

Abstract
It is assumed that MQ are central to glucose sensor bio-fouling and therefore have a major negative impact on continuous glucose monitoring (CGM) performance in vivo. However to our knowledge there is no data in the literature to directly support or refute this assumption. Since glucose and oxygen (O2) are key to glucose sensor function in vivo, understanding and controlling glucose and O2 metabolic activity of MQ is likely key to successful glucose sensor performance. We hypothesized that the accumulation of MQ at the glucose sensor-tissue interface will act as “Cell Based Metabolic Barriers” (CBMB) to glucose diffusing from the interstitial tissue compartment to the implanted glucose sensor and as such creating an artificially low sensor output, thereby compromising sensor function and CGM. Our studies demonstrated that 1) direct injections of MQ at in vivo sensor implantation sites dramatically decreased sensor output (measured in nA), 2) addition of MQ to glucose sensors in vitro resulted in a rapid and dramatic fall in sensor output and 3) lymphocytes did not affect sensor function in vitro or in vivo. These data support our hypothesis that MQ can act as metabolic barriers to glucose and O2 diffusion in vivo and in vitro.
Keywords: Macrophages, diabetes, implantable glucose sensor, continuous glucose monitoring, glucose metabolism
INTRODUCTION
The currently approved usage lifespans for commercial continuous glucose monitor (CGM) in vivo ranges from 3 days to 7+ days. Achieving euglycemia with an artificial pancreas requires a highly accurate CGM. Inflammation is a significant complication of CGM, but the nature of this inflammation and the mechanism involved are not well understood. It is universally accepted that macrophages (MQ) are a histologic hallmark of chronic inflammation including foreign body reactions [1]. Implantable glucose sensors used in CGM of diabetic patients are known to induce foreign body reactions characterized by accumulation of macrophages (MQ) at the sensor-tissue interface. Since MQ are not only pivotal in mediating inflammation as well as wound healing, it is critical to define the role of these cells and their products utilizing biosensor function in vivo. The paucity of information regarding the mechanistic role of MQ, MQ subpopulations and their products in controlling sensor function in vivo represents a critical gap in our knowledge.
Our laboratory previously reported that red blood cells (RBC) serve as a consumptive barrier for glucose sensors by creating “metabolic sinks”, which consume glucose when pooled around glucose sensors in clot formation (hemorrhages) [2, 3]. In these studies, we concluded that RBC accumulation at glucose sensor sites resulted in a marked decrease in local glucose levels. This localized RBC glucose consumption at the site led to a significant reduction in sensor output. Thus, this RBC-based “metabolic sink” mimicked the loss of sensor function when the sensor was correctly reporting the reduced glucose levels in the surrounding microenvironment. Simulation studies reported by Novak et al [4] concluded that inflammatory cells but not RBC at site of sensor location were responsible for increased glucose consumption and the observed “anomalous” sensor behavior. There is no dispute that inflammatory cells are glucophagic. However, we opine that the “anomalous readings” are better explained by the microhemorrhage induced by sensor implantation such that the RBC are responsible for the majority of the glucose consumption when submerged in whole blood. At initial insertion time, the erythrocyte to leukocyte ratio (mainly polymorphonuclear neutrophils (PMN)) is about 1000:1. Nonetheless, erythrocytes in the tissue as a result of hemorrhage are less metabolically active over time as compared to leukocytes. In addition, inflammatory cell removal of RBC from the hemorrhages adjacent to the sensor site, likely increases the degree of sensor-associated inflammation with a resultant decrease in sensor function. Thus, it would be difficult to predict the ratio between RBC and inflammatory cells in this environment. This may be further complicated by hemorrhage formation due to sensor movement hours or days post sensor implantation when a significant number of inflammatory cells are already present at the sensor site.
Notwithstanding, micro-hemorrhages around glucose sensors do not invariably occur. In contrast, accumulation of inflammatory cells characterized by MQ recruitment and accumulation at the sensor-tissue interface are almost invariably seen at sensor implantation sites [5]. This occurs with both acute and chronic inflammatory processes including foreign body reactions. Moreover, macrophages are highly metabolically active cells that consume significant quantities of glucose and oxygen in order to generate their responses to tissue injury and invasive pathogens [6]. We recently reported that MQ accumulate at the sensor-tissue site over time and form a barrier that surrounds the implanted sensor with resultant impairment of continuous glucose sensor performance [5]. This publication also reported that MQ deficient or depleted animals demonstrated enhanced sensor performance [5].
