

Abstract
Heme-regulated inhibitor kinase (HRI), an eukaryotic translation initiation factor 2 alpha (eIF2α) kinase, plays critical roles in cell proliferation, differentiation, and adaptation to cytoplasmic stress. HRI is also a critical modifier of hemoglobin disorders such as β-thalassemia. We previously identified N,N′-diarylureas as potent activators of HRI suitable for studying biology of this important kinase. To expand the repertoire of chemotypes that activate HRI we screened a ~1,900 member N,N′-disubstituted urea library in the surrogate eIF2α phosphorylation assay identifying N-aryl,N′-cyclohexylphenoxyurea as a promising scaffold. We validated hit compounds as a bona-fide HRI activators in secondary assays and explored contributions of substitutions on the N-aryl and N′-cyclohexylphenoxy groups to their activity by studying focused libraries of complementing analogs. We tested these N-aryl,N′-cyclohexylphenoxyureas in the surrogate eIF2α phosphorylation and cell proliferation assays, demonstrating significantly improved bioactivities and specificities. We consider these compounds to represent lead candidates for the development of potent and specific HRI activators.
Keywords: SAR study; N,N′-disubstituted ureas; inhibition of translation initiation; ternary complex; phosphorylation of eIF2α
Introduction
The eukaryotic translation initiation factor 2 (eIF2) forms ternary a complex with initiator methionine transfer RNA (Met-tRNAi) and GTP, which is critically required for recognition of translation start codon. Hydrolysis of GTP in the eIF2·GTP·Met-tRNAi ternary complex and release of inorganic phosphate, Pi, are coupled to start-codon recognition and initiation of translation. The exchange of GDP in the eIF2·GDP binary complex for GTP is catalyzed by eIF2B, a guanine nucleotide exchange factor. This GDP-GTP exchange is the rate-limiting step for the formation of the ternary complex and initiation of a new round of translation. Phosphorylation of alpha subunit of eIF2 (eIF2α) on S51 by eIF2α kinases- HRI, RNA-dependent-protein-kinase/protein kinase R (PKR), pancreatic eIF2α kinase/PKR-like endoplasmic reticulum kinase (PERK), and general control non-derepressible-2 (GCN2)- is the major mechanism that regulates the GDP-GTP exchange. More specifically, S51 phosphorylation on eIF2α concomitantly increases its affinity for eIF2B and inhibits eIF2B’s guanine nucleotide exchange activity. Because the eIF2 is present in excess over eIF2B (low eIF2B/eIF2 stoichiometry), even partial phosphorylation of eIF2α results in sequestration of eIF2B thereby reducing the amount of the eIF2·GTP·Met-tRNAi ternary complex, and inhibiting translation initiation1, 2.
Formation of the eIF2·GTP·Met-tRNAi ternary complex is tightly coupled to cell physiology and plays critical roles in normal- and patho-biology3–9. Proliferating cells synthesize proteins at much higher rate than quiescent cells of similar types. The lower rate of translation in quiescent cells is achieved in part by higher rate of eIF2α phosphorylation compared to proliferating cells. Phosphorylation of eIF2α is essential for coupling protein synthesis to heme availability in red blood cells progenitors, to the folding capacity of ER-Golgi network in the secretory cells, and to the nutrient and oxygen availability in almost all cells.
Consistent with its role in the normal-biology, deregulation of eIF2α phosphorylation is implicated in the patho-biology of various human disorders. For example, inactivating mutations of PERK cause Wolcott-Rallison syndrome, a rare autosomal recessive disease characterized by neonatal/early-onset non-autoimmune insulin-requiring diabetes associated with skeletal dysplasia and growth retardation syndrome10, 11. Insufficiency of eIF2α phosphorylation in red blood cells progenitors, i.e. those deficient in HRI, increases the severity of hemolytic disorders such as β-thalassemia12–14. Deregulation of eIF2α phosphorylation has also been implicated in the pathogenesis of neurodegenerative disorders such as Alzheimer’s disease and proliferative disorders such as cancer15, 16. Forced expression of eIF2α-S51A, a non-phosphorylatable mutant, increases the amount of the ternary complex, renders the translation initiation unrestricted, and causes transformation of normal cells17, 18. Similarly, overexpression of MettRNAi, which increases the amount of the ternary complex, causes cellular transformation19. In contrast, induction of eIF2α phosphorylation pharmacologically or by over-expressing eIF2α kinases inhibits proliferation of cancer cells in vitro and tumor growth in vivo 20–24.
These findings clearly indicate that small molecule activators of eIF2α kinases, particularly of HRI, are important tools for studying the patho-biology of certain human disorders and/or may be deployed for their treatment. This task is highly dependent on discovery of HRI activators whose potency, specificity, and physico-chemical properties can be improved by structure-activity relationship (SAR) studies. Such hits should ideally be based on druggable molecular scaffolds suitable for facile synthesis. We previously reported discovery and preliminary SAR studies of N,N′-diarylureas as activators of HRI25, 26. N,N′-diaryl- as well as N-aryl,N′-alkyl-ureas, such as 12-(3-adamantane-1- ylureido)dodecanoic acid (AUDA), were reported to be potent inhibitors of soluble epoxide hydrolase (sEH)27, 28. We therefore advanced the hypothesis that N,N′-disubstituted urea analogs other than N,N′-diarylureas may also activate HRI and induce eIF2α phosphorylation. If correct, this will allow greater structural versatility that will lend itself to the discovery and development of more potent and selective HRI activators.
