da to 2 kDa. Calibrations were performed with a peptide calibration standard mix (Bruker Daltonics). The laser was fired 100 times at each of ten locations for each sample well on a 96 well plate for a cumulative 1000 shots per sample well taken at 30% intensity. More specific details of sample well preparation and quantification with internal standard are provided in the supplemental materials.
2.4 MALDI Spot Preparation and Quantification with IS
Ten μl of sample were thoroughly mixed with 1 μl of the appropriate concentration of internal standard (30, 40, 300 or 400 μg/L, as stated), followed by the addition of 10 μl of CHCA solution. Following thorough mixing, 2 μl of the solution was placed on the MALDI sample plate. Unless otherwise specified, eight 2 μl replicate spots were aliquoted and allowed to air dry at room temperature. Ten shots were taken at each spot on the 96 well stainless steel plate with the laser firing 100 times at each shot for a cumulative 1000 shots per well at 30% intensity. The data point (intensity or area) was determined as an arithmetic mean. Initially, ratios of both peak intensity and peak area of microcystin congener divided by the equivalent intensity and area of internal standard peak were determined for each spot in order to account for spot-to-spot variation across the plate and heterogeneity of crystal formation. The stainless steel plate was thoroughly cleaned between analyses according to manufacturer instructions, and was routinely checked to avoid any carry over contamination.
2.5 Calibration Curves and Detection Limits
To study the linear range of the method, MC-LR, MC-RR and MC-YR were dissolved in 100% methanol (1mg/mL) and further diluted in MilliQ water to twelve concentrations ranging between 0 and 5000 μg/L. These samples were prepared for analysis as described (Supporting information) using IS-1 and IS-2 at 300 μg/L and 400 μg/L, respectively, in 100% methanol. Standard solutions were prepared in MilliQ water for determination of a method detection limit (MDL) or and in a reference cyanobacterial-bloom water sample (ref-CB) for minimum quantification limit (MQL). At least seven concentrations of the standards were prepared in the 0 to 100-μg/L range, using IS-1 and IS-2 at a working concentration of 30 μg/L and 40 μg/L, respectively (Supporting Information). The ref-CB water sample consisted of a pool of representative water samples from blooms with less than 0.25 μg/L of total MCs, as determined by ELISA (Brena et al., 2006).
2.6 Surface Water Sample Preparation and Spiking Recoveries
Surface water samples collected from three water bodies along the Rio Negro in northern Uruguay, kept chilled under transport and frozen at -20°C until ready for analysis. Following three cycles of freeze/thaw to fully rupture the cyanobacterial cells, 1.0 mL of the water sample was centrifuged at 10,000 × g for 5 minutes, followed by syringe filtration with a 0.22 μm filter (Millipore, MA). An initial survey determination of the concentrations of MC-LR, MC-RR, and MC-YR in the surface water samples was made utilizing IS-2 with the method described in section 2.4. Then nine samples covering a range of MC concentrations were selected and spiked with 100 μg/L of MC-LR, MC-RR and MC-YR, simultaneously, and percent recovery was determined along with total concentrations. A bloom showing extremely high MC concentration was spiked with 1000 μg/L and diluted 10 fold before analysis. Calibration curves were performed for each new analysis. MALDI well spot preparation and laser operation were maintained as for standard curves.
2.7 Safety Considerations
Acutely toxic to mammals, the LD50 (i.p.) for microcystins range between 36-122 μg/kg for mice and rats with an inhalation toxicity of 43 μg/kg. It is much less bioavailable with an oral LD50 of 5 mg/kg.2.3 Proper personal protective equipment was worn (gloves, lab coat) when handling any of the congener standards or handling any bloom samples. Care was also taken to not aerosolize any powder forms of standards or bloom materials.
