

Inflammation: From Tissue Repair to Mechanism of Disease
Inflammation is a coordinated cellular-humoral response to injury. A close interaction between resident cells (i.e. endothelial cells, fibroblasts, dendritic cells) and leukocytes regulates the initiation and resolution of the acute inflammatory response. Constitutive membrane and cytoplasmic receptors function as guardians that ‘signal the alarm’ when activated by products of cell destruction or microbial invasion. This first-line innate immune response initiates a process of leukocyte mobilization from the bone marrow, recruitment to the ‘activated’ endothelium, and migration to the site of tissue injury to prevent infection and facilitate tissue repair. Although critical for many forms of repair, the inflammatory response may also become a mechanism for progressive injury, impaired healing, and disease.
Central Role of Interleukin-1 in the Sterile Inflammatory Response
Interleukin-1 (IL-1) is an apical pro-inflammatory mediator in acute and chronic inflammation, and a powerful inducer of the innate immune response.1,2 The production and activity of IL-1 is finely regulated at multiple levels and very small concentrations of exogenous IL-1 can induce a sepsis-like syndrome and shock.1, 2 IL-1 induces the synthesis and expression of several hundreds of secondary inflammatory mediators.1, 2 IL-1 also induces its own production and processing and this step is key in the pathogenesis of many autoinflammatory diseases.1, 3 Two related genes (IL-1α and IL-1β) code for two different proteins that bind the same IL-1 receptor (type I). IL-1α is synthesized as a fully active peptide that remains membrane-bound or may be released from the cytoplasm during cell death. IL-1α thereby participates more prominently in local response to injury and less in the systemic inflammatory response. 1, 2 IL-1β is the main form of circulating IL-1 and is initially synthesized as a precursor (proIL-1β) that becomes activated by caspase-1 cleavage in the setting of a macromolecular structure known as the inflammasome.1, 4 Caspase-1 also participates in the secretion of active IL-1β that can then bind the membrane IL-1 receptor on the same cell (autocrine), neighboring cells (paracrine), or enter the circulation targeting distant cells (endocrine).1, 2 The constitutive and inducible expression of proIL-1β are highly cell-type specific.1, 2 Both IL-1 isoforms induce proIL-1β synthesis, as do other proinflammatory stimuli such as the numerous Toll-like receptor (TLR) agonists (i.e. cell debris or bacterial products).1, 5 In addition, many potential triggers of the inflammasome have been identified, including microbial antigens, cell debris, ATP, ischemia, cholesterol crystals, and other TLRs ligands such as danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs).4, 6-9
Activation of inflammasome following tissue injury induces a local surge of IL-1β that significantly amplifies the inflammatory response, recruiting more inflammatory cells, stimulating metalloproteinase activities, and ultimately inducing inflammatory cell death (pyroptosis) in leukocytes and resident cells.10-12 The sensitivity to stimuli and the intensity and duration of the ensuing inflammatory response are also highly dependent on genetic variability. Cryopyrin-Associated Periodic Syndromes (CAPS) are rare chronic autoinflammatory disorders in which the cryopyrin (NLRP3) “sensor” of the inflammasome is constitutively active, leading to a debilitating condition of IL-1β overproduction.10 Other polymorphisms in the cryopyrin gene have been linked with increased susceptibility to autoinflammatory diseases.11 Polymorphisms in the IL-1 gene cluster (IL-1α, IL-1β, and IL-1 receptor antagonist) that result in greater expression of the agonists or reduced expression of the antagonist, have been associated with premature onset, increased severity, or worse outcomes in patients with various inflammatory conditions, ranging from rheumatoid arthritis, periodontitis, inflammatory bowel disease, Alzheimer disease, and others.12-14
Heart Disease: an Inflammatory Disease
Cardiovascular disease is the leading cause of death in the US and worldwide.15 Millions of patients succumb to the consequences of myocardial ischemia (primarily triggered by acute coronary syndromes), heart failure (impaired contractility/relaxation), and arrhythmias.15 The epidemics of cardiovascular risk factors such as diabetes, hyperlipidemia, obesity, arterial hypertension, and tobacco use have led to increased numbers of patients at risk for primary and secondary acute coronary events worldwide. While the mortality for acute coronary events has dropped as a consequence of improved interventional treatments, this has resulted in an increased number of survivors at high risk of heart failure, recurrent events, and cardiovascular mortality.16, 17 Cardiovascular disease has classically been considered a metabolic—rather than inflammatory—disease, in which changes in lipid and glucose metabolism are linked to the progression of coronary artery disease and impaired function. Nevertheless, reports of an inflammatory response during heart disease began to emerge several decades ago.18-20 Patients with acute coronary events have increased inflammatory markers, including C reactive protein (CRP), which serves a surrogate for IL-1 activity.21 For example, early studies observed that high CRP levels (>3 mg/L) in patients with unstable angina predicted a significantly worse prognosis.19 Subsequent studies validated that elevated CRP also predicted heart failure related mortality in patients with impaired systolic function (EF<30%) and clinical recurrence after coronary revascularization procedures such as balloon angioplasty, coronary artery stenting, and coronary artery bypass graft surgery,22, 23 as well as in apparently healthy subjects, and even within levels of CRP previously considered normal.24-27 Moreover, genetic polymorphisms in IL-1β and IL-1Ra, and other genes, have been associated with premature onset of atherosclerosis and acute myocardial infarction.28
The natural history of heart disease is not linear.29 Evidence shows that changes in the vessel wall and the heart start decades before the disease becomes clinically evident, and a chronic subclinical inflammation may promote or accelerate the disease (Figure 1).29 The atherothrombotic event leading to acute myocardial infarction is a rather stochastic event occurring on a predisposed substrate: a vulnerable plaque that experiences an intolerable stress leading to rupture and acute thrombosis. An enhanced inflammatory response may predispose the plaques to rupture, and a heightened inflammatory response during myocardial injury may affect the degree of healing and progression to heart failure. Clinically stable patients with prior acute events and symptomatic heart failure display evidence of chronic inflammation, which may further aggravate the cardiac dysfunction and/or predisposing the patient to further decompensation (Figure 1).
Figure 1.
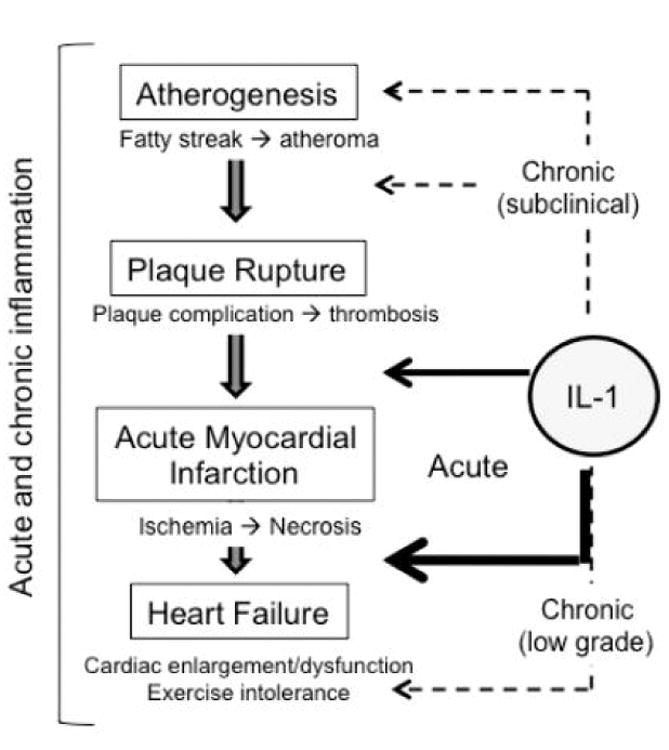
Inflammation and IL-1 in the wide spectrum of ischemic heart disease. The natural history of heart disease is not linear: evidence shows that atherogenesis is a slow process and a chronic subclinical inflammation may promote or accelerate the disease, whereas the atherothrombotic event leading to acute myocardial infarction is more of a sudden and rather stochastic event occurring on a predisposed substrate. An enhanced inflammatory response may predispose the plaques to rupture, and an even more intense response follows the myocardial injury affecting the degree of healing and the progression to heart failure. A chronic inflammation occurs also in many clinically stable patients with prior acute events and symptomatic for heart failure, potentially aggravating the cardiac dysfunction and/or predisposing to further decompensation.
Role of Interleukin-1 in the Progression of Atherosclerosis
Atherosclerosis and atherothrombosis are the underlying mechanisms for the great majority of first and recurrent coronary events.29 The degree to which plaques are formed, progress, and rupture are dependent, at least in part, upon the degree of inflammation in the plaque. In human atherosclerotic plaques, the expression of IL-1α and IL-1β appears to correlate with the progression of atherosclerotic plaques, with minimal expression in healthy coronary arteries, increased expression in simple atherosclerotic plaques, and high expression in complicated plaques.30
Experimental studies
Experimental studies in atherosclerosis-prone animals have consistently shown that genetic deletion or pharmacologic inhibition of IL-1 signaling reduces the formation and progression of atherosclerotic plaques, whereas increased IL-1 activity (by exogenous administration of IL-1β or downregulation of the naturally occurring IL-1 receptor antagonist) favors its progression.31-33 Bone marrow chimeric studies identified IL-1 signaling in the vessel wall—rather than in leukocytes—as a major determinant of atheroma formation.34, 35
IL-1β mediates tissue response to endothelial injury, contributing to the proliferative intimal response.30, 36 Treatment with IL-1 blockers inhibits the progression of atherosclerosis,31 however IL-1 effects may be variable based upon the vascular district and may contribute to both plaque burden and outward vessel remodeling.37 IL-1β also exerts direct effects on thrombosis,38 insulin resistance,39 and obesity-related metabolic derangements.34 Figure 2 shows the central role of IL-1 in atherothrombosis.40
Figure 2.