Herein, we report screening of a ~1,900 member diversity N,N′-disubstituted urea library, originally developed to inhibit sEH29, 30 and discovery of N-phenyl,N′-cyclohexylphenoxy compounds as activators of HRI. We evaluated selected hit compounds for induction of endogenous eIF2α phosphorylation and expression of its downstream effector, CHOP protein and mRNA, and inhibition of the expression of oncogenic proteins. We further determined the dependence of the activity of these agents on HRI at the molecular and cellular level. We then undertook structure-activity relationship study of novel HRI activators as the first step in a hit-to-lead optimization process. To this end, we synthesized focused libraries of N-phenyl,N′-cyclohexylphenoxy ureas to assess structural latitude and improve their potency. These newly synthesized compounds were characterized for induction of eIF2α phosphorylation in a surrogate cell-based reporter assay as well as inhibition of cell proliferation in sulforhodamine B (SRB) binding assay, which assessed the biological consequences of eIF2α phosphorylation. Selected analogs were further evaluated for specificity in functional genetic studies.
Results and discussion
Chemistry
Substituted ureas of interest that were not commercially available and considered to be important for structure-activity relationship studies (I-11o and I-12o), or designed for the initial hit-to-lead optimization (I-14-20 and III-1-5), were synthesized as described in Schemes 1 and 2. Reacting equimolar amounts of the appropriate cyclohexylamine and phenyl isocyantate yielded the anticipated N-phenyl,N′-cyclohexylaryl ureas in good yield. The substituted trans-(4-phenoxy)cyclohexylamines (1–7) were generated from trans-4-aminocyclohexanol and the appropriately substituted fluorobenzenes following previously reported procedures31. The urea incorporating the cis-1,4-disubstituted cyclohexyl moiety, I-11o was obtained by reacting methyl 2-isocyanatobenzoate with a small excess of cis-4-(4- fluorophenoxy)cyclohexanamine, 1b in DCM/DMSO at room temperature (Scheme 1 B). The amine 1b was synthesized from trans-4-aminocyclohexanol and 4-fluorophenol via Mitsunobu coupling reaction as described earlier34.
Scheme 1.

Synthesis of trans- and cis-N-phenyl-N′-(4-phenoxy)cyclohexylureas.#
#Reagents and conditions: i) NaH, DMF, reflux, 2h. ii) Triethylamine, DMF or DCM/DMSO, RT, 16 h. iii) (1) 4-Fluorophenol, PPh3, p-nitrobenzoic acid, DIAD, THF, RT, 12 h; (2) 35% aqueous hydrazine, CH2Cl2, MeOH, RT, 1 day. iv) LiOH, CH3CN/H2O, 24h.
Scheme 2.

Synthesis of substituted cyclohexylamines and their corresponding ureas#.
#Reagents and conditions: i) Boc2O, MeOH, RT, 16 hrs. ii) NaH, 4-fluorobenzyl bromide, THF, RT, 16 hrs. iii) 4-substituted fluorobenzene, NaH, DMF, reflux, 2h. iv) TFA/DCM, 2 hrs, 16 hrs. v) 3- (trifluromethyl)phenyl isocyanate, triethylamine, DMF, RT, 16 hrs
Modifications of position of the phenoxy substituent on the cyclohexyl ring of the ureas (III-1-5) were obtained by reacting the 3-(trifluromethyl)phenyl isocyanate with the properly substituted cyclohexylamines (8–10), which were either commercially available or synthesized as described in Scheme 2. The substituted cyclohexylamines were obtained by the O-alkylation of m-aminocyclohexanol using 4-substituted fluorobenzenes, as in 8–9, and the trans-4-Boc-aminocyclohexanol with alkyl halides followed by deprotection to obtain 10, the free amine. Reaction of the free amines with 3-(trifluromethyl)phenyl isocyanate produced ureas III-1-5 as described in Scheme 2 and the Experimental Section.
Based on analytical reverse-phase high performance liquid chromatography (RP-HPLC) analysis the purity of all final N-aryl,N′-cyclohexylarylureas submitted to biological characterization and reported inhere equaled or exceeded 95%. Some of the pure compounds comprise of racemic mixtures originating from the non-symmetrically disubstituted cyclohexyl moieties. Their structural identity and integrity was confirmed by HR- or LC-MS, 1H-, 13C-, and 19F-NMR.
Identification of N-phenyl,N′-cyclohexylarylureas
We screened a diversity library of ~1900 urea compounds in the dual luciferase (DLR) surrogate eIF2α phosphorylation (ternary complex) assay. This library was originally assembled and screened for inhibition of sEH by Hammock’s group at UC-Davis. We developed surrogate dual-luciferase eIF2α phosphorylation assay in order to identify compounds that activate HRI25, 32, taking advantage of the fact that activated HRI phosphorylates eIF2α thereby reducing the amount of the eIF2·GTP·Met-tRNAi ternary complex that inhibits translation of many mRNAs but paradoxically increases translation of some mRNAs that contain multiple upstream ORF (uORF) in their 5′ untranslated regions (UTRs). In our assay, firefly (F) luciferase mRNA is fused to 5′UTR of activating transcription factor 4 (ATF-4) mRNA 5′UTR that has multiple uORFs while renilla (R) luciferase mRNA is fused to a 5′UTR lacking any uORFs. Compounds that reduce the amount of the ternary complex, such as those that activate HRI, would increase F luciferase while decreasing the R luciferase expression, resulting in an increased F/R luciferase ratio. We calculated the activity scores as the F/R ratios for every compound-treated wells and normalized these for F/R ratio in vehicle-treated (DMSO) wells that was arbitrarily set at 1 (F/R = 1).
We screened compound library at 33μM concentration in 384-well plate format. The activity of all tested compounds in the primary screening assay is shown in Table S1. Interestingly, there is no correlation (r2 < 0.15) between induction of eIF2α phosphorylation and the previously reported inhibition of sEH by these compounds30 suggesting distinct structure-activity relationship. Of interest in this library were the N-phenyl-N′-(4-(4-fluorophenoxy)cyclohexyl)urea (I) and N-phenyl-N′-(4-((2,6- difluorobenzyl)oxy)cyclohexyl)urea (II, see supporting information) scaffolds; they were re-evaluated by dose-response studies in the dual luciferase surrogate eIF2α phosphorylation25 and SRB cell proliferation33 assays (Table 1, Figure 1, and Table S2). This re-testing served the dual purpose: confirming the results of the primary screen and establishing the pharmacologically required dose-response relationships. Importantly, screening the library we had few pairs of cis- and trans-isomers of the same compound for which we did not see significant difference in biological activity. The greater commercial availability of the trans-aminocyclohexanols was the predominant reason we decided to focus our studies on the trans-configuration.