3. Results and Discussions
3.1 Preparation of Internal Standards
We initially optimized the conditions for the reaction of β-mercaptoethylamine and β-mercaptoethanol with the Mdha group of MC-LR. The reaction proceeded very rapidly at high pH as determined by MALDI analysis of time course samples; prepared in borate buffer pH 10.5, all MC-LR was converted to the S-aminoethyl- or S-hydroxyethyl–Cys(7))-MC-LR derivative within seconds (data not shown). Initial experiments suggested that both compounds performed equally well as IS but the hydroxyethyl conjugate (IS-1, MWaverage = 1073.30 Da, MH+ = 1073.7) showed better reproducibility and thus was selected. A second internal standard, the β-mercaptoethanol derivative of MC-RR (IS-2, MWaverage = 1116.33 Da, MH+ = 1116.6), was prepared similarly, and the eventual fragmentation of both IS was studied under different conditions of laser energy. Preliminary tests showed that dilutions of ISs of 30 to 300 μg/L were suitable for quantitative analysis of MCs across a broad range of concentrations. Using these concentrations, a tradeoff value of 30% laser intensity was determined to have maximal sensitivity at low concentrations of MCs, while minimizing the fragmentation of the internal standards, Figure 2.
Figure 2. Mass Spectra of S-hydroxyethyl – Cys(7) microcystin LR (IS-1) and S-hydroxyethyl – Cys(7) microcystin RR (IS-2).
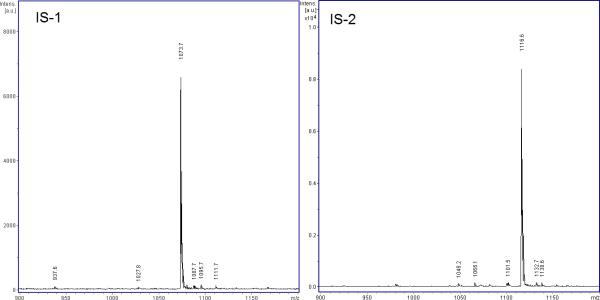
Both standards (40 μg/L) were spotted on the MALDI target as previously described (2 μl per spot), and were analyzed at 30% of laser intensity. We then focused our study on the three most common polar congeners found in cyanobacterial blooms, namely MC-LR, MC-RR, and MC-YR, which produced peaks at 995.6 (m/z), 1038.6 (m/z), and 1045.6 (m/z), respectively. Figure 3 shows their spectra at trace concentrations, in the presence of 40 μg/L of IS-2. In all cases, there were peaks proportional to the concentration of MC, no interference of IS fragments were observed in the case of MC-LR and MC-YR, despite the fact that a fragment, or a low concentration contaminant of the IS, occurred at 1048.5 (m/z). In the case of MC-RR a trace peak corresponding to this MC was observed in the blank, presumably as a consequence of marginal IS-2 fragmentation, but the signal was very weak and did not interfere with the quantification of MC-RR. Similar results with even less interference of contaminant peaks were obtained when IS-1 was used (data not shown).
3.2 MDL and MQL with IS-1
IS-1 generated calibration curves (Supporting Information, Figures A.1-A.3) of standards with excellent linearity in the 0-5000 μg/L range and R2 values > 0.99. As previously described (Welker et al., 2002), the polar congeners generated strong [M+H]+ signals likely because of protonation of the basic arginine side chains. Not surprisingly, MC-RR calibration curves generated a much greater signal (approximately twice the slope) as compared with MC-LR and MC-YR, confirming the role of protonation of the basic arginine side chains; this increased signal probably explains the improve MDL observed for MC-RR as compared with the two other congeners. Table 1 summarizes calculated limit of detection and quantification for IS-1 (Supporting Information, Figures A.1-A.3 and B.1-B.3) using standard solutions prepared in MilliQ water.
Table 1.