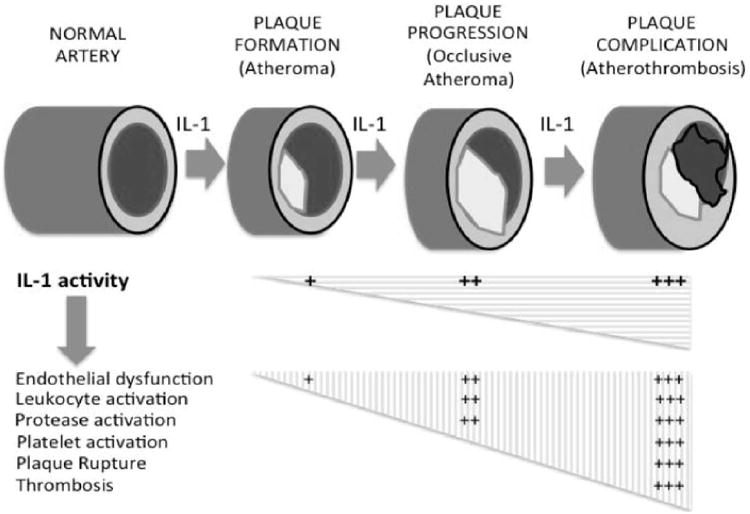
IL-1 and atherosclerosis. The natural history of atherosclerosis is characterized by plaque formation, progression (growth) and complication. IL-1 plays a key role in the formation, progression and complication of the atherosclerotic plaque. An increase in IL-1 activity causes a destabilization of the plaque, rupture and superimposed thrombus formation. Data excerpted from Abbate et al.40
Clinical studies
Due to the challenges of direct measurement of plasma IL-1β levels (often undetectable even in diseases with clear evidence of increased IL-1 activity), only few studies have measured levels and shown that IL-1β levels were increased in patients with a greater atherosclerotic burden41-43 or that IL-1β levels were associated with less favorable prognosis after acute coronary syndromes.44-46 The Interleukin-1 family of cytokines includes several different proteins with agonistic pro-inflammatory activity (such as IL-1β, IL-18 and IL-33) and others with antagonist anti-inflammatory activity (IL-1 receptor antagonist [IL-1Ra], IL-18 binding protein, and soluble ST2).1 Prospective and retrospective clinical studies have described that the plasma levels of IL-1Ra and ST2 (as surrogate markers for IL-1 activity) predict adverse outcome in patients with acute coronary syndromes.47-50 Increasing levels of IL-1Ra and of IL-6 (a secondary downstream mediator of IL-1β) predicted adverse outcome in patients with unstable angina.50 Similar results have been described more recently with the soluble ST2 biomarker.51 Clinical trials testing the effects of IL-1 blockers on atherosclerosis or atherothrombosis are lacking.
Role of Interleukin-1 in the Healing Process Following Acute Myocardial Infarction
Injury to the myocardium induces a sterile inflammatory response to promote healing,51 but unfortunately, restitutio ad intregrum does not occur in the heart and dead tissue is ultimately replaced by a non-functional scar. Healing is a dynamic process lasting for at least several weeks after myocardial injury. Cell death is initially necrotic in the infarct core due to an abrupt decrease in cellular ATP levels, but progresses in the borderzone and non-ischemic myocardium through a more programmed form of cell death. This process is generally classified as apoptosis due to the characteristic DNA fragmentation and caspase-3 activation, but also share features of inflammatory cell death or “pyroptosis”.52, 53
As in all forms of sterile inflammation, IL-1 is largely involved in the recruitment of the leukocytes and coordination of the inflammatory response to infarction.51 Upon cell death, local cardiac resident endothelial cells, cardiomyocytes, and fibroblasts release cytoplasmic IL-1α and pro-IL-1β into the tissue that is susceptible to cleavage/activation IL-1β by extracellular enzymes such as neutrophil elastase.1 Viable injured resident cells also release active IL-1β following activation of the inflammasome. Once leukocytes have been recruited to the infarct, the major source of IL-1β is likely to be the activated leukocyte. The formation of an active inflammasome in the heart follows and regulates the healing process. In preclinical models, genetic deletion of the scaffold component of the inflammasome (apoptosis-associated speck-like protein containing a caspase recruitment domain [ASC]), lead to a significant improvement in cardiac healing after ischemia-reperfusion.54 Similarly, silencing or genetic deletion of cryopyrin (NLRP3), one of the sensor components of the inflammasome, prevented the formation of an active inflammasome (active caspase-1) and protected the heart from ischemic injury.53, 55,56 These data are highly consistent with prior descriptions of a central role for caspase-1.57, 58 The mechanisms leading to the formation of the inflammasome in the heart and the consequences of caspase-1 activation in the heart are not completely characterized. Silencing or pharmacologic inhibition of the purinergic ATP/ADP receptor P2X7 prevented caspase-1 activation during AMI, suggesting that extracellular ATP is an important trigger for the formation of the inflammasome.53 Nevertheless, it cannot be excluded that other triggers may be also important and/or that P2X7 is important for other mechanisms such as release of mature IL-1β. Other purinergic receptors, such as the adenosine A2B receptor, may also be involved in the formation of the inflammasome.59 The expression of the components of the inflammasome also appears to be tissue- and cell-type specific. One study reported the fibroblast (and not the cardiomyocyte) as the prevalent cell type forming the inflammasome, based on the finding of maturation of IL-1β in fibroblasts but not cardiomyocytes.54 A second study, however, demonstrated that the cardiomyocytes expressed all the components of the inflammasome (ASC, cryopyrin, and active caspase-1), but found no evidence of mature IL-1β in cardiomyocytes, suggesting that the processes of inflammasome formation, caspase-1 activation, and IL-1β maturation may be different and disconnected in different cell types.55 Moreover, the activation of caspase-1 in the cardiomyocyte—and perhaps in other cells as well—may lead to IL-1β-independent effects such as inflammatory cell death by pyroptosis (Figure 3).53, 56 In rare genetic diseases such as CAPS, the inflammasome is constitutively active and patients suffer from the consequences of uncontrolled sterile inflammation.60 The finding that CAPS patients were highly responsive to IL-1β blockers has lead to the theory that most inflammasome-mediated effects are mediated by increased IL-1β activity.61
Figure 3.
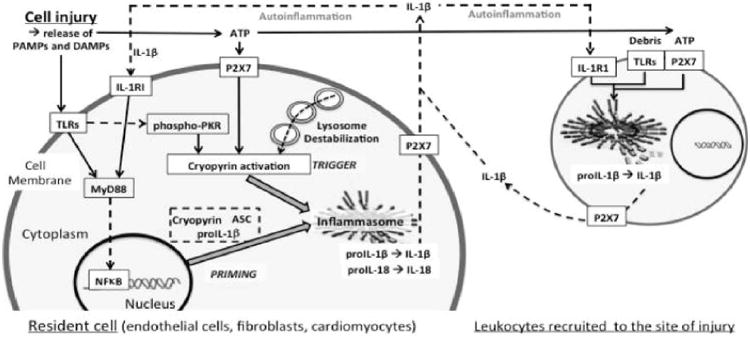
Cryopyrin activation and inflammasome formation. The figure shows a simplified scheme of cryopyrin activation and inflammasome formation in tissue resident cells and leukocytes recruited at the site of injury. TLR/IL-1RI agonists induce the inflammasome components (priming) through MyD88 signaling. ATP (through P2X7) or intracellular signals associated with lysosome destabilization activate cryopyrin triggering the formation of the inflammasome. The inflammasome processes and releases active IL-1β which further enhance the inflammatory response (autoinflammation). Data excerpted from Abbate et al.40. Abbreviations: ASC=apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain; ATP=adenosine triphosphate; DAMP= danger-associated molecular patterns; IL-1=Interleukin-1; IL-1RI=IL-1 receptor type I; MyD88=myeloid differentiation factor-88; NF-κB=nuclear factor-κB; PAMP=pathogen-associated molecular pattern; P2X7=purinergic receptor 2X7; PKR=RNA dependent protein kinase; TLR=Toll-like receptor.
Experimental studies
Genetic deletion of the IL-1 signaling receptor (IL-1R1) was protective in models of AMI due to ischemia/reperfusion62 and permanent ligation,63 as shown by smaller infarct size, reduced left ventricular enlargement, and reduced left ventricular dysfunction. Conversely, mice lacking the naturally occurring IL-1 receptor antagonist (IL-1Ra), developed a severe form of cardiomyopathy characterized by increased cardiomyocyte apoptosis and ventricular dilatation after AMI.63 Moreover, overexpression of IL-1Ra appeared to consistently improve the cardiac response to AMI.63, 64 Administration of the recombinant form of the IL-1 receptor antagonist, after the onset of ischemia, lead to a significant reduction in cardiomyocyte apoptosis and left ventricular dilatation, independent of changes in infarct size,65, 66 whereas pretreatment with anakinra induced a significant reduction in infarct size.69 These findings suggest that IL-1 signaling may be involved in myocardial infarction healing by multiple mechanisms. An initial surge of IL-1 activity early during acute myocardial infarction may be related to the release of intracellular IL-1α,67 which is active and is not processed by the inflammasome, or may be related to the release of pro-IL-1β, which can be cleaved to IL-1β extracellularly and independent of the inflammasome. A second wave of increased IL-1 activity occurs later in the course as a result of leukocyte recruitment to the injured site and activation of the inflammasome. Genetic deletion of IL-1R1 or administration of anakinra block signaling of both IL-1β and IL-1α, rendering it difficult to determine which mechanism is predominant. Studies assessing signaling downstream of the IL-1 receptor, such as the Myeloid Differentiation Factor-88 (MyD88),68 further strengthen the evidence of a critical role of IL-1 activity but do not allow discernment of the exact contribution of the isoforms. An additional anti-apoptotic role of the intracellular IL-1Ra isoform in cardiomyocytes has been described.72 The contribution of IL-1 signaling in infiltrating leukocytes is strengthened by data obtained in experiments of bone-marrow chimeric mice with genetic deletion of the MyD88 or the Interleukin-1 Receptor Associated Kinase-4 (IRAK-4).69, 70 More recent data suggest that IL-1 signaling in the bone marrow affects also the regenerative potential in the heart.71
Several additional IL-1 blockers have been developed recently that have provided opportunities to validate the effect IL-1 blockade in pre-clinical models.40 The use of a recombinant chimeric protein composed of the IL-1 receptor, the IL-1 receptor associated protein, and the Fc fragment of an immunoglobulin constitutes the “IL-1 trap”. The beneficial effects on cardiac remodeling seen with the use of a murine IL-1 trap in the mouse provided the initial evidence that IL-1 blockade in cardiac remodeling is a class effect.72 The IL-1 trap also blocks the effects of endogenous IL-1 receptor antagonist without significant adverse effects. In none of these experiments there was evidence of impaired healing or increased risk of cardiac rupture, suggesting that IL-1 signaling may not be necessary for cardiac healing after acute myocardial infarction.
The evidence supporting the role of IL-1β vs IL-1α in cardiac remodeling after acute myocardial infarction is less straightforward. An initial report had suggested that blockade of IL-1β impaired healing in the heart and favored cardiac rupture in a model of severe non-reperfused myocardial infarction in the mouse.73 The investigators used a commercial hamster anti-mouse IL-1β antibody developed for immunohistochemistry. The characteristics of the antibody (IgG class), the presence or absence of stimulatory properties, and the ability to activate complement or antibody mediated cell death were not reported.73 The deleterious effects of IL-1β blockade in this study were based on a reduction of collagen deposition and increased incidence of cardiac rupture 5-7 days after surgery. This pattern of impaired healing and increased rupture was not seen in the studies using genetic models of IL-1 receptor deletion nor in the studies using pharmacologic agents to non-selectively block IL-1α and IL-1β (i.e. anakinra or IL-1 trap)62-66, 68, 72, 74, 75 and thus raised the question of whether selective IL-1β blockade would elicit adverse effects. More recent studies using well-described IL-1β antibodies specifically developed for in vivo use have shown a protective effect of IL-1β blockade.76, 77 A mouse engineered IL-1β antibody of IgG1 isoclass that shows powerful modulation of IL-1 activity and was developed for in vivo use showed significant limitation of cardiac enlargement and dysfunction following experimental non-reperfused myocardial infarction in the mouse.76 Similar results were seen in the same model with another mouse monoclonal anti-IL-1β.77 Both of these antibodies had shown to have powerful inhibitory effects on IL-1β signaling without cell toxicity. The discrepancies between these recent studies compared with the original antibody study are difficult to reconcile, but likely relate to differences in the characteristics of the antibody used.78 The favorable data obtained with IL-1β blockers specifically developed for in vivo use are in line with the data from studies that inhibited caspase-1 or silenced components of the inflammasome.