Table 1.
Activity of N-phenyl-N′-(4-(4-fluorophenoxy)cyclohexyl)ureas, I, in the ternary complex and cell proliferation assay.
| Compound | R1 | CLogP | IC50 (μM) | Emax (F/R) | C3Xc (μM) |
|---|---|---|---|---|---|
| I-1o | o-F | 4.15 | 17.5±1.1 | 2.10 | N.A. |
| I-1m | m-F | 4.60 | 8.3±0.9 | 6.54 | 13.5 |
| I-1p | p-F | 4.60 | 20±2.1 | 0.98 | N.A. |
| I-2o | o-Cl | 4.61 | 9.1±1.0 | 5.37 | 18.5 |
| I-2m | m-Cl | 5.17 | 5.9±0.6 | 8.76 | 6.4 |
| I-2p | p-Cl | 5.17 | >20 | 1.65 | N.A. |
| I-3o | o-Br | 4.76 | 7.7±0.1 | 5.23 | 16.4 |
| I-3m | m-Br | 5.32 | 6.2±0.3 | 9.52 | 5.7 |
| I-3p | p-Br | 5.32 | >20 | 1.29 | N.A. |
| I-4o | o-I | 4.92 | >20 | 3.25 | 18.5 |
| I-4m | m-I | 5.58 | 5.2±0.2 | 10.32 | 5.6 |
| I-4p | p-I | 5.58 | 10.2±1.5 | 3.77 | 6.3 |
| I-5o | o-CF3 | 4.69 | >20 | 1.56 | N.A. |
| I-5ma | m-CF3 | 5.57 | 2.6±0.3 | 11.84 | 4.5 |
| I-5p | p-CF3 | 5.57 | 2.8±0.1 | 13.99 | 1.6 |
| I-6o | o-CF3O | 5.36 | >20 | 1.41 | N.A. |
| I-6p | p-CF3O | 3.81 | 2.7±0.1 | 15.32 | 0.7 |
| I-13 | H | 4.15 | 20±2.0 | 1.65 | N.A. |
| I-7o | o-Me | 4.09 | >20 | 0.98 | N.A. |
| I-7m | m-Me | 4.65 | 14.4±0.5 | 4.73 | 24.1 |
| I-7p | p-Me | 4.65 | >20 | 1.05 | N.A. |
| I-8o | o-MeO | 4.26 | >20 | 1.51 | N.A. |
| I-8m | m-MeO | 4.26 | 14±1.3 | 3.81 | 26.0 |
| I-8p | p-MeO | 4.26 | >20 | 0.83 | N.A. |
| I-9o | o-NO2 | 4.54 | 3.5±0.3 | 2.23 | N.A. |
| I-9m | m-NO2 | 4.54 | 6.7±0.7 | 7.47 | 11.4 |
| I-9p | p-NO2 | 4.54 | 2.3±0.1 | 9.00 | 6.7 |
| I-10o | o-MeS | 4.71 | >20 | 0.92 | N.A. |
| I-10m | m-MeS | 4.71 | 11.1±1.1 | 6.67 | 12.2 |
| I-10p | p-MeS | 4.71 | >20 | 1.07 | N.A. |
| I-11oa,b | o-CO2Me | 4.67 | 14.5±1.6 | 3.79 | 26.2 |
| I-11m | m-CO2Me | 4.67 | 9.2±0.8 | 6.83 | 17.1 |
| I-11p | p-CO2Me | 4.67 | 11.6±1.2 | 3.97 | 12.8 |
| I-12oa,b | o-CO2H | 4.87 | >20 | 1.20 | N.A. |
| I-12m | m-CO2H | 4.24 | >20 | 0.88 | N.A. |
| I-12p | p-CO2H | 4.24 | 11.6±1.8 | 3.49 | 21.2 |
Reported as a cis-isomer.
Compounds synthesized as described in Scheme 1, all other compounds were part of UC-Davis Library.
C3X: Calculated concentration at which F/R=3.
Figure 1.

Activity of N-phenyl-N′-(4-phenoxy)cyclohexylureas I in the surrogate eIF2α phosphorylation assay. Activity of compounds was measured by DLR assay, the F/R ratio normalized to vehicle treated cells, and expressed as a function of the compounds’ concentration. N-phenyl,N′-cyclohexylarylureas were sorted into three groups based on the nature of R1 on the N-phenyl substituent: (Panel A) halogen substituent; (Panel B) electron donating groups; and (Panel C) mostly electron withdrawing groups. The experiment was conducted in triplicate and each experiment was independently performed three times; data are shown as Mean±S.E.M.