These studies and the observations that MQ are highly metabolically active support our hypothesis that the recruitment and accumulation of MQ at the sensor-tissue site create “Cell Based Metabolic Barriers” (CBMB) to glucose, which results in impaired glucose sensor performance (see Figure 1). Additionally MQ consume significant amounts of oxygen as part of their anti-microbial functions. MQ induced local oxygen consumption occurs in the production of superoxides, which can adversely affect oxygen dependent glucose sensors [7, 8] (see Figure 1). This was assessed with an in vivo murine model of CGM that characterized MQ-sensor interactions. MQ impacts on glucose sensors in vitro were also assessed with newly developed in vitro cell culture based system. The studies demonstrated the following: direct injections of MQ at sensor implantation sites decreased sensor function as represented by the fall in sensor output; addition of MQ to glucose sensors in vitro resulted in a rapid reduction in sensor output and that lymphocytes did not affect sensor function in vitro or in vivo. These data support our hypothesis that the accumulation of MQ at the sensor-tissue interface acts as a metabolic barrier to glucose diffusion from the interstitial tissue compartment to the implanted glucose sensor (Figure 1). This process results in an artificially low sensor output, which compromises CGM. These data could be incorporated into future therapeutic interventions and new sensor designs that ought to lead to acceptable sensor performance and CGM accuracy.
Figure 1. Monocyte related cells create “cell based metabolic barriers” (CBMB) to glucose and oxygen diffusion at sites of glucose sensor implantation and compromise CGM.

Based on the literature and recent data from our laboratories, we hypothesized that the recruitment and accumulation of monocyte related cells (i.e. MQ and FBGC) at the sensor-tissue interface create a “Cell Based Metabolic Barrier” (white arrow) to glucose and oxygen that is diffusing from the vasculature (V) toward implanted glucose sensors (sensor).
MATERIALS AND METHODS
Murine continuous glucose monitoring (CGM): glucose sensors, implantation and mice
All modified Navigator glucose sensors used in these in vivo studies were obtained from Abbott Diabetes Care. Glucose sensors were implanted into mice and continuous glucose monitoring (CGM) was undertaken as previously reported [9]. Implanted sensors were secured to the mouse skin with a mesh, and CGM was initiated per protocol [9]. Blood glucose reference measurements were obtained at least daily using blood obtained from the tail vein of the mouse and a FreeStyle® Blood Glucose Monitor. All C57BL/6J mice used in these studies where obtained from Jackson Laboratories, Bar Harbor Maine. The Institutional Animal Care and Use Committee of the University of Connecticut Health Center (Farmington, CT) approved all the studies involving mice.
Macrophage injections at glucose sensor implantation sites
To investigate the ability of MQ to suppress glucose sensor function in vivo isolated mouse macrophages were directly injected at the sensor implantation site in normal C57BL/6 mice (Figure 2). Specifically, thioglycolate induced peritoneal MQ from normal C57BL/6 mice were obtained as previously described [10]. Glucose sensors were implanted and after a sensor run-in time of about 24 hours either saline or peritoneal MQ were directly injected at site of sensor location. For these studies 105–107 MQ per injection site were used (arrow in Figure 2). To determine “cell specificity/MQ dependence” of these reactions mouse spleen-derived lymphocytes were also tested in place of MQ at sites of sensor implantation.
Figure 2. Cell injection site at glucose sensor implantation site in murine model of CGM.

For in vivo cell injection studies, cells (MQ or spleen lymphocytes) we injected subcutaneously on the back of mice (red arrow) used in our murine model of CGM.
Histopathologic analysis of tissue reactions at glucose sensor implantation sites
In order to evaluate the tissue responses to macrophage injections at glucose sensor implantation sites, individual mice were euthanized and the tissue containing the implanted sensors was removed, fixed in 10% buffered formalin for 24 hours, followed by standard processing, embedded in paraffin and sectioned. The resulting 4–6 um sections were then stained using standard protocols for hematoxylin/eosin stain (H/E). The tissue samples were examined for signs of inflammation, including necrosis, fibrosis, and vessel regression. Resulting tissue sections were evaluated directly and documented by digitized imaging using an Olympus Digital Microscope.
Immunohistchemical staining of macrophages using F4/80
To confirm the observations of the presence of macrophages in tissue sections, we utilized a mouse macrophage specific antibody designated anti-mouse F4/80. Anti-mouse F4/80 (@F4/80) (Invitrogen Catalog # A14800) was validated using mouse spleen tissue and standard immunohistochemical (IHC) techniques.