Determination of MDL and MQL for MC-LR, MC-RR, MC-YR Standard Solutions Using Both Peak Height and Peak Area Ratios with IS-1.
| Congener | Ratio Parameter | Regression Equation | R2 | CV1 | MDL (μg/L)2 | MQL (μg/L)3 |
|---|---|---|---|---|---|---|
| MC-LR | peak height | y = 0.0095 x + 0.04 | 0.989 | 6.7 | 2.8 | 4.5 |
| peak area | y = 0.0093 x + 0.06 | 0.991 | 8.6 | 2.6 | 4.3 | |
| MC-RR | peak height | y = 0.0222 x - 0.005 | 0.996 | 10.3 | 1.6 | 2.6 |
| peak area | y= 0.0214 x - 0.01 | 0.994 | 8.5 | 0.87 | 3.6 | |
| MC-YR | peak height | y = 0.0096 x - 0.0001 | 0.991 | 4.3 | 4.5 | 6.2 |
| peak area | y = 0.0092 x - 0.009 | 0.996 | 6.2 | 3.9 | 4.2 |
CV, coefficient of variation determined for 100 μg/L of MCs
MDL was determined based on standard solutions of 5 μg/L for MC-LR and MC-RR and 10 μg/L for MC-YR
MQL was determined with standard solutions prepared in MilliQ water.
Good correlation was observed between values generated by ratio of intensities (peak height) of microcystin congener to internal standard and the corresponding ratio of areas (peak area). Previously, it has been reported that the peak height ratio is more suitable for MALDI quantification because the integration of a selected baseline and range can be highly variable, particularly when using a linear, nonflectron mode (Duncan et al., 2008; Howard and Boyer, 2007). Previous reports indicated little difference between R2 values for MC standard solutions when using non microcystin internal standards, but a higher coefficient of variation (CV) for peak area ratios, a higher limit of detection, and a smaller linearity of range (Howard and Boyer, 2007). We did not observe a substantial difference in limit of detection between peak area and peak height minimum detection limits; however, the linear range obtained using peak area appeared to decrease with increasing microcystin standard solution indicated by the divergence of the standard curves at higher concentrations, so peak intensity was adopted for quantification.
3.3 MDL and MQL with IS-2
We expected similar results with respect to linearity of range and minimum detection limit. Likely due to the presence of two arginine side chains, IS-2 exhibited greater ionization than IS-1, resulting in decreased slopes for the calibration standard curves for all three microcystin congeners as compared with IS-1 (Supporting Information, Figures C.1-C.3 and D.1-D.3). However, the limit of detection was not affected, MDL = 2.8, 1.4, and 4.5 μg/L, respectively for MC-LR, MC-RR and MC-YR, Table 2, and were very close to the values obtained with IS-1.These data suggest the limit of detection operates independently of ionization of internal standard. The use of lower concentrations of IS, which produced peaks of similar size to those of the MC standards, did not significantly affect the detection limit (data not shown). To study the potential influence of the sample matrix, this time MQL was quantified using MC standards diluted in the ref-CB water sample. Under these conditions, the calculated MQL were 6.5, 4.6, and 6.4 μg/L, for MC-LR, MC-RR and MC-YR, respectively. Contributing to the robustness of this method, sample matrix effects appear to have minimal effect on MQLs. While the peak height intensity and peak area curves appear to diverge a bit, this discrepancy had minimal effect on quantification. MDL curves in MilliQ water yielded similar results (data not shown). While these detection limits remain above the provisional guideline provided by the WHO for drinking water, it is worth noting that a concentration step was not employed prior to the screening of samples. The method has the potential to be adapted for screening of drinking water with an additional concentration step. Regardless, a major advantage is that it allows screening of a large number of untreated surface water samples almost simultaneously.
Table 2.