Structural-activity relationship
In the N-phenyl-N′-(4-phenoxy)cyclohexylureas I, the nature and position of the substitution on the N-phenyl moiety have significant impact on the compound’s activity in both the surrogate eIF2α phosphorylation and cell proliferation assays (Table 1 and Figure 1). The unsubstituted N-phenyl derivative, I-13 is devoid of measurable activity in both assays. All the mono-substituted N-phenyl compounds show substituent position-dependent activity that varies with the nature of the substituent. In general, there is a very good correlation between the mechanistic surrogate eIF2α phosphorylation and the cell proliferation assays. The most potent compounds are those substituted with electron withdrawing groups p-OCF3 (I-6p, F/R@30μM = 15.32; IC50=2.7 μM), m- and p-CF3 (I-5m and I-5p, F/R@30μM = 11.84 and 13.99; IC50=2.6 and 2.8 μM, respectively) and p- and m-NO2 (I-9p and I-9m, F/R@30μM = 9.0 and 7.47; IC50=2.3 and 6.7 μM, respectively), m- and p-CO2Me (I-11m and I-11p, F/R@30μM = 6.83 and 3.97; IC50=9.7 and 11.6 μM, respectively), and p-CO2H (I-12p, F/R@30μM = 3.49). The m-OCF3 analog was generated with purity short of the required ≥ 95% and was therefore not tested. The halogen substituted ureas I-1-to-I-4 (12 analogs, R1=halogen) comprises a second sub-set of compounds active in both assays. In this sub-set, substitution in either ortho- or meta-position result in active compounds with the meta being the most active for every halide (e.g. I-1m and I-1o, m- and o-F, F/R@30μM = 6.54 and 2.10; IC50=8.3 and 17.5 μM, respectively, and I-4m and I-4o, m- and o-I, F/R@30μM = 7.63 and 3.25; IC50=5.2 and >20 μM, respectively). In the entire series presented in Table 1, the ortho position is the least favorable one. Electron donating substituents (I-7 CH3-, I-8 CH3O-, and I-10 CH3S-) in the meta-position yields the most active ureas (I-7m, I-8m, and I-10m, m-CH3, m-OCH3, and m- SCH3, F/R@30μM = 4.73, 3.81, and 6.67; IC50=14.4, 14, and 11.1 μM, respectively). On the other hand, for electron withdrawing substituents the most active ones are the para-substituted urea (p-CF3/I-5p, p- CF3O/I-6p, p-NO2/I-9p, and p-CO2H/I-12p). The trifluoromethoxy group that has been referred to as a super-34 or a pseudo-halogen35 and is best represented as a partially polarized function carrying partial positive and negative charges on the oxygen and fluorines, respectively36. Comparison of I-1p, I-5p, and I-6p show the great advantage of p-trifluoromethoxy and p-trifluoromethyl over the p-fluoro substituent in terms of potency both in the cell proliferation and surrogate eIF2α phosphorylation assay, latter expressed by the ~15 fold higher Emax. Therefore, combination of the highly polarized C-F bonds, the low polarizability of the most electronegative fluorine atom, and the high lipophilicity of the trifluoromethyl and trifluoromethoxy groups may be responsible for this interesting SAR. Evidently, I-9p that is substituted by highly polar p-NO2 group characterized by a strong negative inductive effect is less potent in the mechanistic surrogate eIF2α phosphorylation assay than the p-CF3 substituted I-5p and the p-CF3O substituted I-6p (cf. F/R@30=9.00 vs. 13.99 and 15.32, respectively). Apparently, high lipophilicity parameters (πx) of the substituent combined with a strong negative inductive effect contribute to high potency (πx = −0.02, 0.14, 0.56, 0.88, and 1.04 for X = OCH3, F, CH3, CF3, and OCF3, respectively35. There is no correlation between the biological potency and cLogP for either the cell proliferation or the ternary complex assays.
Importantly, a parallel SAR study carried out on N-phenyl-N′-(4-((2,6- difluorobenzyl)oxy)cyclohexyl)ureas II (27 analogs, Table S2), which differ from ureas in series I only by replacing the 4-fluorophenoxy with the (2,6-difluorobenzyl)oxy, demonstrated to be devoid of activity in both the surrogate eIF2α phosphorylation and cell proliferation assays. We were quite encouraged by this observation suggesting a remarkable level of scaffold-target specificity.
Activities in secondary mechanistic assays
To confirm that the N-phenyl,N′-(4-phenoxy)cyclohexylureas activate HRI and thereby induce phosphorylation of eIF2α, we selected representative compounds from Table 1, (N-phenyl,N′-(4- phenoxy)cyclohexylureas (I), and tested them in secondary mechanistic assays, namely endogenous eIF2α phosphorylation and expression of the transcription factor C/EBP homologous (CHOP) protein and mRNA. The surrogate eIF2α phosphorylation assay utilized for screening is a reporter gene assay that relates to a downstream event, namely up-regulating translation of a reporter fused to the 5′UTR of ATF-4 mRNA in response to the reduced abundance of the ternary complex. Phosphorylation of endogenous eIF2α, which inhibits recycling of the eIF2·GDP to eIF2·GTP is the direct target of HRI and upstream regulator of the ternary complex abundance while the expression of CHOP is a downstream effector of ternary complex abundance. The N-phenyl,N′-(4-phenoxy)cyclohexylureas (I) selected for these secondary mechanistic studies cover a range of potencies in the surrogate eIF2α phosphorylation and cell proliferation assays. We first determined their effect on the phosphorylation of endogenous eIF2α in CRL-2813 cells by the Western blot analysis in which expression of the total (T-eIF2α) and the phosphorylated eIF2α (P-eIF2α) was determined using specific antibodies (Figure 2). We also studied by Western blot analysis expression of CHOP protein and by real-time PCR expression of CHOP mRNA. Similarly, we studied the expression of cyclin D1 protein and mRNA, an oncogenic protein whose expression is reduced in response to eIF2α phosphorylation (Figure 3). As anticipated, all compounds caused phosphorylation of endogenous eIF2α (Figure 2) and increased the expression of CHOP protein and mRNA roughly proportional to their activity in the surrogate eIF2α phosphorylation assay (Figure 3A and B). Consistently, N-phenyl,N′-(4-phenoxy)cyclohexylureas reduced the expression of cyclin D1 protein with minimal effect on the expression of its mRNA (Figure 3A and C), most likely owing to their specific effects on translation initiation. The compounds’ activity on cyclin D1 protein expression correlated well with their activity in the surrogate eIF2α phosphorylation assay. These data show a direct correlation between the activity of the compounds in the surrogate eIF2α phosphorylation assay, their ability to induce the phosphorylation of endogenous eIF2α, expression of CHOP protein and mRNA, and inhibit expression of oncogenic proteins.