Evaluation of the impact of macrophages on glucose sensor function in vitro
An in vitro cell model system was developed in order to better understand the interaction of cells with glucose sensors (Figure 3a and 3b). For this system, a standard nylon washer was utilized in which a V shaped-well was drilled into the disk holding any candidate cells as well as the sensing element of the glucose sensor (Sensor 2 in Figure 3a and 3b). The nylon washer was then adhered to the bottom of a standard tissue culture petri dish and one glucose sensor was inserted into the V-well thru a small hole in the wall of the petri dish (Sensor 2 in Figure 3a and 3b). An additional glucose sensor was inserted distal to the V-well inserted sensor. Sensor 1 measures glucose levels in the entire tissue culture dish (i.e. 50 ml of tissue culture media) and Sensor 2 measures the glucose levels in the V-well containing the added cells. The tissue culture dish containing the 2-sensor system was placed in a bench top incubator (Incu-Shaker Mini) and operated at 37°C. Using this system, an initial baseline sensor response was established for both sensors for low (50 mg/dL), high (350 mg/dL) and standard (200 mg/dL) glucose containing media. Once baselines were established, various numbers of test cells were added directly into V-well containing media. Generally, 105 to 107 cells were added to the V-well and CGM was run for both sensors for 5–7 days at 37°C. All CGM data is expressed as nano-Ampere (nA). Media glucose levels in the petri dish were evaluated at various time points using a standard Abbott Freestyle external glucose monitor. At the end of the incubations, the media was removed and the cells present in the V-well were washed out. Sensor performance was followed for the same low, standard and high glucose containing media in order to determine if there was any cell related damage to the sensor incubated in the V-well containing cells (Sensor 2) compared to the sensor incubated in the media (Sensor 1).
Figure 3. Sensor-cell culture system used to evaluate the impact of cell populations on glucose sensor function in vitro.
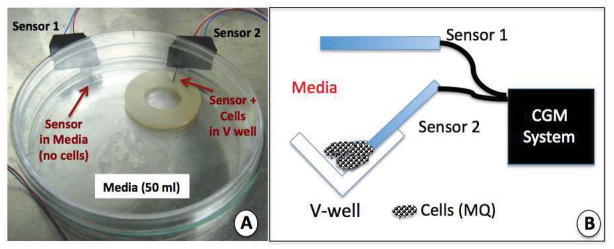
The sensor-cell culture system presented in Figure 3a and 3b were developed to mimic the cell sensor interactions that occur at the sensor-tissue interface at glucose sensor implantation sites.
Figure 3a is a photograph of our dual sensor cell culture system for evaluation of the impact of cell populations on glucose sensor function and CGM in vitro. The Cell culture system is composed of a standard tissue culture petri dish in which a nylon disk with a V-well drilled into one edge of the nylon disk. Additionally, 2 sensors are inserted into the petri dish thru predrilled holes, and sealed with aquarium silicone. For this system 50 ml of standard cell culture media is added to the petri dish, and candidate cells are pipetted into the V-well containing glucose sensor 2 (thin red arrow).
Figure 3b is a diagram of the V-well area in the nylon disk used in the dual sensor-cell culture system presented in Figure 3a. Also included is the location of the dual sensor system and the link to the CGM electronics.
RESULTS
Characterization of the presence of MQ at tissue implantation sites of glucose sensors
The presence and distribution of macrophages at sites of sensor implantation in the mouse were characterized using @F4/80 IHS. Using @F4/80 antibodies and standard IHC technology, scattered MQ were detected as early as 1-day post sensor implantation (Figure 4 modified from Figure 3B [5]). Accumulation of MQ at the sensor-tissue interface continued to increase, ultimately forming a large MQ “band/barrier” which creates a “Cell Based Metabolic Barrier” of macrophages surrounding the sensor by 14-days post implantation and beyond. As a control, normal Ig [DEFINE THIS] was tested at all time-points. It was negative at all time-points and did not react with any of the cells at the sensor implantation site.
Figure 4. Characterization of macrophage distribution at glucose sensor-tissue interface at sites of glucose sensor implantation using anti-F4//80 antibodies.
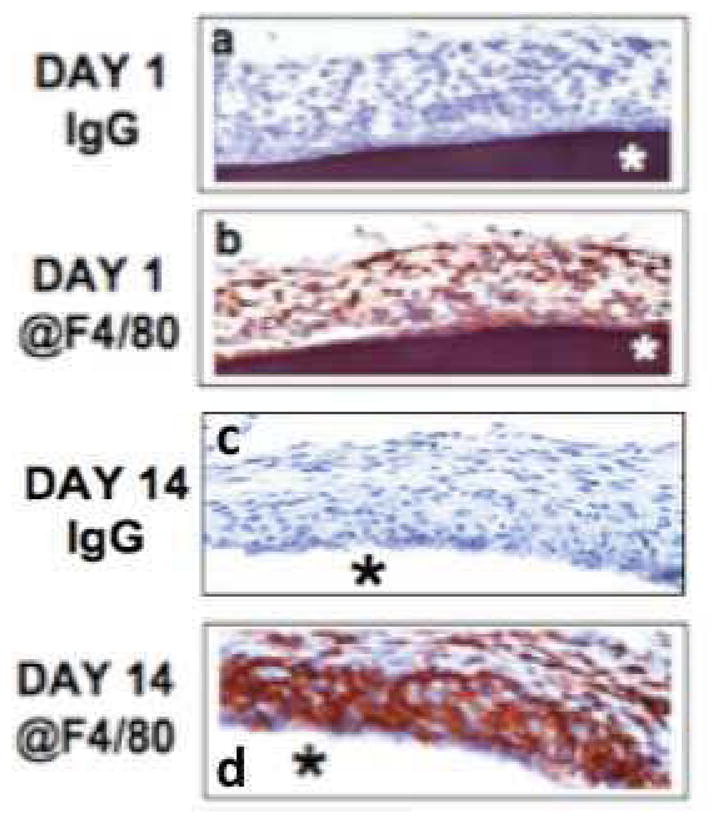
Figures 4a–d are photomicrographs of IHC analysis of sensor-tissue interface at sites of glucose sensor implantation in our murine model of CGM. The goal of these studies was to characterized the presence and distribution of macrophages at sites of sensor implantation in the mouse using @F4/80 IHC. Using @F4/80 antibodies and standard IHC technology, scattered MQs were detected as early as 1-day post senor implantation (Figure 4b). Accumulation of MQ at the sensor-tissue interface continued to increase, ultimately forming a large MQ “band/barrier” which creates “Cell Based Metabolic Barrier” of macrophages surrounding the sensor at day 14-post implantation and beyond (Figure 4b). As a control normal Ig was tested at all time points and was negative for all time-points (Figure 4a and 4c).