Determination of MDL and MQL for MC-LR, MC-RR, MC-YR Standard Solutions Using Both Peak Height and Peak Area Ratios with IS-2.
| Congener | Ratio Parameter | Regression Equation | R2 | CV1 | MDL (μg/L)2 | MQL (μg/L)3 |
|---|---|---|---|---|---|---|
| MC-LR | peak height | y = 0.0033 x - 0.017 | 0.984 | 18.5 | 2.8 | 6.5 |
| peak area | y = 0.0031 x - 0.010 | 0.991 | 25.3 | 1.8 | 4.4 | |
| MC-RR | peak height | y = 0.0124 x - 0.057 | 0.991 | 6.2 | 1.4 | 4.6 |
| peak area | y = 0.0116 x - 0.050 | 0.992 | 3.1 | 2.3 | 4.3 | |
| MC-YR | peak height | y = 0.0025 x - 0.013 | 0.992 | 7.7 | 4.5 | 6.4 |
| peak area | y = 0.0021x - 0.0087 | 0.985 | 9.8 | 3.6 | 4.5 |
CV, coefficient of variation determined for 100 μg/L of MCs
MDL was determined based on standard solutions of 5 μg/L for MC-LR and MC-RR and 10 μg/L for MC-YR; to study matrix effect
MQL was determined with standard solutions prepared in the reference water sample ref-CB.
3.4 Selection of IS
The utility of any IS may be severely threatened by the occurrence of common compounds of similar mass in the sample, as demonstrated in the case of the bloom samples taken from three water bodies in northern Uruguay along the Rio Negro. Upon an initial screening without internal standard, a number of the samples were determined to contain an unknown peak at 1072.6 (m/z). Post source decay (PSD) analysis of this peak did not show the distinctive fragmentation products (ADDA fragment [PhCH2CH(OCH3)]+ =135 (m/z), [Mdha-Ala + H]+ = 155 (m/z), [Glu-Mdha + H]+ = 213 (m/z), [ADDA-fragment-Glu-Mdha]+ = 375.0 (m/z)) of the MC core, ruling out that this was a MC congener. Due to the close proximity to the peak of IS-1 (1073.6 m/z) and the high signal intensity of the 1072.6 m/z compound IS-1 could not be used with our particular set of samples. Nevertheless, IS-1 may prove useful for surface water analysis in other regions of the world, with different contaminants and should not be discounted. This result actually prompted the synthesis of IS-2 after an additional screening of up to 20 raw surface water samples showed that no interfering compounds with similar m/z value to the calculated mass of IS-2 (1116.6 m/z) occurred in the samples. This case highlights the importance of a simple and flexible IS synthesis strategy that makes it possible to design the m/z of the IS to maximally avoid interference present in samples. In particular, blooms in this region of the world have been shown to produce uncommon MCs (Andrinolo et al., 2007; Neumann et al., 2000), so we emphasize the importance of pre-screening bloom samples for potential interfering peaks. In particular, the presence of [DAsp3,ADMAdda5, Dhb7] MC-HtyR (MW =1073)(Beattie et al., 1998) could interfere with IS-1, and [LMeLan7] MC-LR (MW=1115)(Namikoshi et al., 1995) could interfere with IS-2, if present in the bloom sample.
3.5 Spike Recoveries for Surface Bloom Samples
Table 3 shows the results and spike recoveries of samples taken during the blooming season from three reservoirs in northern Uruguay along the Rio Negro, utilizing peak height ratios. The results and spike recoveries quantified utilizing peak area ratio are available in Supporting Information, Table A. Fortification with congeners was performed simultaneously, using 100 μg/L of the respective congener (except for wbB4 that was spiked with 1000 μg/L). The concentration of the spiked MCs was chosen to be of the same order than its original concentration in the sample, in order to fulfill our goal of developing a method that could be used for direct screening of unprocessed raw water samples. All spike recoveries were between 80 and 120%. Determined concentrations were highly reproducible and values by peak height and peak area corresponded closely throughout the spiking experiments. As mentioned, for each spiking set, a calibration curve with three to four data points was established with each microcystin congener standard and IS-2. We did all initial experiments using eight replicate well spots, but data analysis showed that three replicates might be used without altering estimated concentrations or recovery of spiked samples (data not included).