In auto-inflammatory diseases, IL-1β induces the synthesis, processing, and release of more IL-1β.79 When tested in the myocardial infarction model, IL-1β blockade with an IL-1β antibody ameliorated the cardiac remodeling process but did not affect the intensity of the inflammatory response or the caspase-1 activity in the tissue,77 suggesting that in acute myocardial infarction in the mouse, IL-1 related inflammation may not follow an autoinflammatory pattern. Other triggers (i.e. cell debris, ATP) must therefore be equally or more important in determining the intensity of the sterile inflammatory response. Figure 4 is a schematic representation of the mediators involved in the pathway of the sterile inflammation following AMI, the formation of the inflammasome, and the subsequent release of IL-1. The figure also presents treatments that have been demonstrated to blunt the inflammatory response and ameliorate the remodeling in preclinical models of AMI.
Figure 4.
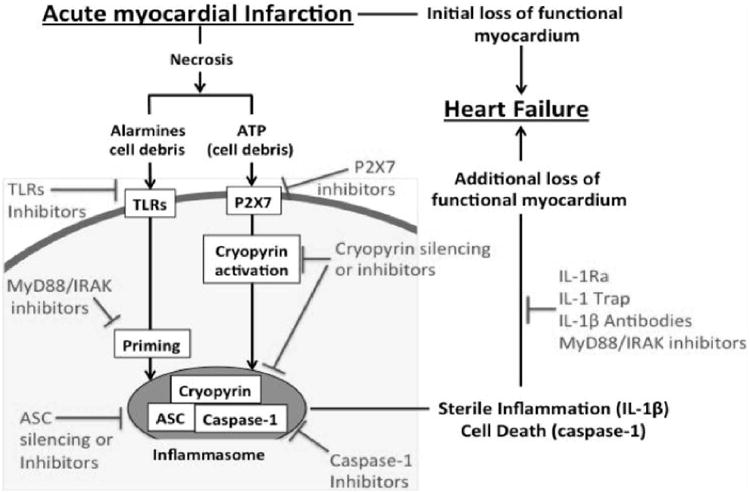
Inflammasome, IL-1 and heart failure following AMI. The formation of the cryopyrin inflammasome and subsequent caspase-1 activation promotes heart failure following acute myocardial infarction. The receptors which contribute to the inflammasome activation (TLR; P2X7; cryopyrin), the inflammasome components (cryopyrin, ASC and Caspase-1) and the inflammasome product IL-1β, and also contribute to the amplification of the inflammatory response and to cell death, are all potential targets for the treatment of acute myocardial infarction and the prevention or treatment of heart failure. Data excerpted from Mezzaroma et al.53. Abbreviations: ASC=apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain; AMI= acute myocardial infarction; ATP=adenosine triphosphate; IL-1=Interleukin-1; P2X7=purinergic receptor 2X7; TLR=Toll-like receptor; MyD88=myeloid differentiation factor-88; IRAK= interleukin-1 receptor associated kinase; IL-1Ra=Interleukin-1 receptor antagonist.
Clinical studies
The MRC-ILA-HEART study80 randomized 186 patients with non-ST segment elevation acute myocardial infarction within 48 hours of symptom onset in 3 centers in UK to either anakinra 100 mg daily for 14 days or placebo. The study was prompted by preclinical data from the same group showing that inappropriate arterial response to injury in animal models was IL-1β mediated.32 The primary end-point of the study was the area-under-the-curve for CRP over the first 7 days.80 The results of the study are not available.
The VCU-ART and VCU-ART2 pilot studies81, 82 included 10 and 30 clinically stable patients, respectively, with reperfused ST-segment elevation acute myocardial infarction that were randomized to anakinra 100 mg daily for 14 days (as in the MRC-ILA-HEART study) or placebo.80 All 40 patients were followed for 3 months with paired cardiac magnetic resonance analyses. Mortality and event rates were low in both groups, likely reflecting a selection bias toward lower risk STEMI patients that were stable enough to undergo initial cardiac MRI at 24 hours. Overall, anakinra showed an acceptable safety profile with only 2 patients (10%) experiencing adverse event requiring discontinuation of therapy (versus 2 patients [10%] in placebo). Injection site reactions were the most commonly encountered side effect occurring in 5 patients (25%) in the anakinra group and 2 patients (10%) in the placebo group. Anakinra was associated with a numerically greater incidence of infections (25% vs 15%), but not of serious infections (10% in each group).81, 82 Nine adverse cardiac events (1 death, 2 recurrent MI, 6 new onset heart failure) occurred in 6 placebo patients, whereas 3 adverse cardiac events (2 recurrent MI and 1 new onset heart failure) occurred in 3 anakinra treated patients (P=0.25). Anakinra-treated patients were significantly less likely to experience new onset heart failure (1 patient, 5%) vs placebo-treated patients (6 patients, 30%, P=0.035, Figure 5).81, 82 Treatment with anakinra 100 mg daily significantly blunted—but did not eliminate—the inflammatory response at 72 hours (CRP plasma levels of 8.4 mg/dL [anakinra] vs 18.9 mg/dL [placebo], P<0.001, Figure 5).81, 82 Paired changes in left ventricular end-systolic volume index (LVESVi) and left ventricular ejection fraction (LVEF) in the entire cohort were rather small over the 3 month follow-up, reflecting a group of STEMI patients at low risk for adverse cardiac remodeling, yet at significant risk for heart failure. Anakinra-treated patients showed a trend toward more favorable changes in LVESVi and LVEF, but placebo-corrected differences were not statistically significant (Figure 5).81, 82 Anakinra had no effects on infarct size measured with cardiac magnetic resonance.81, 82
Figure 5.
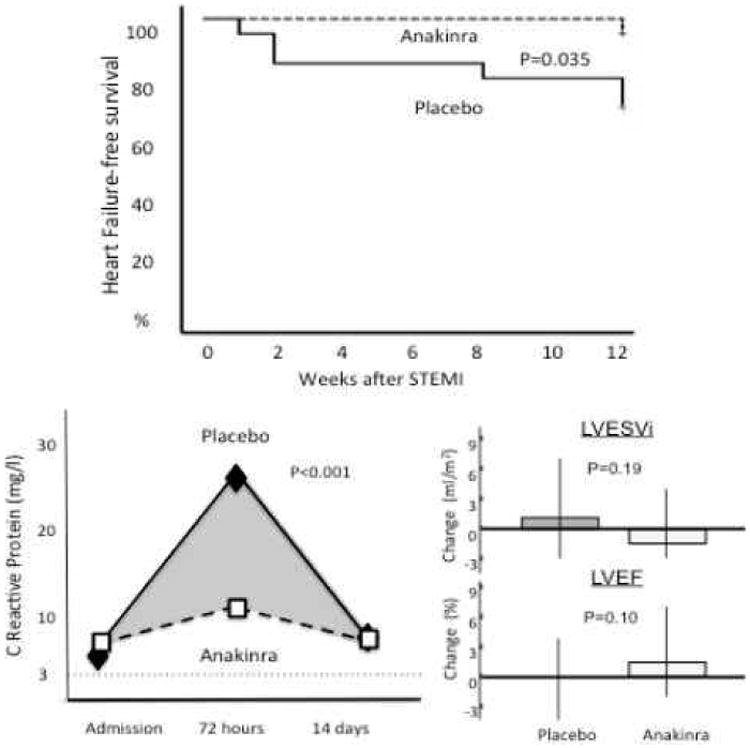
Interleukin-1 blockade in acute myocardial infarction. The VCU-ART and VCU-ART2 pilot studies showed significantly blunting of the inflammatory response with anakinra 100 mg daily during ST-segment elevation acute myocardial infarction and a numerically lower incidence of heart failure at 3 months. Data excerpted from Abbate et al.82
Role of Interleukin-1 in Heart Failure
Heart failure is a clinical condition of impaired cardiac contraction or relaxation at rest or with exercise, associated with symptoms of shortness of breath, fatigue, or exercise intolerance. Heart failure represents a final common pathway for many forms of cardiac injury (ischemia, pressure overload, toxicity) and leads to increased morbidity and mortality.
Experimental studies
IL-1 was identified one of the ‘soluble myocardial depressant factors’ in the plasma of patients with sepsis.83, 84 Without affecting the β-adrenergic receptor (β-AR) density or the affinity for its ligands, IL-1 impairs the coupling of the adenylyl cyclase (responsible for the production of cAMP) with the β-AR, thus reducing the response to the endogenous and exogenous β-AR agonists (i.e. isoproterenol).85, 86 The mechanisms for this observation are not entirely clear.86, 87 The Ca++ current (Ica) of the L-type calcium channel is also affected by IL-1β. This channel is responsible for the increase of the Ica following β-AR stimulation and IL-1 uncouples this channel from the receptor independent of cAMP concentration or β-AR density.88 Some reports show that nitric oxide (NO) produced in response to IL-1 affected the production of ATP (in the mitochondria) and the coupling of the calcium channel with the β-AR.89-92 IL-1 also regulates Ca++ re-uptake in the sarcoplasmic reticulum through down-regulation of phospholamban and sarco/endoplasmic reticulum calcium-ATPase (SERCA) mRNA and protein levels.90, 93 More recently, a role for phospho-inositide-3-kinase-γ (PI3Kγ) in IL-1 signaling has been identified as potentially linked to impaired contractility, phosphodiesterase-3 activitiy, and β-AR desensitization.94-98 A separate report describes the role of IL-1 in ischemia-induced systolic dysfunction.99 Figure 6 summarizes the proposed effects of IL-1 on cardiac contractility.
Figure 6.