Impact of macrophage and lymphocyte injections at glucose sensor implantation sites
Given that we had demonstrated the accumulation of MQ at the tissue-sensor interface, we hypothesized that MQ contribute directly to sensor biofouling by local glucose metabolism at the sensor implantation site. Therefore, we predicted that direct injections of metabolically active MQ to sites of sensor implantation (“MQ additions”) will accelerate loss of sensor function in vivo. To determine “cell specificity/MQ dependence” of these reactions, we injected spleen-derived lymphocytes, in place of MQ, at sites of sensor implantation. For these studies, MQ or lymphocytes (2.4x10^6 cells/30 ul) were injected at the sensor site and CGM was evaluated for the subsequent 48 hrs. Figure 5 represents sensor response of triplicates (3 sensor in 3 separate mice) with each sensor output as a separate solid line. As depicted, sensor output declined significantly within hours post MQ cell injection. Notably, sensor output did not decline following lymphocyte injection at sites of sensor location Figure 5. These studies lend further support to the critical role of MQ in glucose metabolism at the senor implantation site. Given these data, we believe that this “metabolic effect” results from the biofouling of glucose sensors in vivo. We opine that these same metabolic effects would likely be observed when MQ are recruited to sites of sensor implantation in vivo.
Figure 5. Impact of macrophage or lymphocytes injections at glucose sensor implantation sites on sensor function and CGM in vivo.

Presented in Figure 5 are the results of in vivo injection of MQ (Figure 5a) or spleen derived lymphocytes (Figure 5b) at the glucose sensor implantation in our murine model of CGM. For these studies MQ or lymphocytes (2.4x10^6 cells/30 ul) were injected in a volume of 30 ul at tip of sensor and CGM evaluated for the subsequent 48 hrs. Results presented in Figure 5 represents sensor response of triplicates (i.e. for each cell type 3 sensors were implanted in 3 separate mice) for MQ (Figure 5a) or lymphocyte (Figure 5b) injections. Sensor output for each sensor is represented as a separate solid line. Timing of cell injections is noted with the vertical red arrow.
Impact of injected MQ at sensor implantation sites to intra-peritoneal injections of glucose
Based on the hypothesis that MQ create metabolic barriers for glucose sensors, we next determined whether the negative effects of MQ injection on CGM could be overcome by artificially elevating blood glucose levels using i. p. injection of glucose in mice. Figure 6 represents the results of 3 independent studies. For these studies, sensors were implanted in mice as previously described. After the sensor signal stabilized, 30 ul of saline containing 2.5x10^6 MQ were injected at the sensor implantation site (red vertical arrow) of 3 separate mice. As expected, injection of MQ induced a profound drop in sensor output (blue line). Once MQ induced loss of sensor output had stabilized, sterile glucose was injected i. p., and serum glucose levels were recorded with an external glucose monitor (orange diamonds) (Figure 6a). In selected cases, there were multiple i. p. glucose injections (Figure 6b and Figure 6c). In all three studies, MQ injections induced the loss of sensor function as reflected by a reduction in sensor output. In addition, i. p. injections of glucose caused an elevation of serum glucose levels and sensor output. Finally, as serum glucose levels returned to normal, sensor output diminished again. These studies lend further credence to our hypothesis that MQ at sensor implantation sites can bio-foul glucose sensors by acting as a metabolic barrier preventing glucose diffusion from the interstitial space to the sensor itself (Figure 1). This artificially elevated i. p. induced blood glucose level oversaturates the interstitial tissue space with glucose. MQ in the periphery of the implanted sensor are unable to metabolize this increased tissue glucose. This allows the residual glucose to diffuse to the implanted sensor.
Figure 6. Impact of injection of macrophages at glucose sensor implantation sites on sensor response to intra-peritoneal injection of glucose.