Figure 2.
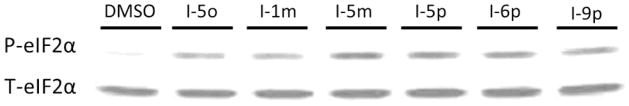
N-phenyl,N′-(4-phenoxy)cyclohexylureas cause phosphorylation of eIF2α. CRL-2813 human melanoma cells were incubated with selected N-phenyl,N′-(4-phenoxy)cyclohexylureas and phosphorylated (P-eIF2α) and total eIF2α (T-eIF2α) were analyzed by Western blot using pS51-eIF2α– specific rabbit monoclonal antibodies or total eIF2α-specific mouse monoclonal antibodies, respectively. The experiment was independently performed three times.
Figure 3. Effect of N-phenyl,N′-(4-phenoxy)cyclohexylureas on protein and mRNA expressions of CHOP and cyclin D1.

(A) Human CRL-2813 melanoma cells were incubated with 10 μM of each compound for 6 hours. Expression of CHOP and cyclin D1 proteins was determined by Western blot analysis. A representative gel from three independent experiments is shown. (B and C) CRL-2813 cells were incubated with 7.5 or 15 μM of each compound for 6 hours. Expression of CHOP and (B) cyclin D1 (C) mRNAs was determined by real time PCR analysis. Each experiment was carried out in triplicate, data shown are mean of three independent experiments, and error bars are ±S.E.M.
Dependence of activity of N-phenyl,N′-(4-phenoxy)cyclohexylureas on HRI
To determine if the molecular and cellular effects of N-phenyl,N′-(4-phenoxy)cyclohexylureas are mediated by HRI, we knock downed its expression by transfecting reporter CRL-2813-pBISA-DL(ATF-4) cells used in the surrogate eIF2α phosphorylation assay with siRNA targeting HRI or non-targeting siRNA, treated them with selected compounds and measured F/R ratio. As shown in Figure 4, siRNA targeting HRI significantly blunted activity of N-phenyl,N′-(4-phenoxy)cyclohexylureas in the surrogate eIF2α phosphorylation assay compared to non-targeting siRNA.
Figure 4. N-phenyl,N′-(4-phenoxy)cyclohexylureas (I) reduce the abundance of the ternary complex by activating HRI.
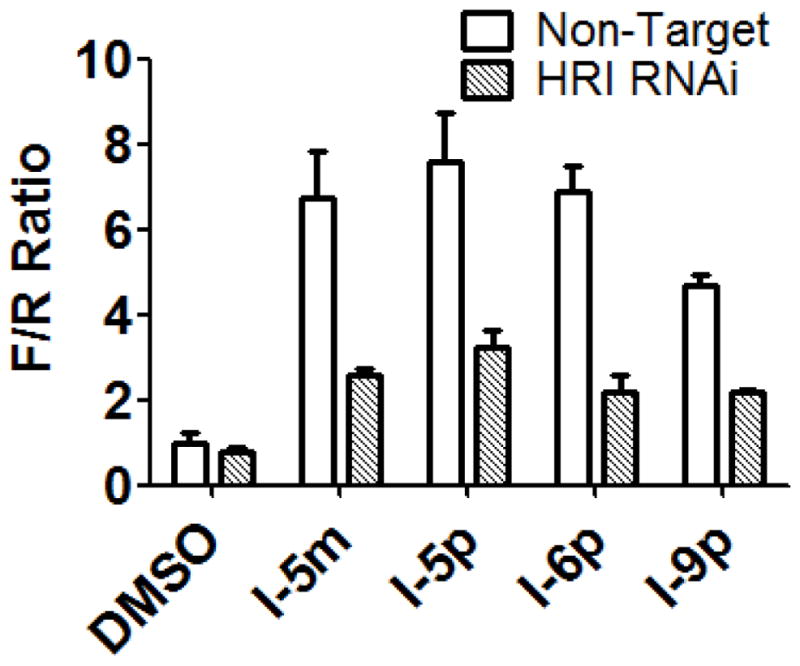
Stably transfected CRL-2813-pBISA-DL(ATF-4) cells were transiently transfected with either non-targeting siRNA, or siRNA targeting HRI for 48 hours. Cells were treated with N-phenyl,N′-phenoxycyclohexylurea compounds or DMSO for 10 hours and the normalized F/R ratio was determined by DLR assay. The experiment was conducted in triplicate and each experiment was independently performed three times; Data are shown as Mean±S.E.M.
To further determine the specificity of N-phenyl,N′-(4-phenoxy)cyclohexylureas for HRI, we utilized cell proliferation as a biological response paradigm. Cell proliferation is a very meaningful read-out for determining the specificity of a compound as it captures both on-target and off-target effects of the compound under study. If N-phenyl,N′-(4-phenoxy)cyclohexylureas specifically activate HRI and this activity is required for the inhibition of cell proliferation, then reducing the expression of HRI should blunt compounds’ effect on cell proliferation. For this purpose, we knocked down expression of HRI in CRL-2813 human melanoma (~70% knockdown efficiency) and MCF-7 human breast cancer cells (~90% knockdown efficiency). These two cell lines were chosen because the HRI knockdown efficiency is lower in CRL-2813 than in MCF-7 cells providing a sort of dose-response data that may correlate knockdown efficiency with the resistance of cells to inhibition of cell proliferation by activators of HRI. Data shown in Figure 5 indicate that inhibition of cell proliferation by the Nphenyl, N′-(4-phenoxy)cyclohexylureas is mostly dependent on HRI and that it correlates with the HRI knockdown efficiency. Taken together, these data demonstrates that the N-phenyl,N′-(4- phenoxy)cyclohexylureas activate HRI, induce eIF2α phosphorylation and reduce the amount of the ternary complex thereby inhibiting translation initiation and cell proliferation.