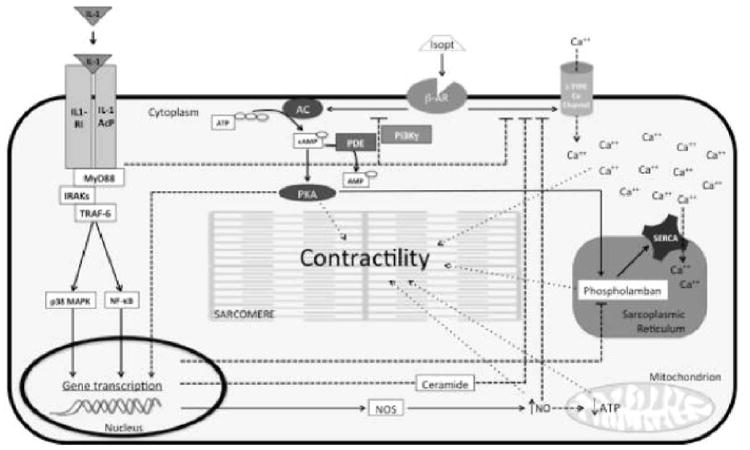
Proposed mechanisms of IL-1 induced contractile dysfunction. The diagram represents the intracellular pathways initiated by IL-1 and describes the proposed mechanisms by which IL-1 impairs the contractility of the cardiomyocyte. IL-1 (-α or -β) binds to the IL-1 receptor type I (IL-1RI) and the IL-1R accessory protein (IL-1AcP) and recruits the intracellular adaptor protein myeloid differentiation factor 88 (MyD88). This interaction activates the interleukin-1 receptor associated kinases (IRAKs) and the tumor necrosis factor (TNF) receptor associated factor 6 (TRAF-6) and leads to the activation of the p38 mitogen activated protein kinase (p38 MAPK) and the nuclear translocation of the nuclear factor kB (NF-kB), causing transcriptional changes of several genes. The IL-1 signaling induces uncoupling of the β-adrenergic receptor (β-AR) from the adenylyl cyclase (AC),85, 86 and from the L-type calcium (Ca++) channels,87, 88 thus reducing the cardiomyocytes responsiveness to the β-AR agonist (such as isoproterenol [isopt]). The cyclic AMP (cAMP)-dependent protein kinase A (PKA) is sensitive to increasing concentration of cAMP and regulates the function of several proteins, including sarcomere components, phospholamban and the calcium re-uptake in the sarcoplasmic reticulum, and the transcription of several genes. The changes in the gene transcription downstream of the IL-1 receptor affect the expression of phospholamban and the sarco/endoplasmic reticulum Ca++-ATPase (SERCA),90, 93 and increase the production of ceramide,90 which contributes to the uncoupling of the Ca++ channel from the β-AR. A similar effect has been described by increasing amounts of nitric oxide (NO) secondary to augmented expression of the nitric oxide synthase (NOS), 89-92 which not only contributes to the uncoupling of the Ca++ regulation from the β-AR91, 93 but also affects the production of ATP in the mitochondria.90 Altered levels of Ca++, cAMP (through PKA) and ATP affect the contractility of the sarcomeres, the cardiomyocytes and the heart as a whole. Class 3 cAMP phosphodiesterases (PDE-3) oppose the effects of AC by hydrolyzing cAMP into AMP. Phosphoinositide-3 kinase gamma (PI3Kγ) regulates the activity of β-AR and PDE-3. PI3Kγ is also activated downstream of the IL-1 receptor 95 and may be involved in the impairment of contractility by activating PDE-3 97, 98 and inducing β-AR desensitization.95
The impairment in contractility is experimentally reproducible in vivo by challenging the mouse with recombinant mouse IL-1β. A single injection of IL-1β (3 mcg/kg) induces systolic dysfunction and impaired response to isoproterenol, and repeated injections of IL-1β give a reversible non-ischemic cardiomyopathy.100, 101 Interestingly, the injection of plasma from human patients with systolic heart failure (and elevated CRP) is sufficient to induce systolic dysfunction and impaired response to isoproterenol in the mouse, while the pretreatment of the mice with IL-1 blockers preserves the systolic function and the contractile reserve.103
IL-1 has also been shown to effect myocardial relaxation. In vitro studies describe impaired relaxation of cardiomyocytes.92, 102 In our own laboratory, we have observed that a single injection of recombinant murine IL-1β (3 mcg/kg) in healthy adult mice leads to an increase in left ventricular end diastolic pressure (4 mmHg in controls vs 8 mmHg in IL-1β-treated mice) accompanied by an overall reduction in contractility and relaxation, measured as +dP/dT and –dP/dT, and an increase in isovolumetric relaxation measured at Doppler echocardiography (personal communication by the authors).
Clinical studies
The first study to show an improvement in cardiac function with IL-1 blockade enrolled a series of patients with rheumatoid arthritis, in which a single dose of anakinra 100 mg improved myocardial contractility and relaxation, coronary flow reserve, and brachial artery flow mediated dilatation within 3 hours of administration.103 The AIR-HF pilot study showed tolerability of anakinra in 7 patients with symptomatic systolic heart failure and elevated CRP levels (>2 mg/L).100 Anakinra lead to a significant increase in peak VO2 (+2.9 mL/kg/min), decrease in VE/VCO2 (-3.2, reflecting greater ventilator efficiency), and increase in exercise time (+2.9 min).100 CRP and Interleukin-6 levels dropped by nearly 90%, suggesting that IL-1 activity may regulate the systemic inflammatory response in heart failure. Furthermore, the observation that IL-1 blockade also reduced the plasma concentration of IL-1β by nearly 90% suggests that IL-1 does follow a positive feedback loop in heart failure wherein “IL-1 induces IL-1” and implicates heart failure as an auto-inflammatory phenotype.79 Another pilot study in patients with heart failure and preserved ejection fraction (DHART) is ongoing.104
Role of Interleukin-1 in Myocardial and Pericardial Disease
Auto-inflammatory diseases are often manifested with serositis (such as pericarditis) and occasionally with myocarditis. Isolated pericarditis without apparent cause is referred to as idiopathic recurrent pericarditis.105 Different studies have reported successful treatment of pericarditis with anakinra in patients with idiopathic recurrent pericarditis,106-109 pericarditis caused by a mutation in the TNFRSF1a gene,110 pericarditis associated with Still's disease,111 myocarditis associated with Still's disease,112 and parvovirus B19 associated myocardio-pericarditis.113 Prospective clinical trials are lacking and much needed. In the mean time, considering its safety profile, anakinra may be considered for the treatment of refractory or recurrent pericarditis.105
Preclinical studies also support a pathogenetic role of IL-1 in myocardial disease due to doxorubicin toxicity, radiation injury, or genetic mutations leading to dilated cardiomyopathy.114-117
Overview of the Clinically Available Interleukin-1 Blockers
Anakinra (Kineret™)
Anakinra, recombinant human IL-1 receptor antagonist, was originally approved for the treatment of rheumatoid arthritis in 2001.118 As a recombinant version of the naturally occurring IL-1 receptor antagonist, anakinra binds to the IL-1 receptor to exert powerful IL-1α and IL-1β blocking activity without observable off-target effects.119, 120 Anakinra is highly effective in the treatment of several inflammatory diseases 1-3 and preliminary studies show potential benefits in ischemic stroke, hemorrhagic stroke, and diabetes.121-123 The favorable safety profile of anakinra is demonstrated by the hundreds of thousands of patients who have been treated in the past decades.120, 124 Anakinra is associated with an increased risk of infections (mainly upper respiratory infections). A review of clinical trials in patients with rheumatoid arthritis who were also receiving other immunomodulating agents—and therefore at greater risk of infection—found that anakinra was associated with a 2- to 3-fold increase in serious infections (5-6% vs 1-2% in placebo), but not with increased infection-related mortality.119, 120, 124 Three randomized controlled trials of anakinra in patients with severe sepsis showed a favorable trend toward reduced mortality.125-127 In contrast, TNF-α blockers are considered to be immunosuppressants and were associated with worse outcomes in sepsis trials.128 Moreover, anakinra prevented immunosuppression in a recent study of stroke patients122 and does not inhibit the production of interferon-γ.1, 128 Nevertheless, given that IL-1 mediates fever and leukocytosis, it is expected that patients treated with IL-1 blockers (such as anakinra) may not develop fever or leukocytosis during infection. Close monitoring of treated patients is therefore warranted to identify more subtle symptoms of infection and to prevent potential delays in antimicrobial treatment.
The most common side effect of anakinra is injection site reaction presenting as erythematous skin reactions. These reactions are generally mild and self-limiting, occur within few days of initiation of therapy, and subside spontaneously within one or two weeks of continued treatment or within days upon cessation of therapy. Rotation of injection site may alleviate minor reactions and topical corticosteroids may be helpful for severe reactions.
Anakinra is given as a fixed dose of 100 mg subcutaneously every 24 hours in adults, or every 48 hours in patients with severely impaired kidney function.119 Nevertheless, the pharmacokinetic characteristics of anakinra following 1 mg/kg subcutaneous injection (half-life = 4-6 hours) suggest that suboptimal IL-1 blockade may occur with this regimen. In many inflammatory diseases physicians often use a higher dose of anakinra (up to 5 mg/kg) to achieve superior results.129, 130 A much higher dose of anakinra (2 mg/kg/hour infusion for 72 hours) was used in the clinical trials of patients with sepsis,125, 126 while maintaining a favorable safety profile. Pilot clinical trials evaluating the use of anakinra in stroke patients have employed the same high doses (2 mg/kg/hour infusion for 72 hours).122
Prior to the approval of rilonacept (Arcalyst, 2008) and canakinumab (Ilaris, 2009), anakinra was the only available IL-1 blocker in clinical practice. Due to its safety record and short half-life, anakinra has been considered the ideal drug for pilot proof-of-concept studies. Compliance issues with daily injections and injection-site reactions may theoretically limit prolonged treatment courses; however, thousands of rheumatoid arthritis patients receive prolonged treatment of daily injections without significant problems. The pharmacokinetic characteristics of the newer agents (reviewed below) present the possibility of simplified dosing regimens. Based upon current labeling, however, anakinra remains the only agent approved for a non-orphan indication and therefore remains the mostly widely utilized IL-1 blocking agent in clinical practice.
Rilonacept (Arcalyst™)
Rilonacept is a recombinant fusion protein based upon elements of the IL-1 type I receptor and the IL-1 receptor accessory protein.131, 132 Circulating rilonacept effectively functions as an “IL-1 Trap” that binds with high affinity to circulating IL-1α and IL-1β and with less affinity also to IL-1 receptor antagonist. In 2008, rilonacept received FDA approval for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS).10 Rilonacept does offer the potential advantage over anakinra of weekly injections. A murine analogue of rilonacept showed significant benefits versus vehicle on cardiac remodeling after permanent coronary ligation in a mouse model of myocardial infarction.72 Notably, the benefits appeared to follow a U-shaped distribution, as moderate doses (5 mg/kg) appeared to be more efficacious than lower (1 mg/kg) or higher doses (30 mg/kg).72 A small pilot study showed preliminary safety and efficacy in 10 patients with active gouty arthritis, but no follow-up studies have been published.133 As with anakinra, precautions are necessary regarding infections, and the combination of rilonacept with other immunomodulating drugs has not been adequately tested. Minor infections are generally more common with rilonacept than placebo, and the risk of serious infections with rilonacept cannot be assessed due to limited data. Injection site reaction are also common, but unlikely to lead to drug discontinuation.131
Canakinumab (Ilaris™)
Canakinumab is a monoclonal IL-1β antibody (immunoglobulin G1/κ isotype) that binds irreversibly to circulating human IL-1β and prevents activation of the IL-1 receptor.134-138 Unlike anakinra and rilonacept, canakinumab is specific for IL-1β blockade. Like rilonacept, canakinumab is only FDA approved for the treatment of CAPS, however, multiple reports already describe the use of canakinumab in other inflammatory disease states such as diabetes, gouty arthritis, rheumatoid arthritis, juvenile arthritis, uveitis, Behcet's disease, and familial Mediterranean fever.139-144 Moreover, canakinumab is currently being investigated for prevention of recurrent cardiovascular events in the Cardiovascular Risk Reduction Study (CANTOS) in the largest anti-cytokine study ever conducted. CANTOS began enrollment in 2011 and will target a population of 17,200 patients with a history of acute myocardial infarction and elevated CRP.145 A murine analogue of canakinumab showed significant benefits on cardiac remodeling and LV function in the mouse after myocardial infarction due to coronary artery ligation.77
Canakinumab offers the potential advantage over anakinra (and rilonacept) of a 3-month dosing interval (terminal elimination half-life of 26 days) that may be preferable for the treatment of chronic diseases.145 The prolonged duration of action, however, may be counterproductive in patients who experience significant adverse events.