Figure 6 represents the results of 3 independent studies to determine whether the loss of glucose sensor CGM after MQ injection at sensor implantation sites using our murine model of CGM. For these studies sensor were implanted in mice as previously described. After the sensors signal stabilized 30 ul of saline containing 2.5X10^6 MQ was injected into the sensor tip at the sensor implantation site (red vertical arrow) of 3 separate mice. As expected the injection of the MQs caused a dramatic drop in sensor output (blue line). Once MQ induced loss of sensor output had stabilized 80 μl of 50% sterile glucose in saline was injected i. p. (green arrows) and blood glucose levels were followed using an external glucose monitor (orange diamonds) (Figure 6a). In selected cases there were multiple i. p. glucose injections (Figure 6b and Figure 6c). In all 3 studies 1) the MQ injections induced loss of sensor function as reflected by drop in sensor output and 2) i. p. injections of glucose caused spiking elevation of blood glucose levels and sensor output; and 3) as blood glucose levels returned to normal sensor output also dropped to below normal output levels.
Histopathologic evaluation of MQ/lymphocyte in vivo injection sites
The studies presented above demonstrate that injections of MQ, but not lymphocytes, cause an apparent loss of glucose sensor function in vivo. We hypothesize that this loss in sensor function is the result of MQ consuming the interstitial glucose and preventing it from reaching the implanted sensor; e.g. creating a “metabolic barrier” around the implanted sensor. To evaluate the distribution of the injected MQ and lymphocytes in the mouse skin, we undertook histologic evaluations of sensor implantation sites with or without MQ or lymphocyte injections. As can be seen in Figure 7, sensors implanted in mouse skin with either no injections (Figure 7A) or saline injections (Figure 7B), displayed no macrophage or lymphocyte accumulation at the sensor implantation site. In contrast, implantation sites that were injected with MQ (Figure 7C–D) or lymphocytes (Figure 7E–F) demonstrated a clear presence of the inflammatory cell injected, MQ or lymphocytes, surrounding the implanted sensors (*). These data demonstrate that MQ or lymphocytes injected at sensor implantation sites surround the implanted sensor. Nevertheless, sensor data demonstrated that only MQ cause a drop in the sensor output. The combination of our functional and histologic data related to leukocyte injections at sensor implantation sites supports our hypothesis that highly metabolically active cells, such as MQ, can create metabolic barriers to glucose diffusion to the sensor site and thus adversely affect glucose sensor function in vivo.
Figure 7. Histopathology of glucose sensor implantation sites injected with macrophage or lymphocyte.

Presented in Figure 7 are representative photomicrographs of histopathology (H&E) of glucose sensor implantation sites including: non-injected (A), saline injected (B), macrophage (MQ) injected (C, D) and lymphocyte (LYM) injected (E, F).
Evaluation of the impact of MQ and lymphocytes on glucose sensor function in vitro
The studies presented in Figure 5 and 6 support the hypothesis that MQ can directly cause loss of glucose sensor function in vivo. MQ metabolize the interstitial glucose thus making it unavailable to diffuse to the implanted glucose sensor. It could be argued that confounding variables or additional biofouling effects are responsible for this loss of sensor function in vivo. In order to exclude these possibilities, we developed an in vitro model system that would allow evaluation of only MQ-sensor interactions, thus eliminating tissue effects of biofouling. As depicted in Figure 8, MQ cell numbers at 3x10^5 or below did not have any significant impact on either the glucose sensor or media glucose levels. However, MQ cell numbers of 1x10^6 to 3x10^6 resulted in a dose dependent loss of sensor function for the sensor located in the V-wells. It should be noted that at higher cell numbers, sensors submerged in media indicated a fall in the total media glucose levels as reflected by external glucose monitoring of the media (red diamonds in Figure 8). The return of sensor function that occurred between days 2–3 in culture was associated with loss of MQ viability as reflected by direct microscopic evolution of residual cells in the V-wells using trypan blue dye. It should be noted that addition of fresh glucose containing media to the cell containing media reversed the loss of sensor function (data not depicted). This in vitro observation was similar to our in vivo observation of i. p. injected glucose into mice previously treated with MQ injected at sensor sites (Figure 6). Comparison of sensor function post MQ addition to sensor function pre MQ addition indicated that MQ did not have any significant impact on sensor function in the absence of MQ. Finally, as was the case for the in vivo studies, the addition of spleen-derived lymphocytes at comparable numbers did not induce any significant loss of sensor function in this in vitro system (see Figure 8).
Figure 8. Evaluation of the impact of macrophages and lymphocytes on glucose sensor function in vitro.