Table 3.
Spike Recoveries for Surface Water Bloom Samples Calculated with Peak Height Ratio using the Internal Standard IS-2
| MC-LR | MC-RR | MC-YR | ||||
|---|---|---|---|---|---|---|
| Sample | Initial conc. | Recovery | Initial conc. | Recovery | Initial conc. | Recovery |
| μg/L | % | μg/L | % | μg/L | % | |
| wbA 1 | 307 | 116 | 234 | 119 | 439 | 107 |
| wbA 2 | 157 | 93.9 | 41.6 | 87.7 | 55.7 | 80.2 |
| wbB 1 | 235 | 94.5 | 198 | 80.0 | 347 | 97 |
| wbB 3 | 76.3 | 84.5 | < MQL | 82.1 | 45.0 | 109 |
| wbB 4 | 12000 | 84.4 | 1470 | 99.8 | 1240 | 79.0 |
| wbB 5 | 35.3 | 84.2 | ND | 86.2 | ND | 108 |
| wbC 1 | 49.5 | 81.1 | < MQL | 90.3 | < MQL | 108 |
| wbC 2 | 417 | 80.4 | 53.5 | 93.5 | 95.8 | 94 |
| wbC I | 27.4 | 86.2 | ND | 89.9 | ND | 110 |
All samples were spiked with 100 μg/L of each congener except for sample wbB 4 4 that was spiked with 1000 μg/L
4. Conclusions
Previous work on the quantitative use of MALDI-TOF for MC analysis showed that the best sensitivity was obtained when nodularin was used as IS (Howard and Boyer, 2007; Puddick et al., 2011). This is probably due to the high similitude of nodularin with the common structure of MCs. However, nodularin has a smaller amino acid ring, its size (m/z 825.4) is close to interfering peaks of the MALDI matrix; in addition, it is a cyanobacterial toxin and could become target of the analysis. As an alternative, the reaction of sulfhydryl groups with the Mdha moiety of commercial or in-house purified MCs offers a simple and versatile method to prepare ISs for MC quantification that preserve the physicochemical characteristics of these toxins. Additionally, the combination of different MC congeners paired with different sulfhydryl compounds allows for adjustment of the m/z of the IS to the particular pattern of common interfering compounds in the samples, as demonstrated by the advantageous use of IS-2 versus IS-1 when a frequent interfering compound of 1072.6 m/z was found in surface water blooms in Uruguay. The combination of ISs highly similar to the analyte with the high sensitivity and resolution of the reflectron mode MALDI-TOF allowed direct analysis of raw water samples with increased sensitivity. Although still not sufficient for direct screening of drinking water, the method fulfills our purpose of developing a rapid screening tool for monitoring environmentally relevant concentrations of MCs using small volumes of untreated samples. Indeed, the MQL for untreated surface water obtained in this study was 6.5, 4.6 and 6.4 μg/L for MC-LR, MC-RR and MC-YR, respectively, while Howard and Boyer using a linear MALDI-TOF instrument found a MQL for cell extracts of 190, 230 and 190 μg/L, respectively. With this 30 fold increase in sensitivity, the method performs within a linear range of 7 – 5.000 μg/L, which is of great relevance for the intended application of the technique. Certainly, this range covers the reference values of < 15-25 μg/L that has been adopted by many countries as an immediate public health action level for recreational waters (Chorus, 2012), and it also covers the vast majority of the concentrations of these congeners found in fresh water reservoirs and needed to be monitored to manage the progression of cyanobacterial blooms (Pirez et al., 2013). High throughput spatial and temporal monitoring could substantially enhance our understanding of human health risks presented by cyanobacterial blooms and inform public health recommendations. In addition, spatial and temporal monitoring could help identify potential factors or environmental triggers for enhanced toxin production in hopes of future prevention. Considering that the new method allows for quantification of some of the major MC congeners from few microliters of unprocessed sample with a rapid per sample analysis that allows processing of numerous samples at a time with minimal instrument maintenance and per-sample cost, together with rapid turnaround time for results, the method offers a salient option with which researchers, treatment water facilities or public health agencies can address cyanobacterial blooms, an ever increasing human and animal health concern.