In published clinical trials, canakinumab has shown potential safety and efficacy in the treatment of gouty arthritis, rheumatoid arthritis, and diabetes.136, 141, 146 In two studies of canakinumab in patients with acute gouty arthritis, canakinumab significantly improved pain and prevented new flares versus triamcinolone.146 Patients randomized to canakinumab also experienced increased rates of infectious adverse events (20.4% vs 12.2%) and serious adverse events (7.6% vs 3.1%) versus traimcinolone, accompanied by slight increases in serum urate and serum creatinine (decreased eGFR), and slight reductions in platelet, neutrophil, and leukocyte counts. Notably, however, no opportunistic infections were observed. As seen with other antibodies, canakinumab may induce an antibody reaction toward the drug, but this has not been shown to be clinically relevant. In patients with diabetes, canakinumab provided a highly significant reduction in CRP levels, thus proving that the elevation in CRP seen in such patients is highly IL-1β-dependent. Canakinumab had no significant effect on hemoglobin A1c, fasting glucose, insulin, insulin resistance, LDL cholesterol, or HDL cholesterol at 4 months, however, patients receiving canakinumab experienced a mild rise in triglyceride concentration. Neutropenia (ANC<1,500/mm3) occurred in 10.7% of canakinumab patients versus 4.6% of placebo patients, however, no serious adverse events were reported and no patients experienced life-threatening consequences or need for intervention. The large effect of canakinumab on CRP as surrogate for cardiovascular risk provided justification for the design of the large secondary prevention CANTOS study. Sub-studies of CANTOS study will also address the effect of canakinumab on carotid plaque burden, insulin secretion, and exercise capacity (among heart failure patients).
Gevokizumab
Gevokizumab (XOMA052) is a humanized monoclonal antibody (immunoglobulin G2 isotype) that binds to human IL-1β.147 Gevokizumab is not currently approved for any indications in the US, but has undergone preliminary testing in arthritis, uveitis, and diabetes.147-149 In comparison to the FDA-approved IL-1 blockers, gevokizumab appears to be most similar to canakinumab due to IL-1β selectivity and a pharmacokinetic profile (terminal elimination half-life = 23 days) that allows for extended dosing intervals up to 4 weeks. However, similar to rilonacept, gevokizumab also displays a U-shaped dose response, in which moderate doses may exert more favorable anti-inflammatory effects in vivo.150 In contrast to other agents, the IL-1β/gevokizumab complex appears to retain some degree of agonist activity at the IL-1 receptor.151 Whether the residual agonistic activity represents an advantage or a disadvantage from an efficacy and safety point of view is unclear at this time.
Table 1 summarizes characteristics of the different IL-1 blockers.
Table 1.
Comparison of IL-1 blocking agents.
| Name | Trade | Mechanism | Blockade | FDA approval | Dose | Route | Frequency | ||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| IL-1α | IL-1β | IL-1Ra | |||||||
| Anakinra | Kineret | Receptor Antagonist | Y | Y | N | Rheumatoid arthritis | 100 mg | SC | Daily |
| Rinalocept | Arcalyst | IL-1 Trap | Y | Y | Y | CAPS | 160 mg | SC | Weekly |
| Canakinumab | Ilaris | IL-1β antibody | N | Y | N | CAPS | 150 mg | SC | 1-3 months |
| Gevokizumab | -- | IL-1β antibody | N | Y | N | -- | 0.3 mg/Kg | IV | Monthly |
Abbreviations: CAPS=cryopyrin-associated periodic syndromes; IV=intravenous; SC=subcutaneous.
Data excerpted from Abbate et al.40
Current Indications of Interleukin-1 Blockers
As of May 2013, IL-1 blockers are approved only for the treatment of rheumatoid arthritis and cryopyrin-associated periodic syndromes. The favorable safety profile of anakinra in patients with rheumatoid arthritis who are at increased risk of infections, cancer, and cardiovascular disease provides some reassurance and justification for the many off-label use of IL-1 blockers (as witnessed by the numerous reports in the literature) and the many ongoing experimental studies in areas of medicine ranging from rheumatology, oncology, neurology, dermatology, gastroenterology and now cardiology.2
Perspectives
While IL-1 blockade in cardiovascular disease is still in its infancy, it may represent a unique opportunity to quench the inflammatory response following tissue injury by selectively inhibiting a single apical mediator in the cascade. Observational data, pilot studies, and preclinical models suggest a beneficial role of IL-1 blockade in a variety of pathologic processes including atherosclerosis, atherothrombosis, acute myocardial infarction, heart failure, and pericarditis. While pivotal clinical trial data are lacking, small pilot studies have shown promising safety and efficacy signals and a large cytokine inhibition study is ongoing to test IL-1β blockade in secondary prevention of patients with AMI.145
It remains unknown whether IL-1 blockade will significantly affect the morbidity and mortality of patients with cardiovascular disease, and whether such potential benefit will be obtained with an acceptable risk and cost profile. The availability of multiple different IL-1 blockers offers potential benefits to patients, but also additional challenges to determine whether any specific agent will provide superior benefits in individual disease states. It is also unclear how to best measure the pharmacodynamics or pharmacogenetic responses to IL-1 blockers, and whether a ‘one-size-fits-all’ approach will suffice or tailored therapy will be necessary.
Acknowledgments
Funding Sources: Dr. Abbate is supported by an American Heart Association Scientist Development Grant and funds from the VCU Pauley Heart Center, Dr. Van Tassell is supported by an institutional National Institute of Health KL2TR000057 grant, Dr. Mezzaroma is supported by an American Heart Association Post-Doctoral Grant.
Footnotes
Conflict of Interest Disclosures: During the last 36 months, Dr. Abbate has received research grants from Gilead, Novartis and XOMA; he has lectured for Glaxo-Smith-Kline, Novartis, XOMA; and he has consulted for Gilead, Janssen, Omni Biopharma, Swedish Orphan Biovitrum, and XOMA. Dr. Van Tassell has received research grants from Gilead and Novartis, and has consulted for Novartis. The other authors have no potential conflicts of interests to disclose.
References
- 1.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doria A, Zen M, Bettio S, Gatto M, Bassi N, Nalotto L, Ghirardello A, Iaccarino L, Punzi L. Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev. 2012;12:22–30. doi: 10.1016/j.autrev.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 4.Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. 2009;119:3502–3511. doi: 10.1172/JCI40599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 6.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol. 2010;30:628–631. doi: 10.1007/s10875-010-9440-3. [DOI] [PubMed] [Google Scholar]
- 8.Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 10.Terkeltaub R, Sundy JS, Schumacher HR, Murphy F, Bookbinder S, Biedermann S, Wu R, Mellis S, Radin A. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68:1613–1617. doi: 10.1136/ard.2009.108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol. 2010;42:792–795. doi: 10.1016/j.biocel.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurme M, Lahdenpohja N, Santtila S. Gene polymorphisms of interleukins 1 and 10 in infectious and autoimmune diseases. Ann Med. 1998;30:469–473. doi: 10.3109/07853899809002488. [DOI] [PubMed] [Google Scholar]
- 13.Tabrizi AR, Zehnbauer BA, Freeman BD, Buchman TG. Genetic markers in sepsis. J Am Coll Surg. 2001;192:106–117. doi: 10.1016/s1072-7515(00)00748-1. quiz 145-106. [DOI] [PubMed] [Google Scholar]
- 14.Witkin SS, Gerber S, Ledger WJ. Influence of interleukin-1 receptor antagonist gene polymorphism on disease. Clin Infect Dis. 2002;34:204–209. doi: 10.1086/338261. [DOI] [PubMed] [Google Scholar]
- 15.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Keefe JH, Carter MD, Lavie CJ. Primary and secondary prevention of cardiovascular diseases: a practical evidence-based approach. Mayo Clin Proc. 2009;84:741–757. doi: 10.4065/84.8.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D'Agostino RB, Levy D, Kannel WB, Vasan RS. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–2062. doi: 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kushner I, Broder ML, Karp D. Control of the acute phase response. Serum C-reactive protein kinetics after acute myocardial infarction. J Clin Invest. 1978;61:235–242. doi: 10.1172/JCI108932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB, Maseri A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N Engl J Med. 1994;331:417–424. doi: 10.1056/NEJM199408183310701. [DOI] [PubMed] [Google Scholar]
- 20.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 21.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 22.Abbate A, Biondi-Zoccai GG, Brugaletta S, Liuzzo G, Biasucci LM. C-reactive protein and other inflammatory biomarkers as predictors of outcome following acute coronary syndromes. Semin Vasc Med. 2003;3:375–384. doi: 10.1055/s-2004-815695. [DOI] [PubMed] [Google Scholar]
- 23.Biasucci LM, Bellocci F, Landolina M, Rordorf R, Vado A, Menardi E, Giubilato G, Orazi S, Sassara M, Castro A, Massa R, Kheir A, Zaccone G, Klersy C, Accardi F, Crea F. Risk stratification of ischaemic patients with implantable cardioverter defibrillators by C-reactive protein and a multi-markers strategy: results of the CAMI-GUIDE study. Eur Heart J. 2012;33:1344–1350. doi: 10.1093/eurheartj/ehr487. [DOI] [PubMed] [Google Scholar]