Figure 5. HRI mediates inhibition of cancer cell proliferation by N-phenyl,N′-(4- phenoxy)cyclohexylureas.
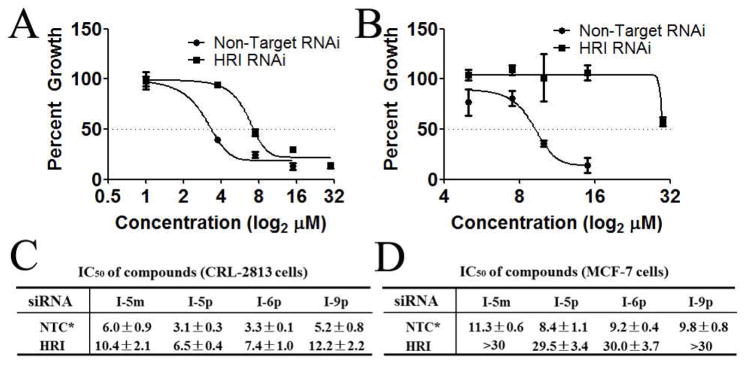
(A) CRL-2813 human melanoma cancer cells and (B) MCF-7 human breast cancer cells were transfected with HRI targeted or non-targeted siRNA, treated with the indicated concentrations of I-6p and cell proliferation was measured by SRB assay. (C and D) Calculated IC50 for all four compounds tested in CRL-2813 human melanoma cancer cells (C) and MCF-7 human breast cancer cells (D) transfected with non-targeting siRNA control or HRI targeting siRNA. The experiment was conducted in triplicate and each experiment was independently performed three times. Data are shown as Mean±S.E.M, *NTC = non-targeting siRNA control.
Preliminary Hit-to-Lead Optimization
Based on the screening and characterization of the Diversity Library described above we carried out a preliminary hit-to-lead optimization on the N-phenyl,N′-(4-phenoxy)cyclohexylurea scaffold I in which the N′-(4-phenoxy)cyclohexyl part of the molecule was modified while the N-(m-CF3-phenyl) part was maintained. This process included two steps: 1) modification of substituents on the N-phenoxy ring (I-14-20, Table 2) and 2) exploration of substitution permutation on the cyclohexyl moiety (III-1-5, Table 3).
Table 2.
Activity of the phenoxy substituted 1-(4-phenoxycyclohexyl)-3-(3-(trifluoromethyl)phenyl)ureas, I-14-20, in the surrogate eIF2α phosphorylation and cell proliferation assays.
| Compound | Structure | CLogP | IC50 (μM) | Emax (F/R) | C3Xa (μM) |
|---|---|---|---|---|---|
| I-5m |
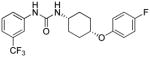
|
5.57 | 2.6±0.6 | 11.84 | 4.5 |
| I-14 |
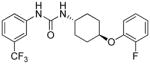
|
5.37 | 2.45±0.9 | 7.98 | 3.2 |
| I-15 |
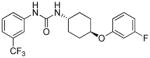
|
5.57 | 2.4±0.6 | 7.23 | 2.6 |
| I-16 |
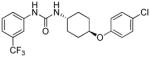
|
6.14 | 2.0±0.1 | 6.73 | 1.6 |
| I-17 |
|
6.48 | 0.69±0.1 | 15.42 | 1.5 |
| I-18 |
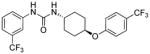
|
6.41 | 0.46±0.1 | 25.12 | 0.3 |
| I-19 |
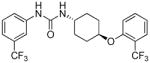
|
6.41 | 1.4±0.3 | 15.74 | 1.2 |
| I-20 |
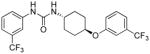
|
6.41 | 1.3±0.1 | 26.00 | 1.1 |
C3X: Calculated concentration at which F/R=3.
Table 3.
SAR study to identify the minimum pharmacophore.
| Compound | Structure | CLogP | IC50 (μM) | Emax (F/R) | C3Xa (μM) |
|---|---|---|---|---|---|
| I-18 |
|
6.41 | 0.46±0.1 | 25.12 | 0.3 |
| III-1 |
|
4.54 | 13.2±2.3 | 1.87 | N.A. |
| III-2 |
|
2.46 | >20 | 0.89 | N.A. |
| III-3 |
|
5.16 | 2.65±0.8 | 9.58 | 4.4 |
| III-4 |
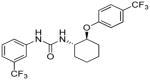
|
6.98 | 3.1±1.1 | 5.89 | 1.7 |
| III-5 |
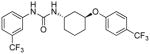
|
6.85 | 3.8±1.2 | 4.52 | 2.7 |
C3X: Calculated concentration at which F/R=3.
Similar to the SAR observed for the substituents on the N′-(4-phenoxy)cyclohexyl moiety (see Table 1) where the lipophilic electron withdrawing substituents CF3- and CF3O- and to a lesser extent halogen substituents resulted in the most potent analogs in both the surrogate eIF2α phosphorylation and cell proliferation assays, their substitution on the phenoxy ring yielded more potent ureas I-17-20 (Table 2) (cf. I-17 and I-18; p-OCF3 and p-CF3; F/R@30μM = 14.42 and 25.12; SRB IC50=0.69 and 0.46μM, respectively, with I-5m and I-16; p-F, and p-Cl; F/R@30μM = 11.84 and 6.73; SRB IC50=2.6 and 2.0μM, respectively). The analogs with highest maximum activity in the surrogate eIF2α phosphorylation assay were I-18 and I-20 which displayed ~25-fold increase in F/R relative to vehicle. I-18 also inhibited CRL-2813 human melanoma cell proliferation at sub-μM concentrations. Note that even though Emax of I-20 is marginally higher, I-18 has much higher activity across the entire concentration range tested (Figure 6).
Figure 6. Dose-response studies of the phenoxy substituted 1-(4-phenoxycyclohexyl)-3-(3- (trifluoromethyl)phenyl)ureas, I-14-20, in the surrogate eIF2α phosphorylation assay.