Figure 8 represents in vitro cell data to directly determine whether MQ can directly inhibit (bio-foul) sensor in vitro and whether this effect is MQ specific. As can be seen in Figure 8a at 3x10^5 MQ or below (data not shown) did not have any significant impact on either glucose sensor or media glucose levels. Addition of 1x10^6 (Figure 8b) or 3x10^6 (Figure 8b) MQ to the v-well in this in vitro system cause a dose dependent loss of sensor function for the sensor present in the V-wells (blue line in Figure 8a, 8b and 8c). Comparison of sensor function post MQ addition to sensor function pre MQ addition indicated that the MQ did not have any significant impact on sensor function in the absence of the MQ (Figure 8a–8c). Finally, as was the case in vivo, the addition of spleen-derived lymphocytes at comparable numbers did not cause any significant loss of sensor function in this in vitro system (see blue line in Figure 8d).
DISCUSSION
Dramatic fluctuations of serum glucose levels in diabetic patients (i.e. hyperglycemia and hypoglycemia) effect changes in the vascular system resulting in numerous diabetes related complications including death [11]. A highly effective method proven to reduce the risk of diabetic complications is the maintenance of a normal serum glucose level (i.e. euglycemia) [11, 12]. External glucose monitors are typically utilized in order to optimize serum glucose levels in patients with diabetes. However, many diabetic patients do not consistently monitor their serum glucose levels. Even patients who frequently test their blood glucose levels struggle to maintain euglycemia. One alternative to external blood monitoring uses implantable glucose sensors and CGM [13, 14]. CGM technology allows the patients with diabetes to monitor their glucose level in real-time in order to alert the user when glucose are out of range [15, 16]. This knowledge can help the user to prevent potential harmful hyperglycemic or hypoglycemic events. Crucial to effective CGM performance, and the ultimate development of the artificial pancreas, is an accurate glucose sensor. Suboptimal CGM performance has been attributed to the tissue reactions caused by the implanted sensor, i.e. biofouling [5, 17–20]. This biofouling of glucose sensors is thought to be driven by the tissue reaction triad (TRT) of inflammation, fibrosis and blood vessel regression [5, 17, 18]. These tissue reactions compromise sensor performance through a variety of mechanisms alone or in combination such as local glucose metabolism, oxygen consumption, impairment of glucose diffusion and direct damage to sensor components (membranes, glucose oxidase, etc.). However, the precise roles of specific cells, mediators and pathways that induce bio-fouling and poor sensor performance are not well characterized.
Chronic inflammatory reactions to foreign materials and devices such as sensors are known as foreign body reactions (FBR). Histologically, FBR are dominated by macrophages (MQ) and MQ related foreign body giant cell (FBGC) formation. Their central role in controlling bio-fouling, directly and indirectly, is accepted. Nonetheless, there are limited data that demonstrate the direct role of MQ in glucose sensor bio-fouling. In order to address these issues, we investigated the in vivo and in vitro impact of MQ on glucose sensors and CGM. For these studies, we utilized our previously described in vivo murine model of CGM as well as a newly developed in vitro model for evaluating the direct impact of cells, such as MQ, on glucose sensor function.
Glucose sensor biofouling: role of MQ in CGM in vivo and in vitro
MQ are known to be important cells in host defense against foreign objects, including microorganisms, via metabolically intense activities (i.e. glucose metabolism) such as chemotaxis, phagocytosis and generation of reactive oxygen species. Sensors, like microorganisms, are foreign objects and also trigger these same intense metabolic activities. Although it is assumed that MQ have a negative impact on CGM, currently there is no data in the literature to support or refute this assumption Thus, we conducted these investigations by utilizing an in vivo murine model for CGM and an in vitro model to evaluate the impact of macrophages on sensor function. The major conclusions of these MQ/lymphocyte studies are as follows:
Direct injection of MQ, but not lymphocytes, induces an immediate loss of sensor function in vivo
Intra-peritoneal injections of high concentrations of glucose temporarily mitigates the observed loss of sensor function,
In vitro MQ, but not lymphocytes, can induce an apparent loss of glucose sensor function, which is overcome by the addition of fresh glucose to the culture system
These data support the concept that MQ represent “metabolic barriers” to glucose and oxygen diffusion to the sensor with a resultant decrease in the function of the implanted sensor and accuracy of CGM in vivo (Figure 9). Based on these studies, as well as the literature, we have developed a general model of the role of monocyte/macrophages and related cells including the foreign body giant cell in direct and indirect biofouling of glucose sensors in vivo (Figure 10 from [5]).
Figure 9. Theoretical model of “cell based metabolic barrier” responsible for loss of glucose sensor function and loss of accuracy of CGM in vivo.

Presented in Figure 9 is a simple theoretical model of the mechanism by which for “cell based metabolic barriers” deplete diffusing glucose and oxygen levels between the interstitial space and the implanted glucose sensor. These metabolic barriers result in a loss of the apparent sensor function during normal CGM in vivo.
Figure 10. General Hypothetical model of the impact of monocyte related cells (i.e. macrophage and foreign body giant cells (GC)) on direct and indirect bio-fouling of glucose sensors in vivo.