- 24.Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 26.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 27.Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, Gotto AM., Jr Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–1965. doi: 10.1056/NEJM200106283442601. [DOI] [PubMed] [Google Scholar]
- 28.Andreotti F, Porto I, Crea F, Maseri A. Inflammatory gene polymorphisms and ischaemic heart disease: review of population association studies. Heart. 2002;87:107–112. doi: 10.1136/heart.87.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Libby P. The interface of atherosclerosis and thrombosis: basic mechanisms. Vasc Med. 1998;3:225–229. doi: 10.1177/1358836X9800300309. [DOI] [PubMed] [Google Scholar]
- 30.Dewberry R, Holden H, Crossman D, Francis S. Interleukin-1 receptor antagonist expression in human endothelial cells and atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:2394–2400. doi: 10.1161/01.atv.20.11.2394. [DOI] [PubMed] [Google Scholar]
- 31.Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, Michelson K, Hunter JJ, Kantak SS. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. 2011;216:313–320. doi: 10.1016/j.atherosclerosis.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 32.Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D, Francis S. Interleukin-1beta and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168:1396–1403. doi: 10.2353/ajpath.2006.051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isoda K, Sawada S, Ishigami N, Matsuki T, Miyazaki K, Kusuhara M, Iwakura Y, Ohsuzu F. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:1068–1073. doi: 10.1161/01.ATV.0000127025.48140.a3. [DOI] [PubMed] [Google Scholar]
- 34.Chamberlain J, Francis S, Brookes Z, Shaw G, Graham D, Alp NJ, Dower S, Crossman DC. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS One. 2009;4:e5073. doi: 10.1371/journal.pone.0005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devlin CM, Kuriakose G, Hirsch E, Tabas I. Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci U S A. 2002;99:6280–6285. doi: 10.1073/pnas.092324399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chamberlain J, Gunn J, Francis S, Holt C, Crossman D. Temporal and spatial distribution of interleukin-1 beta in balloon injured porcine coronary arteries. Cardiovasc Res. 1999;44:156–165. doi: 10.1016/s0008-6363(99)00175-3. [DOI] [PubMed] [Google Scholar]
- 37.Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, Owens GK. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest. 2012;122:70–79. doi: 10.1172/JCI43713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bevilacqua MP, Pober JS, Majeau GR, Cotran RS, Gimbrone MA., Jr Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J Exp Med. 1984;160:618–623. doi: 10.1084/jem.160.2.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 40.Abbate A, Van Tassell BW, Biondi-Zoccai GG. Blocking interleukin-1 as a novel therapeutic strategy for secondary prevention of cardiovascular events. Biodrugs. 2012;26:217–233. doi: 10.1007/BF03261881. [DOI] [PubMed] [Google Scholar]
- 41.Ikonomidis I, Andreotti F, Economou E, Stefanadis C, Toutouzas P, Nihoyannopoulos P. Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation. 1999;100:793–798. doi: 10.1161/01.cir.100.8.793. [DOI] [PubMed] [Google Scholar]
- 42.Ikonomidis I, Lekakis J, Revela I, Andreotti F, Nihoyannopoulos P. Increased circulating C-reactive protein and macrophage-colony stimulating factor are complementary predictors of long-term outcome in patients with chronic coronary artery disease. Eur Heart J. 2005;26:1618–1624. doi: 10.1093/eurheartj/ehi192. [DOI] [PubMed] [Google Scholar]
- 43.Saitoh T, Kishida H, Tsukada Y, Fukuma Y, Sano J, Yasutake M, Fukuma N, Kusama Y, Hayakawa H. Clinical significance of increased plasma concentration of macrophage colony-stimulating factor in patients with angina pectoris. J Am Coll Cardiol. 2000;35:655–665. doi: 10.1016/s0735-1097(99)00583-5. [DOI] [PubMed] [Google Scholar]
- 44.Correia LC, Andrade BB, Borges VM, Clarencio J, Bittencourt AP, Freitas R, Souza AC, Almeida MC, Leal J, Esteves JP, Barral-Netto M. Prognostic value of cytokines and chemokines in addition to the GRACE Score in non-ST-elevation acute coronary syndromes. Clin Chim Acta. 2010;411:540–545. doi: 10.1016/j.cca.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Kilic T, Ural D, Ural E, Yumuk Z, Agacdiken A, Sahin T, Kahraman G, Kozdag G, Vural A, Komsuoglu B. Relation between proinflammatory to anti-inflammatory cytokine ratios and long-term prognosis in patients with non-ST elevation acute coronary syndrome. Heart. 2006;92:1041–1046. doi: 10.1136/hrt.2005.080382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orn S, Ueland T, Manhenke C, Sandanger O, Godang K, Yndestad A, Mollnes TE, Dickstein K, Aukrust P. Increased interleukin-1beta levels are associated with left ventricular hypertrophy and remodelling following acute ST segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J Intern Med. 2012;272:267–276. doi: 10.1111/j.1365-2796.2012.02517.x. [DOI] [PubMed] [Google Scholar]
- 47.Biasucci LM, Liuzzo G, Fantuzzi G, Caligiuri G, Rebuzzi AG, Ginnetti F, Dinarello CA, Maseri A. Increasing levels of interleukin (IL)-1Ra and IL-6 during the first 2 days of hospitalization in unstable angina are associated with increased risk of in-hospital coronary events. Circulation. 1999;99:2079–2084. doi: 10.1161/01.cir.99.16.2079. [DOI] [PubMed] [Google Scholar]
- 48.Biasucci LM, Vitelli A, Liuzzo G, Altamura S, Caligiuri G, Monaco C, Rebuzzi AG, Ciliberto G, Maseri A. Elevated levels of interleukin-6 in unstable angina. Circulation. 1996;94:874–877. doi: 10.1161/01.cir.94.5.874. [DOI] [PubMed] [Google Scholar]
- 49.Kohli P, Bonaca MP, Kakkar R, Kudinova AY, Scirica BM, Sabatine MS, Murphy SA, Braunwald E, Lee RT, Morrow DA. Role of ST2 in non-ST-elevation acute coronary syndrome in the MERLIN-TIMI 36 trial. Clin Chem. 2012;58:257–266. doi: 10.1373/clinchem.2011.173369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patti G, Di Sciascio G, D'Ambrosio A, Dicuonzo G, Abbate A, Dobrina A. Prognostic value of interleukin-1 receptor antagonist in patients undergoing percutaneous coronary intervention. Am J Cardiol. 2002;89:372–376. doi: 10.1016/s0002-9149(01)02254-8. [DOI] [PubMed] [Google Scholar]
- 51.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbate A, Biondi-Zoccai GG, Baldi A. Pathophysiologic role of myocardial apoptosis in post-infarction left ventricular remodeling. J Cell Physiol. 2002;193:145–153. doi: 10.1002/jcp.10174. [DOI] [PubMed] [Google Scholar]
- 53.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 55.Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P, Yndestad A. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischemia-reperfusion injury. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt091. [DOI] [PubMed] [Google Scholar]
- 56.Mezzaroma E, Toldo S, Abbate A. Role of NLRP3 (cryopyrin) in acute myocardial infarction. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt123. in press. [DOI] [PubMed] [Google Scholar]
- 57.Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R, Tracey D, Allen H, Lee RT, Kelly RA. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol. 2003;35:685–694. doi: 10.1016/s0022-2828(03)00113-5. [DOI] [PubMed] [Google Scholar]
- 58.Merkle S, Frantz S, Schon MP, Bauersachs J, Buitrago M, Frost RJ, Schmitteckert EM, Lohse MJ, Engelhardt S. A role for caspase-1 in heart failure. Circ Res. 2007;100:645–653. doi: 10.1161/01.RES.0000260203.55077.61. [DOI] [PubMed] [Google Scholar]
- 59.Toldo S, Zhong H, Mezzaroma E, Van Tassell BW, Kannan H, Zeng D, Belardinelli L, Voelkel NF, Abbate A. GS-6201, a selective blocker of the A2B adenosine receptor, attenuates cardiac remodeling after acute myocardial infarction in the mouse. J Pharmacol Exp Ther. 2012;343:587–595. doi: 10.1124/jpet.111.191288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shinkai K, McCalmont TH, Leslie KS. Cryopyrin-associated periodic syndromes and autoinflammation. Clin Exp Dermatol. 2008;33:1–9. doi: 10.1111/j.1365-2230.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- 61.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, Soroosh P, Watford WT, O'Shea JJ, Kastner DL, Hoffman HM. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–887. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abbate A, Salloum FN, Van Tassell BW, Vecile E, Toldo S, Seropian I, Mezzaroma E, Dobrina A. Alterations in the interleukin-1/interleukin-1 receptor antagonist balance modulate cardiac remodeling following myocardial infarction in the mouse. PLoS One. 2011;6:e27923. doi: 10.1371/journal.pone.0027923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki K, Murtuza B, Smolenski RT, Sammut IA, Suzuki N, Kaneda Y, Yacoub MH. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation. 2001;104:I308–I303. doi: 10.1161/hc37t1.094871. [DOI] [PubMed] [Google Scholar]
- 65.Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, Dobrina A. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117:2670–2683. doi: 10.1161/CIRCULATIONAHA.107.740233. [DOI] [PubMed] [Google Scholar]
- 66.Salloum FN, Chau V, Varma A, Hoke NN, Toldo S, Biondi-Zoccai GG, Crea F, Vetrovec GW, Abbate A. Anakinra in experimental acute myocardial infarction--does dosage or duration of treatment matter? Cardiovasc Drugs Ther. 2009;23:129–135. doi: 10.1007/s10557-008-6154-3. [DOI] [PubMed] [Google Scholar]