Supplementary Material
Highlights.
Easily synthesized ISs for quantitative MALDI-TOF of MCs were developed.
Microliters of untreated raw water samples were analyzed within minutes for MCs.
MDLs were consistently below 5 μg/L with no concentration step.
Recoveries from spiked surface water samples remained between 80 and 119%.
We offer a high-throughput, screening tool for rapid MC detection in surface water.
Figure 3. Mass spectra of low concentrations of MC-LR, MC-RR and MC-YR in presence of IS-2.
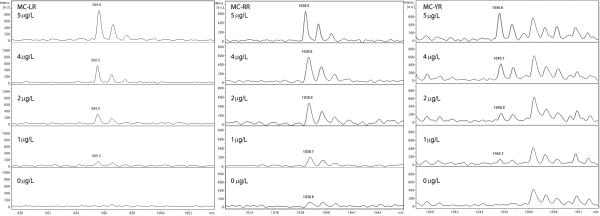
The spectra were prepared with 10 μl of the relevant concentration of microcystin standard prepared in MilliQ water with 1 μl of IS-2 (diluted to a final concentration of 40 μg/L) and then thoroughly mixed with 10 μl of CHCA matrix. The 2 μl spots were allowed to dry and then shot at 30% intensity.
Acknowledgments
Funding: This work was supported by NIH Fogarty International Center Grant TW05718, CSIC 400/2008, and ANII FMV 7263. The U.S. Fulbright Student Research Fellowship (Montevideo, Uruguay) and the USEPA STAR Graduate Student Fellowship provided support for the first author.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors have no conflicts of interest to report.
Protocols: No human subjects or experimental animals were involved with the research presented in this manuscript.
References
- Andrinolo D, Pereira P, Giannuzzi L, Aura C, Massera S, Caneo M, Caixach J, Barco M, Echenique R. Occurrence of Microcystis aeruginosa and microcystins in Río de la Plata river (Argentina). Acta toxicólogica argentina. 2007;15:8–14. [Google Scholar]
- Beattie KA, Kaya K, Sano T, Codd GA. Three dehydrobutyrine (Dhb)- containing microcystins from the cyanobacterium Nostoc sp. Phytochemistry. 1998;47:1289–1292. [Google Scholar]
- Brena BM, Diaz L, Sienra D, Ferrari G, Ferraz N, Hellman U, Gonzalez-Sapienza G, Last JA. ITREOH building of regional capacity to monitor recreational water: development of a noncommercial microcystin ELISA and its impact on public health policy. Int J Occup Environ Health. 2006;12:377–385. doi: 10.1179/oeh.2006.12.4.377. [DOI] [PubMed] [Google Scholar]
- Chorus I. Current approaches to cyanotoxin risk assessment, risk management and regulations in different countries. Federal Environmental Agency (Umweltbundesamt); Dessau-Rosslau: 2012. [Google Scholar]
- Chorus I, Falconer IR, Salas HJ, Bartram J. Health risks caused by freshwater cyanobacteria in recreational waters. J Toxicol Environ Health B Crit Rev. 2000;3:323–347. doi: 10.1080/109374000436364. [DOI] [PubMed] [Google Scholar]
- Codd GA. Cyanobacterial toxins, the perception of water quality, and the prioritisation of eutrophication control. Ecological Engineering. 2000;16:51–60. [Google Scholar]
- de Figueiredo DR, Azeiteiro UM, Esteves SM, Goncalves FJ, Pereira MJ. Microcystin-producing blooms--a serious global public health issue. Ecotoxicol Environ Saf. 2004;59:151–163. doi: 10.1016/j.ecoenv.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dietrich D, Hoeger S. Guidance values for microcystins in water and cyanobacterial supplement products (blue-green algal supplements): a reasonable or misguided approach? Toxicol Appl Pharmacol. 2005;203:273–289. doi: 10.1016/j.taap.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Dodds WK, Bouska WW, Eitzmann JL, Pilger TJ, Pitts KL, Riley AJ, Schloesser JT, Thornbrugh DJ. Eutrophication of U.S. freshwaters: analysis of potential economic damages. Environ Sci Technol. 2009;43:12–19. doi: 10.1021/es801217q. [DOI] [PubMed] [Google Scholar]