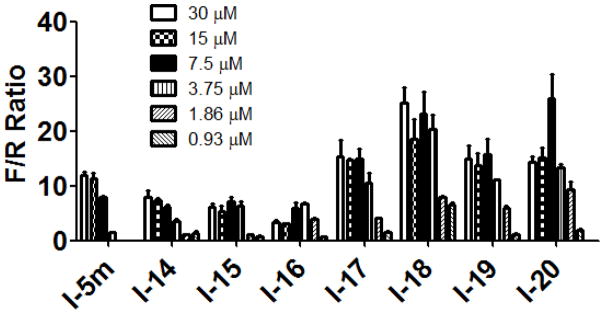
Activity of compounds was measured by determining the F/R normalized to vehicle treated cells, and expressed as a function of the concentration. The experiment was conducted in triplicate and each experiment was independently performed three times; Data are shown as Mean±S.E.M.
Evidently, the substituted phenoxy moiety on the N-cyclohexyl is an integral part of the pharmacophore (Table 2). As presented in urea I-18, it is needed to generate the above high potency (Table 2). Elimination of the 4-trifluoromethylphenoxy (as in III-1) or 4-trifluoromethylphenyl (as in III-2) was sufficient to wipe out activity of these compounds in both the surrogate eF2α phosphorylation and cell proliferation assays (Table 3 and Figure 7). Moreover, insertion of an extra methylene that transforms the phenoxy into a benzyloxy moiety markedly reduced the activity (cf III-3 and I-5m, F/R@30μM = 9.58 and 11.84, respectively). Of interest was the effect of moving the 4- trifluoromethylphenoxy from position 4 to positions 2 or 3 on the cyclohexyl ring (cf. I-18 with III-4 and III-5, respectively) which, lead to a drastic reduction in the activity of compounds in both the surrogate eIF2α phosphorylation and cell proliferation assays (IC50 = 0.46 vs. 3.1 and 3.8 μM, and F/R@30μM = 25.12 vs. 5.89 and 4.52, respectively, Tables 2 and 3 and Figure 7). Taken together, data obtained with this series of ureas suggest that the phenoxy substitution is essential and its position on the cyclohexyl ring plays an important role in the hit-to-lead optimization process.
Figure 7. Activity of compounds in which the phenoxy moiety is either missing or relocated to another position on the cyclohexyl in surrogate eIF2α phosphorylation assays.

Activity of compounds was measured by DLR assay and F/R ratio was normalized to vehicle treated cells, and expressed as a function of the concentration. The experiment was conducted in triplicate and each experiment was independently performed three times; Data are shown as Mean±S.E.M.
Specificity of novel N-phenyl,N′-(4-phenoxy)cyclohexylurea compounds
The improved activity of newly synthesized compounds in the surrogate eIF2α phosphorylation and cell proliferations assays could either be due to better cell-penetration or higher affinity for their molecular target. It is also possible that the increased activity in the cell proliferation assay may be due, at least in part, to the increased off-target effects. We were also concerned that some compounds did not appear to have much activity in the surrogate eIF2α phosphorylation assay at their IC50 concentrations in the cell proliferation assay.
In general, if the affinity of compounds for their molecular target is increased this should result in higher specificity of the compounds in the cell proliferation assay. To demonstrate that this was indeed the case, we determined the dependence of the anti-proliferative effects of five selected compounds in these series on HRI. These five compounds, two with average and three with high potency, were tested in cell proliferation assay using CRL-2813 cells transfected with non-targeted siRNA or HRI-targeted siRNA as described in Figure 5. Evidently, IC50 values in these studies cannot be directly compared to the corresponding values in Table 2 as the assay duration is reduced from five to three days, optimal time for suppression of gene expression by siRNA. As shown in Figure 8, two average potency compounds, I-14 and I-15, displayed a significant dependence on HRI for inhibition of cell proliferation (~7 fold higher IC50 values in cells transfected with HRI-targeting siRNA vs. in those transfected with non-targeting siRNA). This is a significant improvement in specificity toward the molecular target over initial hits shown in Figure 5 that displayed 1.7–2.2 fold higher IC50 values in CRL-2813 cells transfected with HRI-targeting siRNA vs. those transfected with non-targeting siRNA. Compounds with higher potency in the surrogate eIF2α phosphorylation and SRB assays also showed a very good target dependencies/specificities (~6, 7.5, and 5 fold higher IC50 values in cells transfected with HRI-targeting siRNA vs. in those transfected with non-targeting siRNA for I-17, I-18, and I-20, respectively). These data indicate that our SAR studies resulted in the generation of more specific compounds. These data suggest that compounds’ Emax in the surrogate eIF2α phosphorylation assay alone may not be sufficient to predict their specificity and that additional parameters such as C3X may also need to be considered.
Figure 8. Newly synthesized 1-(4-phenoxycyclohexyl)-3-(3-(trifluoromethyl)phenyl)ureas display higher HRI dependence for inhibition of cell proliferation.

CRL-2813 human melanoma cancer cells were transfected with HRI targeting or non-targeting siRNA, treated with the indicated concentrations of (A) I-14, (B) I-15, (C) I-17 and (D) I-18 and cell proliferation was measured by SRB assay. Calculated IC50 values for library compound I-5m and I-14, I-15, I-17, I-18, and I-20 in CRL-2813 human melanoma cancer cells transfected with non-targeting siRNA or HRI-targeting siRNA are shown in (E). The experiment was conducted in triplicate and each experiment was independently performed three times. Data are shown as Mean±S.E.M. *NTC = non-targeting control.