- 67.Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A. 2010;107:2574–2579. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Tassell BW, Seropian IM, Toldo S, Salloum FN, Smithson L, Varma A, Hoke NN, Gelwix C, Chau V, Abbate A. Pharmacologic inhibition of myeloid differentiation factor 88 (MyD88) prevents left ventricular dilation and hypertrophy after experimental acute myocardial infarction in the mouse. J Cardiovasc Pharmacol. 2010;55:385–390. doi: 10.1097/FJC.0b013e3181d3da24. [DOI] [PubMed] [Google Scholar]
- 69.Feng Y, Zou L, Si R, Nagasaka Y, Chao W. Bone marrow MyD88 signaling modulates neutrophil function and ischemic myocardial injury. Am J Physiol Cell Physiol. 2010;299:C760–769. doi: 10.1152/ajpcell.00155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maekawa Y, Mizue N, Chan A, Shi Y, Liu Y, Dawood S, Chen M, Dawood F, de Couto G, Li GH, Suzuki N, Yeh WC, Gramolini A, Medin JA, Liu PP. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin-1 receptor-associated kinase-4 signaling: a regulator of bone marrow-derived dendritic cells. Circulation. 2009;120:1401–1414. doi: 10.1161/CIRCULATIONAHA.109.865956. [DOI] [PubMed] [Google Scholar]
- 71.Wang X, Takagawa J, Lam VC, Haddad DJ, Tobler DL, Mok PY, Zhang Y, Clifford BT, Pinnamaneni K, Saini SA, Su R, Bartel MJ, Sievers RE, Carbone L, Kogan S, Yeghiazarians Y, Hermiston M, Springer ML. Donor myocardial infarction impairs the therapeutic potential of bone marrow cells by an interleukin-1-mediated inflammatory response. Sci Transl Med. 2011;3:100ra190. doi: 10.1126/scitranslmed.3002814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Tassell BW, Varma A, Salloum FN, Das A, Seropian IM, Toldo S, Smithson L, Hoke NN, Chau VQ, Robati R, Abbate A. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J Cardiovasc Pharmacol. 2010;55:117–122. doi: 10.1097/FJC.0b013e3181c87e53. [DOI] [PubMed] [Google Scholar]
- 73.Hwang MW, Matsumori A, Furukawa Y, Ono K, Okada M, Iwasaki A, Hara M, Miyamoto T, Touma M, Sasayama S. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J Am Coll Cardiol. 2001;38:1546–1553. doi: 10.1016/s0735-1097(01)01591-1. [DOI] [PubMed] [Google Scholar]
- 74.Grothusen C, Hagemann A, Attmann T, Braesen J, Broch O, Cremer J, Schoettler J. Impact of an interleukin-1 receptor antagonist and erythropoietin on experimental myocardial ischemia/reperfusion injury. Scientific World Journal. 2012;2012:737585. doi: 10.1100/2012/737585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toldo S, Schatz AM, Mezzaroma E, Chawla R, Stallard TW, Stallard WC, Jahangiri A, Van Tassell BW, Abbate A. Recombinant human interleukin-1 receptor antagonist provides cardioprotection during myocardial ischemia reperfusion in the mouse. Cardiovasc Drugs Ther. 2012;26:273–276. doi: 10.1007/s10557-012-6389-x. [DOI] [PubMed] [Google Scholar]
- 76.Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, Salloum FN, Smithson L, Dinarello CA. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail. 2010;12:319–322. doi: 10.1093/eurjhf/hfq017. [DOI] [PubMed] [Google Scholar]
- 77.Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, Abbate A. Interleukin-1beta blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol. 2013;98:734–745. doi: 10.1113/expphysiol.2012.069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaminski MS, Kitamura K, Maloney DG, Campbell MJ, Levy R. Importance of antibody isotype in monoclonal anti-idiotype therapy of a murine B cell lymphoma. A study of hybridoma class switch variants. J Immunol. 1986;136:1123–1130. [PubMed] [Google Scholar]
- 79.Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, Libby P. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139:1902–1910. [PubMed] [Google Scholar]
- 80.Crossman DC, Morton AC, Gunn JP, Greenwood JP, Hall AS, Fox KA, Lucking AJ, Flather MD, Lees B, Foley CE. Investigation of the effect of Interleukin-1 receptor antagonist (IL-1ra) on markers of inflammation in non-ST elevation acute coronary syndromes (The MRC-ILA-HEART Study) Trials. 2008;9:8. doi: 10.1186/1745-6215-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, Gelwix CC, Salloum FN, Hastillo A, Dinarello CA, Vetrovec GW. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–1377. e1371. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 82.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, Abouzaki NA, Rengel LR, Varma A, Gambill ML, Falcao RA, Voelkel NF, Dinarello CA, Vetrovec GW. Effects of Interleukin-1 Blockade With Anakinra on Adverse Cardiac Remodeling and Heart Failure After Acute Myocardial Infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study] Am J Cardiol. 2013 doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183:949–958. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Muller-Werdan U, Buerke M, Ebelt H, Heinroth KM, Herklotz A, Loppnow H, Russ M, Schlegel F, Schlitt A, Schmidt HB, Soffker G, Werdan K. Septic cardiomyopathy - A not yet discovered cardiomyopathy? Exp Clin Cardiol. 2006;11:226–236. [PMC free article] [PubMed] [Google Scholar]
- 85.Chung MK, Gulick TS, Rotondo RE, Schreiner GF, Lange LG. Mechanism of cytokine inhibition of beta-adrenergic agonist stimulation of cyclic AMP in rat cardiac myocytes. Impairment of signal transduction. Circ Res. 1990;67:753–763. doi: 10.1161/01.res.67.3.753. [DOI] [PubMed] [Google Scholar]
- 86.Liu SJ, Zhou W, Kennedy RH. Suppression of beta-adrenergic responsiveness of L-type Ca2+ current by IL-1beta in rat ventricular myocytes. Am J Physiol. 1999;276:H141–148. doi: 10.1152/ajpheart.1999.276.1.H141. [DOI] [PubMed] [Google Scholar]
- 87.Schreur KD, Liu S. Involvement of ceramide in inhibitory effect of IL-1 beta on L-type Ca2+ current in adult rat ventricular myocytes. Am J Physiol. 1997;272:H2591–2598. doi: 10.1152/ajpheart.1997.272.6.H2591. [DOI] [PubMed] [Google Scholar]
- 88.Liu S, Schreur KD. G protein-mediated suppression of L-type Ca2+ current by interleukin-1 beta in cultured rat ventricular myocytes. Am J Physiol. 1995;268:C339–349. doi: 10.1152/ajpcell.1995.268.2.C339. [DOI] [PubMed] [Google Scholar]
- 89.Tatsumi T, Matoba S, Kawahara A, Keira N, Shiraishi J, Akashi K, Kobara M, Tanaka T, Katamura M, Nakagawa C, Ohta B, Shirayama T, Takeda K, Asayama J, Fliss H, Nakagawa M. Cytokine-induced nitric oxide production inhibits mitochondrial energy production and impairs contractile function in rat cardiac myocytes. J Am Coll Cardiol. 2000;35:1338–1346. doi: 10.1016/s0735-1097(00)00526-x. [DOI] [PubMed] [Google Scholar]
- 90.Combes A, Frye CS, Lemster BH, Brooks SS, Watkins SC, Feldman AM, McTiernan CF. Chronic exposure to interleukin 1beta induces a delayed and reversible alteration in excitation-contraction coupling of cultured cardiomyocytes. Pflugers Arch. 2002;445:246–256. doi: 10.1007/s00424-002-0921-y. [DOI] [PubMed] [Google Scholar]
- 91.Schulz R, Panas DL, Catena R, Moncada S, Olley PM, Lopaschuk GD. The role of nitric oxide in cardiac depression induced by interleukin-1 beta and tumour necrosis factor-alpha. Br J Pharmacol. 1995;114:27–34. doi: 10.1111/j.1476-5381.1995.tb14901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsujino M, Hirata Y, Imai T, Kanno K, Eguchi S, Ito H, Marumo F. Induction of nitric oxide synthase gene by interleukin-1 beta in cultured rat cardiocytes. Circulation. 1994;90:375–383. doi: 10.1161/01.cir.90.1.375. [DOI] [PubMed] [Google Scholar]
- 93.McTiernan CF, Lemster BH, Frye C, Brooks S, Combes A, Feldman AM. Interleukin-1 beta inhibits phospholamban gene expression in cultured cardiomyocytes. Circ Res. 1997;81:493–503. doi: 10.1161/01.res.81.4.493. [DOI] [PubMed] [Google Scholar]
- 94.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 95.Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, Blair SL, Ginsberg MH, Cheresh DA, Hirsch E, Field SJ, Varner JA. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19:715–727. doi: 10.1016/j.ccr.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Van Tassell B, Seropian I, Harrington J, Smithson L, Toldo S, Menna A, Scharf A, Robati R, Abbate A. PI3Kgamma inhibition prevents adverse cardiac remodeling after acute myocardial infarction in the mouse. Circulation. 2010;122:A21531. Abstract. [Google Scholar]
- 97.Marcantoni A, Levi RC, Gallo MP, Hirsch E, Alloatti G. Phosphoinositide 3-kinasegamma (PI3Kgamma) controls L-type calcium current (ICa,L) through its positive modulation of type-3 phosphodiesterase (PDE3) J Cell Physiol. 2006;206:329–336. doi: 10.1002/jcp.20467. [DOI] [PubMed] [Google Scholar]
- 98.Perino A, Ghigo A, Ferrero E, Morello F, Santulli G, Baillie GS, Damilano F, Dunlop AJ, Pawson C, Walser R, Levi R, Altruda F, Silengo L, Langeberg LK, Neubauer G, Heymans S, Lembo G, Wymann MP, Wetzker R, Houslay MD, Iaccarino G, Scott JD, Hirsch E. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol Cell. 2011;42:84–95. doi: 10.1016/j.molcel.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A. 2001;98:2871–2876. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM, Shah K, Canada J, Voelkel NF, Dinarello CA, Abbate A. Enhanced Interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS One. 2012;7:e33438. doi: 10.1371/journal.pone.0033438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm Res. 2013;62:637–640. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 102.Radin MJ, Holycross BJ, Dumitrescu C, Kelley R, Altschuld RA. Leptin modulates the negative inotropic effect of interleukin-1beta in cardiac myocytes. Mol Cell Biochem. 2008;315:179–184. doi: 10.1007/s11010-008-9805-6. [DOI] [PubMed] [Google Scholar]
- 103.Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T, Katsimbri P, Skarantavos G, Soucacos PN, Kremastinos DT. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117:2662–2669. doi: 10.1161/CIRCULATIONAHA.107.731877. [DOI] [PubMed] [Google Scholar]
- 104. [Accessed 3/18/2013]; http://www.clinicaltrials.gov NCT01542502.