- Donohue J, Orme-Zavaleta J, Burch M, Dietrich D, Hawkins B, Lloyd T, Munns W, Steevens J, Steffensen D, Stone D, Tango P. Risk Assessment Workgroup report. Adv Exp Med Biol. 2008;619:759–829. doi: 10.1007/978-0-387-75865-7_35. [DOI] [PubMed] [Google Scholar]
- Duncan MW, Roder H, Hunsucker SW. Quantitative matrix-assisted laser desorption/ionization mass spectrometry. Brief Funct Genomic Proteomic. 2008;7:355–370. doi: 10.1093/bfgp/eln041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhard M, von Dohren H, Jungblut P. Rapid typing and elucidation of new secondary metabolites of intact cyanobacteria using MALDI-TOF mass spectrometry. Nat Biotechnol. 1997;15:906–909. doi: 10.1038/nbt0997-906. [DOI] [PubMed] [Google Scholar]
- Ferranti P, Nasi A, Bruno M, Basile A, Serpe L, Gallo P. A peptidomic approach for monitoring and characterising peptide cyanotoxins produced in Italian lakes by matrix-assisted laser desorption/ionisation and quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2011;25:1173–1183. doi: 10.1002/rcm.4973. [DOI] [PubMed] [Google Scholar]
- Fischer A, Hoeger SJ, Stemmer K, Feurstein DJ, Knobeloch D, Nussler A, Dietrich DR. The role of organic anion transporting polypeptides (OATPs/SLCOs) in the toxicity of different microcystin congeners in vitro: a comparison of primary human hepatocytes and OATP-transfected HEK293 cells. Toxicol Appl Pharmacol. 2010;245:9–20. doi: 10.1016/j.taap.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Howard KL, Boyer GL. Quantitative analysis of cyanobacterial toxins by matrix-assisted laser desorption ionization mass spectrometry. Anal Chem. 2007;79:5980–5986. doi: 10.1021/ac0705723. [DOI] [PubMed] [Google Scholar]
- Meriluoto J, Lawton L, Harada K. Isolation and detection of microcystins and nodularins, cyanobacterial peptide hepatotoxins. Methods Mol Biol. 2000;145:65–87. doi: 10.1385/1-59259-052-7:65. [DOI] [PubMed] [Google Scholar]
- Msagati TA, Siame BA, Shushu DD. Evaluation of methods for the isolation, detection and quantification of cyanobacterial hepatotoxins. Aquat Toxicol. 2006;78:382–397. doi: 10.1016/j.aquatox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Namikoshi M, Sun F, Choi B, Rinehart KL, Carmichael WW, Evans WR, Beasley VR. Seven more microcystins from Homer lake cells: application of the general method for structure assignment of peptides containing,-dehydroamino acid unit(s). J. Org. Chem. 1995;60:3671–3679. [Google Scholar]
- Neumann U, Campos V, Cantarero S, Urrutia H, Heinze R, Weckesser J, Erhard M. Cooccurrence of non-toxic (cyanopeptolin) and toxic (microcystin) peptides in a bloom of Microcystis sp. from a Chilean lake. Syst Appl Microbiol. 2000;23:191–197. doi: 10.1016/S0723-2020(00)80004-1. [DOI] [PubMed] [Google Scholar]