Based on the data here as well as the literature related to role of sensor induced tissue reactions and CGM we have developed the following hypothetical model of the role on monocyte related cells in controlling tissue reactions and sensor function in vivo.
Hypothetical model of direct and indirect bio-fouling of glucose sensors in vivo by monocytes, macrophage and related cells
The following hypothetical model on the role of monocyte related cells in controlling tissue reactions and sensor function in vivo is depicted in (Figure 10 from [5]). We hypothesized that monocyte related cells such as MQ and foreign body giant cells (FBGC) play a central role in both direct and indirect biofouling of glucose sensors in vivo. MQ and GC directly cause glucose sensor biofouling by creating cellular barriers that surround the implanted sensor, which attenuates glucose and oxygen diffusion to the implanted sensor site. MQ and GC can also indirectly biofoul glucose sensors by metabolizing glucose adjacent to the sensor. This creates an artificially low glucose “microclimate” as compared to the general interstitial glucose levels that exist distant from the implanted sensor. Our data from Figure 8 demonstrate that the metabolic effects of MQ are the main source for the observed poor sensor performance. Our in vitro data demonstrate that once MQ cease to function, sensor performance returns to its original state (Figure 8). Unlike what is observed in vivo, MQ debris that has accumulated at the sensor site will remain at that location. Restated, protein biofouling from MQ debris accumulation has different effects in vitro in that sensor output will not return to its initial performance level once MQ lose their metabolic activity.
Given the proximity of monocyte related cells to the implanted sensor, MQ can also produce relatively high concentrations of agents, such as oxygen radicals, that can damage the glucose sensors [21]. Monocyte related cells can produce extremely potent inflammatory mediators that promote additional monocyte recruitment from the vasculature via vascular endothelial cells (VEC) activating monocytes into specific macrophage subpopulations (e.g. M1 or M2 macrophages). M1 macrophages can further amplify the inflammatory reactions through expression of pro-inflammatory mediators including cytokines. M2 macrophages can stimulate wound-healing processes including angiogenesis and fibrosis. Although angiogenesis can be beneficial to sensor function, it also promotes fibroblast proliferation and collagen synthesis resulting in fibrosis or scar tissue. Additionally, FBGC can also elaborate a variety of mediators including transforming growth factor beta (TGFB), which would promote fibrosis. As fibrosis increases at the sensor implantation site, it induces blood vessel regression as well as slowing glucose diffusion toward the sensor. In summary, we believe that monocytes and monocyte related cells such as macrophages and foreign body giant cells are main targets for the therapeutic intervention in order to attenuate the tissue reactions that bio-foul glucose sensors. A more comprehensive understanding of the specific cell sensor interactions that occur at the tissue sensor interface should provide a more rational approach when designing a future generation of glucose sensors used in CGM.
CONCLUSION
Implantable glucose sensors used in CGM of patients with diabetes are known to induce foreign body reactions characterized by accumulation of macrophages (MQ) at the sensor-tissue interface. Although it is assumed that these MQ have a negative impact on CGM, currently there is a paucity of data in the literature to support or refute this assumption. We addressed this issue by utilizing our CGM mouse model and an in vitro model in order to determine the impact of cell-sensor interactions on CGM. For the in vivo studies, we determined the impact of direct injection of mouse MQ or lymphocytes at sites of sensor implantation on CGM in normal mice. For the in vitro studies, we compared addition of these same cells to sensor function. In vivo studies demonstrated that direct injection of MQ, but not lymphocytes, adversely affected sensor function in vivo resulting in aberrant glucose readings. This could be overcome by i. p. injections of glucose. The in vitro model of CGM also demonstrated that the addition of MQ, but not lymphocytes, induced a significant drop in sensor output (nA) and could be reversed by the addition of glucose as was noted with the in vivo studies. We believe that our present studies demonstrate the critical role of MQ and glucose metabolism in the apparent loss of sensor function in vivo. The creation of a dense wall of MQ surrounding the sensor creates a “metabolic barrier” to the diffusion of glucose from the vasculature. These “metabolic barriers” can prevent detection of hypoglycemic events and can compromise CGM, which could result in life threatening consequences in “brittle” diabetic patients. Future studies that target MQ and FBGC may achieve the goal of extending glucose sensor function and CGM as well as contributing to the advancement of “closed loop technology”.