- 105.Seferovic PM, Ristic AD, Maksimovic R, Simeunovic DS, Milinkovic I, Seferovic Mitrovic JP, Kanjuh V, Pankuweit S, Maisch B. Pericardial syndromes: an update after the ESC guidelines 2004. Heart Fail Rev. 2012 doi: 10.1007/s10741-012-9335-x. [DOI] [PubMed] [Google Scholar]
- 106.Camacho-Lovillo M, Mendez-Santos A. Successful Treatment of Idiopathic Recurrent Pericarditis With Interleukin-1 Receptor Antagonist (Anakinra) Pediatr Cardiol. 2013 doi: 10.1007/s00246-013-0663-y. [DOI] [PubMed] [Google Scholar]
- 107.Picco P, Brisca G, Traverso F, Loy A, Gattorno M, Martini A. Successful treatment of idiopathic recurrent pericarditis in children with interleukin-1beta receptor antagonist (anakinra): an unrecognized autoinflammatory disease? Arthritis Rheum. 2009;60:264–268. doi: 10.1002/art.24174. [DOI] [PubMed] [Google Scholar]
- 108.Scardapane A, Brucato A, Chiarelli F, Breda L. Efficacy of an Interleukin-1beta Receptor Antagonist (Anakinra) in Idiopathic Recurrent Pericarditis. Pediatr Cardiol. 2012 doi: 10.1007/s00246-012-0532-0. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 109.Vassilopoulos D, Lazaros G, Tsioufis C, Vasileiou P, Stefanadis C, Pectasides D. Successful treatment of adult patients with idiopathic recurrent pericarditis with an interleukin-1 receptor antagonist (anakinra) Int J Cardiol. 2012;160:66–68. doi: 10.1016/j.ijcard.2012.05.086. [DOI] [PubMed] [Google Scholar]
- 110.Cantarini L, Lucherini OM, Cimaz R, Galeazzi M. Recurrent pericarditis caused by a rare mutation in the TNFRSF1A gene and with excellent response to anakinra treatment. Clin Exp Rheumatol. 2010;28:802. [PubMed] [Google Scholar]
- 111.Baszis KW, Singh G, White A, Thatayatikom A. Recurrent cardiac tamponade in a child with newly diagnosed systemic-onset juvenile idiopathic arthritis. J Clin Rheumatol. 2012;18:304–306. doi: 10.1097/RHU.0b013e3182685857. [DOI] [PubMed] [Google Scholar]
- 112.Raffeiner B, Botsios C, Dinarello C, Ometto F, Punzi L, Ramonda R. Adult-onset Still's disease with myocarditis successfully treated with the interleukin-1 receptor antagonist anakinra. Joint Bone Spine. 2011;78:100–101. doi: 10.1016/j.jbspin.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 113.Butin M, Mekki Y, Phan A, Billaud G, Di Filippo S, Javouhey E, Cochat P, Belot A. Successful Immunotherapy in Life-Threatening Parvovirus B19 Infection in a Child. Pediatr Infect Dis J. 2013;32:789–792. doi: 10.1097/INF.0b013e31828df4d1. [DOI] [PubMed] [Google Scholar]
- 114.Abbate A. The heart on fire: inflammasome and cardiomyopathy. Exp Physiol. 2013;98:385. doi: 10.1113/expphysiol.2012.069021. [DOI] [PubMed] [Google Scholar]
- 115.Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, Jabagi H, Wright JR, Jr, Macdonald JA, Lees-Miller JP, Roach D, Semeniuk LM, Duff HJ. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1beta. Exp Physiol. 2013;98:462–472. doi: 10.1113/expphysiol.2012.068338. [DOI] [PubMed] [Google Scholar]
- 116.Zhu J, Zhang J, Xiang D, Zhang Z, Zhang L, Wu M, Zhu S, Zhang R, Han W. Recombinant human interleukin-1 receptor antagonist protects mice against acute doxorubicin-induced cardiotoxicity. Eur J Pharmacol. 2010;643:247–253. doi: 10.1016/j.ejphar.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 117.Mezzaroma E, Toldo S, Van Tassell BW, Graves P, Gewirtz DA, Abbate A. Interleukin-1 receptor antagonism amelioartes radiation-induced cardiomyopathy in the mouse. Eur Heart J. 2011;32:277. Abstract. [Google Scholar]
- 118.Furst DE. Anakinra: review of recombinant human interleukin-I receptor antagonist in the treatment of rheumatoid arthritis. Clin Ther. 2004;26:1960–1975. doi: 10.1016/j.clinthera.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 119.Biovitrum AB. Version. Stockholm; Sweden: Dec, 2012. Kineret prescribing information. [Google Scholar]
- 120.Fleischmann RM, Schechtman J, Bennett R, Handel ML, Burmester GR, Tesser J, Modafferi D, Poulakos J, Sun G. Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: A large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 2003;48:927–934. doi: 10.1002/art.10870. [DOI] [PubMed] [Google Scholar]
- 121.Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 2009;32:1663–1668. doi: 10.2337/dc09-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smith CJ, Emsley HC, Udeh CT, Vail A, Hoadley ME, Rothwell NJ, Tyrrell PJ, Hopkins SJ. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine. 2012;58:384–389. doi: 10.1016/j.cyto.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 123.Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fleischmann RM, Tesser J, Schiff MH, Schechtman J, Burmester GR, Bennett R, Modafferi D, Zhou L, Bell D, Appleton B. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:1006–1012. doi: 10.1136/ard.2005.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fisher CJ, Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, Reines HD, Shelly MP, Thompson BW, LaBrecque JF, Catalano MA, Knaus WA, Sadoff JC. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836–1843. [PubMed] [Google Scholar]
- 126.Opal SM, Fisher CJ, Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, Sadoff JC, Slotman GJ, Levy H, Balk RA, Shelly MP, Pribble JP, LaBrecque JF, Lookabaugh J, Donovan H, Dubin H, Baughman R, Norman J, DeMaria E, Matzel K, Abraham E, M S. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25:1115–1124. doi: 10.1097/00003246-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 127.Fisher CJ, Jr, Slotman GJ, Opal SM, Pribble JP, Bone RC, Emmanuel G, Ng D, Bloedow DC, Catalano MA. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med. 1994;22:12–21. doi: 10.1097/00003246-199401000-00008. [DOI] [PubMed] [Google Scholar]
- 128.Fisher CJ, Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, Abraham E, Schein RM, Benjamin E. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996;334:1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- 129.Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, Wittkowski H, Bialkowski A, Tzaribachev N, Lohse P, Koitchev A, Deuter C, Foell D, Benseler SM. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. 2011;63:840–849. doi: 10.1002/art.30149. [DOI] [PubMed] [Google Scholar]
- 130.Neven B, Marvillet I, Terrada C, Ferster A, Boddaert N, Couloignier V, Pinto G, Pagnier A, Bodemer C, Bodaghi B, Tardieu M, Prieur AM, Quartier P. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010;62:258–267. doi: 10.1002/art.25057. [DOI] [PubMed] [Google Scholar]
- 131.Version. Regeneron Pharmaceuticals Inc; Tarrytown, NY: Apr, 2010. Arcalyst prescribing information. [Google Scholar]
- 132.Stahl N, Radin A, Mellis S. Rilonacept--CAPS and beyond. Ann N Y Acad Sci. 2009;1182:124–134. doi: 10.1111/j.1749-6632.2009.05074.x. [DOI] [PubMed] [Google Scholar]
- 133. [Accessed 3/18/2013]; http://www.clinicaltrials.gov NCT00417417.
- 134.Version. Novartis Pharaceutical Corp; East Hannover, NJ: Oct, 2012. Ilaris prescribing information. [Google Scholar]
- 135.Curran MP. Canakinumab: in patients with cryopyrin-associated periodic syndromes. Biodrugs. 2012;26:53–59. doi: 10.2165/11208450-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 136.Alten R, Gomez-Reino J, Durez P, Beaulieu A, Sebba A, Krammer G, Preiss R, Arulmani U, Widmer A, Gitton X, Kellner H. Efficacy and safety of the human anti-IL-1beta monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, phase II, dose-finding study. BMC Musculoskelet Disord. 2011;12:153. doi: 10.1186/1471-2474-12-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schlesinger N, Mysler E, Lin HY, De Meulemeester M, Rovensky J, Arulmani U, Balfour A, Krammer G, Sallstig P, So A. Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind, randomised study. Ann Rheum Dis. 2011;70:1264–1271. doi: 10.1136/ard.2010.144063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.So A, De Meulemeester M, Pikhlak A, Yucel AE, Richard D, Murphy V, Arulmani U, Sallstig P, Schlesinger N. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis Rheum. 2010;62:3064–3076. doi: 10.1002/art.27600. [DOI] [PubMed] [Google Scholar]
- 139.Cantarini L, Vitale A, Borri M, Galeazzi M, Franceschini R. Successful use of canakinumab in a patient with resistant Behcet's disease. Clin Exp Rheumatol. 2012;30:S115. [PubMed] [Google Scholar]
- 140.Hacihamdioglu DO, Ozen S. Canakinumab induces remission in a patient with resistant familial Mediterranean fever. Rheumatology (Oxford) 2012;51:1041. doi: 10.1093/rheumatology/kes021. [DOI] [PubMed] [Google Scholar]
- 141.Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, Thuren T. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- 142.Rissanen A, Howard CP, Botha J, Thuren T. Effect of anti-IL-1beta antibody (canakinumab) on insulin secretion rates in impaired glucose tolerance or type 2 diabetes: results of a randomized, placebo-controlled trial. Diabetes Obes Metab. 2012 doi: 10.1111/j.1463-1326.2012.01637.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 143.Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, Brik R, McCann L, Kasapcopur O, Rutkowska-Sak L, Schneider R, Berkun Y, Calvo I, Erguven M, Goffin L, Hofer M, Kallinich T, Oliveira SK, Uziel Y, Viola S, Nistala K, Wouters C, Cimaz R, Ferrandiz MA, Flato B, Gamir ML, Kone-Paut I, Grom A, Magnusson B, Ozen S, Sztajnbok F, Lheritier K, Abrams K, Kim D, Martini A, Lovell DJ. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2396–2406. doi: 10.1056/NEJMoa1205099. [DOI] [PubMed] [Google Scholar]
- 144.Simonini G, Xu Z, Caputo R, De Libero C, Pagnini I, Pascual V, Cimaz R. Clinical and transcriptional response to the long-acting interleukin-1 blocker canakinumab in Blau syndrome-related uveitis. Arthritis Rheum. 2013;65:513–518. doi: 10.1002/art.37776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 146.Schlesinger N, Alten RE, Bardin T, Schumacher HR, Bloch M, Gimona A, Krammer G, Murphy V, Richard D, So AK. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann Rheum Dis. 2012;71:1839–1848. doi: 10.1136/annrheumdis-2011-200908. [DOI] [PubMed] [Google Scholar]
- 147.Geiler J, McDermott MF. Gevokizumab, an anti-IL-1beta mAb for the potential treatment of type 1 and 2 diabetes, rheumatoid arthritis and cardiovascular disease. Curr Opin Mol Ther. 2010;12:755–769. [PubMed] [Google Scholar]
- 148.Gul A, Tugal-Tutkun I, Dinarello CA, Reznikov L, Esen BA, Mirza A, Scannon P, Solinger A. Interleukin-1beta-regulating antibody XOMA 052 (gevokizumab) in the treatment of acute exacerbations of resistant uveitis of Behcet's disease: an open-label pilot study. Ann Rheum Dis. 2012;71:563–566. doi: 10.1136/annrheumdis-2011-155143. [DOI] [PubMed] [Google Scholar]
- 149.Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, Solinger AM, Mandrup-Poulsen T, Dinarello CA, Donath MY. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care. 2012;35:1654–1662. doi: 10.2337/dc11-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Owyang AM, Maedler K, Gross L, Yin J, Esposito L, Shu L, Jadhav J, Domsgen E, Bergemann J, Lee S, Kantak S. XOMA 052, an anti-IL-1{beta} monoclonal antibody, improves glucose control and {beta}-cell function in the diet-induced obesity mouse model. Endocrinology. 2010;151:2515–2527. doi: 10.1210/en.2009-1124. [DOI] [PubMed] [Google Scholar]
- 151.Roell MK, Issafras H, Bauer RJ, Michelson KS, Mendoza N, Vanegas SI, Gross LM, Larsen PD, Bedinger DH, Bohmann DJ, Nonet GH, Liu N, Lee SR, Handa M, Kantak SS, Horwitz AH, Hunter JJ, Owyang AM, Mirza AM, Corbin JA, White ML. Kinetic approach to pathway attenuation using XOMA 052, a regulatory therapeutic antibody that modulates interleukin-1beta activity. J Biol Chem. 2010;285:20607–20614. doi: 10.1074/jbc.M110.115790. [DOI] [PMC free article] [PubMed] [Google Scholar]