- Pan C, Xu S, Zhou H, Fu Y, Ye M, Zou H. Recent developments in methods and technology for analysis of biological samples by MALDI-TOF-MS. Anal Bioanal Chem. 2007;387:193–204. doi: 10.1007/s00216-006-0905-4. [DOI] [PubMed] [Google Scholar]
- Pearson L, Mihali T, Moffitt M, Kellmann R, Neilan B. On the chemistry, toxicology and genetics of the cyanobacterial toxins, microcystin, nodularin, saxitoxin and cylindrospermopsin. Mar Drugs. 2010;8:1650–1680. doi: 10.3390/md8051650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirez M, Gonzalez-Sapienza G, Sienra D, Ferrari G, Last M, Last JA, Brena BM. Limited analytical capacity for cyanotoxins in developing countries may hide serious environmental health problems: simple and affordable methods may be the answer. J Environ Manage. 2013;114:63–71. doi: 10.1016/j.jenvman.2012.10.052. [DOI] [PubMed] [Google Scholar]
- Puddick J, Prinsep MR, Wood SA, Craig Cary S, Hamilton DP. Enhanced Sample Preparation for Quantitation of Microcystins by Matrix-assisted Laser Desorption/Ionisation-Time of Flight Mass Spectrometry. Phytochem Anal. 2011:285–291. doi: 10.1002/pca.1356. [DOI] [PubMed] [Google Scholar]
- Pusch W, Kostrzewa M. Application of MALDI-TOF mass spectrometry in screening and diagnostic research. Curr Pharm Des. 2005;11:2577–2591. doi: 10.2174/1381612054546932. [DOI] [PubMed] [Google Scholar]
- Qin B, Zhu G, Gao G, Zhang Y, Li W, Paerl HW, Carmichael WW. A drinking water crisis in Lake Taihu, China: linkage to climatic variability and lake management. Environ Manage. 2010;45:105–112. doi: 10.1007/s00267-009-9393-6. [DOI] [PubMed] [Google Scholar]
- Rinehart K, Namikoshi M, Choi B. Structure and biosynthesis of toxins from blue-green algae (cyanobacteria). J Appl Phycol. 1994;6:159–176. [Google Scholar]
- Runnegar M, Berndt N, Kong SM, Lee EY, Zhang L. In vivo and in vitro binding of microcystin to protein phosphatases 1 and 2A. Biochem Biophys Res Commun. 1995;216:162–169. doi: 10.1006/bbrc.1995.2605. [DOI] [PubMed] [Google Scholar]
- Smith JL, Boyer GL. Standardization of microcystin extraction from fish tissues: a novel internal standard as a surrogate for polar and non-polar variants. Toxicon. 2009;53:238–245. doi: 10.1016/j.toxicon.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Szajli E, Feher T, Medzihradszky KF. Investigating the quantitative nature of MALDI-TOF MS. Mol Cell Proteomics. 2008;7:2410–2418. doi: 10.1074/mcp.M800108-MCP200. [DOI] [PubMed] [Google Scholar]
- Welker M, Fastner J, Erhard M, von Dohren H. Applications of MALDI-TOF MS analysis in cyanotoxin research. Environ Toxicol. 2002;17:367–374. doi: 10.1002/tox.10073. [DOI] [PubMed] [Google Scholar]
- Wood SA, Rueckert A, Hamilton DP, Cary SC, Dietrich DR. Switching toxin production on and off: intermittent microcystin synthesis in a Microcystis bloom. Environmental Microbiology Reports. 2011;3:118–124. doi: 10.1111/j.1758-2229.2010.00196.x. [DOI] [PubMed] [Google Scholar]
- Znachor P, Jurczak T, Komarkova J, Jezberova J, Mankiewicz J, Kastovska K, Zapomelova E. Summer changes in cyanobacterial bloom composition and microcystin concentration in eutrophic Czech reservoirs. Environ Toxicol. 2006;21:236–243. doi: 10.1002/tox.20176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.