Acknowledgments
The National Institute of Diabetes and Digestive and Kidney Diseases provided funding for this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20(2):86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klueh U, Liu Z, Feldman B, Henning TP, Cho B, Ouyang T, et al. Metabolic biofouling of glucose sensors in vivo: role of tissue microhemorrhages. J Diabetes Sci Technol. 2011;5(3):583–95. doi: 10.1177/193229681100500313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klueh U, Liu Z, Ouyang T, Cho B, Feldman B, Henning TP, et al. Blood-induced interference of glucose sensor function in vitro: implications for in vivo sensor function. J Diabetes Sci Technol. 2007;1(6):842–9. doi: 10.1177/193229680700100607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novak MT, Yuan F, Reichert WM. Predicting glucose sensor behavior in blood using transport modeling: relative impacts of protein biofouling and cellular metabolic effects. J Diabetes Sci Technol. 2013;7(6):1547–60. doi: 10.1177/193229681300700615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klueh U, Qiao Y, Frailey JT, Kreutzer DL. Impact of macrophage deficiency and depletion on continuous glucose monitoring in vivo. Biomaterials. 2014;35(6):1789–96. doi: 10.1016/j.biomaterials.2013.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Critical reviews in immunology. 2012;32(6):463–88. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 7.Feldman B, Brazg R, Schwartz S, Weinstein R. A continuous glucose sensor based on wired enzyme technology -- results from a 3-day trial in patients with type 1 diabetes. Diabetes Technol Ther. 2003;5(5):769–79. doi: 10.1089/152091503322526978. [DOI] [PubMed] [Google Scholar]
- 8.Gerritsen M, Jansen JA, Lutterman JA. Performance of subcutaneously implanted glucose sensors for continuous monitoring. Neth J Med. 1999;54(4):167–79. doi: 10.1016/s0300-2977(99)00006-6. [DOI] [PubMed] [Google Scholar]
- 9.Klueh U, Liu Z, Cho B, Ouyang T, Feldman B, Henning TP, et al. Continuous glucose monitoring in normal mice and mice with prediabetes and diabetes. Diabetes Technol Ther. 2006;8(3):402–12. doi: 10.1089/dia.2006.8.402. [DOI] [PubMed] [Google Scholar]
- 10.Paulnock DM, editor. Macrophages. Oxford New York: Oxford University Press; 2000. [Google Scholar]
- 11.Patton SR, Clements MA. Average daily risk range as a measure for clinical research and routine care. J Diabetes Sci Technol. 2013;7(5):1370–5. doi: 10.1177/193229681300700529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dailey G. Early and intensive therapy for management of hyperglycemia and cardiovascular risk factors in patients with type 2 diabetes. Clinical therapeutics. 2011;33(6):665–78. doi: 10.1016/j.clinthera.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 13.Klonoff DC. Continuous glucose monitoring: roadmap for 21st century diabetes therapy. Diabetes Care. 2005;28(5):1231–9. doi: 10.2337/diacare.28.5.1231. [DOI] [PubMed] [Google Scholar]
- 14.Klonoff DC. The Benefits of Implanted Glucose Sensors. Journal of Diabetes Science and Technology. 2007;1(6):797–800. doi: 10.1177/193229680700100601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desalvo DJ, Keith-Hynes P, Peyser T, Place J, Caswell K, Wilson DM, et al. Remote Glucose Monitoring in Camp Setting Reduces the Risk of Prolonged Nocturnal Hypoglycemia. Diabetes Technol Ther. 2013 doi: 10.1089/dia.2013.0139. [DOI] [PubMed] [Google Scholar]
- 16.Hoss U, Jeddi I, Schulz M, Budiman E, Bhogal C, McGarraugh G. Continuous glucose monitoring in subcutaneous tissue using factory-calibrated sensors: a pilot study. Diabetes Technol Ther. 12(8):591–7. doi: 10.1089/dia.2010.0051. [DOI] [PubMed] [Google Scholar]
- 17.Klueh U, Antar O, Qiao Y, Kreutzer DL. Role of vascular networks in extending glucose sensor function: Impact of angiogenesis and lymphangiogenesis on continuous glucose monitoring in vivo. Journal of biomedical materials research Part A. 2013 doi: 10.1002/jbm.a.35031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klueh U, Antar O, Qiao Y, Kreutzer DL. Role of interleukin-1/interleukin-1 receptor antagonist family of cytokines in long-term continuous glucose monitoring in vivo. J Diabetes Sci Technol. 2013;7(6):1538–46. doi: 10.1177/193229681300700614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh A, Nichols SP, Schoenfisch MH. Glucose sensor membranes for mitigating the foreign body response. J Diabetes Sci Technol. 2011;5(5):1052–9. doi: 10.1177/193229681100500505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts JR, Park J, Helton K, Wisniewski N, McShane MJ. Biofouling of polymer hydrogel materials and its effect on diffusion and enzyme-based luminescent glucose sensor functional characteristics. J Diabetes Sci Technol. 2012;6(6):1267–75. doi: 10.1177/193229681200600605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerritsen M, Jansen JA, Kros A, Vriezema DM, Sommerdijk NA, Nolte RJ, et al. Influence of inflammatory cells and serum on the performance of implantable glucose sensors. J Biomed Mater Res. 2001;54(1):69–75. doi: 10.1002/1097-4636(200101)54:1<69::aid-jbm8>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]