To determine if the discrepancy in the active concentrations of some 1-(4-phenoxycyclohexyl)-3-(3- (trifluoromethyl)phenyl)urea in the surrogate eIF2α phosphorylation and cell proliferation assays is due to differences in the length of these two assays we determined the activity of four compounds which display such discrepancy in the surrogate eIF2α phosphorylation assay after 8, 16, or 32 hour incubation. As shown in Supplemental Figure 1, all four compounds displayed much higher activity after 32 hours of incubation compared to 8 or 16 hours. These data indicate that much of the discrepancy in the activity of some compounds in the two primary assays used in these studies are due to the differences in the duration of the assays and support our contention that there is a very good correlation between the activity of compounds in the surrogate eIF2α phosphorylation and cell proliferation assays. One test of suitability of a compound for anti-cancer therapy is the selectivity for cancer cells compared to normal cells. We choose one moderately active, I-6p, and three highly active compounds, I-17, I-18, I-20, to determine if these series of N-phenyl,N′-cyclohexylphenoxy ureas can selectively inhibit proliferation of cancer cells. Dose-response studies in non-transformed NIH-3T3 fibroblast and CRL- 2813 human melanoma cancer cell lines show that these compounds inhibit proliferation of cancer cells with 2.5- to 7-fold higher potency compared to that of non-transformed cells (Table 4). Among these compound I-18 displayed the highest discrimination between cancer and non-cancerous cells. Taken together, data in Figure 8, Table 4 and Supplemental Figure 1 demonstrate that some of the more potent N-phenyl,N′-cyclohexylphenoxy ureas reported inhere are highly potent and specific inducers of HRI activity.
Table 4.
Selective inhibition of cancer cell proliferation by N-aryl,N′-cyclohexylarylureas.
| IC50 of compounds (μM) | ||||
|---|---|---|---|---|
| I-6p | I-17 | I-18 | I-20 | |
| NIH/3T3 | 13.6±0.8 | 3.4±0.2 | 3.4±0.1 | 3.3±0.2 |
| CRL-2813 | 2.7±0.1 | 0.69±0.1 | 0.46±0.1 | 1.30±0.1 |
Compounds I-6p, I-17, I-18 and I-20 were tested in NIH/3T3 fibroblast and CRL-2813 melanoma cancer cells in cell proliferation assay. The experiment was conducted in triplicate and each experiment was independently performed three times. Data are shown as Mean IC50±S.E.M.
Conclusions
Structure-activity relationship analysis of the focused library of N-phenyl,N′-phenoxycyclohexylureas consistently shows that substitution of the N-phenyl and cyclohexylphenoxy moieties with lipophilic and strong electron withdrawing groups yield highly potent ureas in both the mechanistic surrogate eIF2α phosphorylation and cell proliferation assays. While the para-positon is strongly preferred in the N-phenyl moiety, there seems to be only a slight preference for it the phenoxy moiety. Moreover, substitution of position 4 on the N-cyclohexyl by the phenoxy moiety emerges as the preferred arrangement relative to positions 2- and 3. Compounds I-17 and I-18 are the prototypes that may form the basis of advanced lead optimization efforts.
At the mechanistic level, we demonstrate that N-phenyl-N′-(4-phenoxy)cyclohexylureas induce phosphorylation of eIF2α and expression of its downstream effector, CHOP protein and mRNA. These N,N′-disubstituted-ureas also inhibit expression of oncogenic proteins and cell proliferation. Most importantly, activity of these compounds in mechanistic surrogate eIF2α phosphorylation assay as well as non-mechanistic cell proliferation assay is dependent on HRI. Consistently, there is a very good correlation between the activity of these agents in the mechanistic assays and cell proliferation assay. Taken together, these data indicate that N-phenyl,N′-(4-phenoxy)cyclohexylureas are mechanism specific agents suitable for further lead optimization to generate promising molecular probes for studying the role of HRI and eIF2α phosphorylation in normal- and patho-biology and for pre-clinical leads suitable for development as drug candidates for the treatment of various human ailments such as cancer and α– and β-thalassemia,
Experimental Section
Chemistry
All reagents and solvents were purchased from commercial sources and used without further purification. All the reaction solvents were anhydrous and the reactions were maintained under inert atmosphere. Thin layer chromatography (TLC) were run on pre-coated EMD silica gel 60 F254 plates and observed under UV light at 254 nm and with basic potassium permanganate dip. All the reactions were monitored by LC-MS analysis on reverse phase (column Waters Symmetry C18, 2.1x 100 mm, 3.5 μm particle size) using a Waters Alliance 2695 /Micromass ZQ or Agilent 1200 series system with UV detector (214 nm and 254 nm) and an Agilent 6130 quadrupole mass detector, with the binary system water/acetonitrile containing 0.1 % of formic acid as eluent. The purity of all final compounds was ≥ 95% as determined by analytical HPLC on reverse phase (column XBridge BEH130 C18, 4.6 × 100 mm, 5 μm particle size) using a Waters Alliance 2695 with the binary system water/acetonitrile containing 0.1 % of trifluoracetic acid (TFA) as eluent. Column chromatography was performed with silica gel (230–400 mesh, grade 60, Fisher scientific, USA). Purifications by flash chromatography were performed on Biotage SP1 using silica gel pre-packed columns, (200–400 mesh), and were monitored by UV at 254 and 280 nm. Melting points were determined using a Mel-Temp Electrothermal apparatus and were uncorrected. Proton, carbon, and fluorine NMR analyses were performed on Topspin 300 MHz, Varian-400 or Varian-500 MHz spectrometers using DMSO-d6 or CDCl3 as solvent. Chemical shifts (δ) are reported in ppm relative to TMS as internal standard. The HR-MS analyses were performed at the FAS Center for Systems Biology, Harvard University. Compound 1b was synthesized as described in literature34.
A. General Procedure for the synthesis of N-substituted cyclohexylamines, 1–9
To a stirred solution of aminocyclohexanol, n= 2, 3 or 4, (230 mg, 2 mmol) in DMF (10 mL) at 0°C, was added 60% NaH in mineral oil (240 mg, 6 mmol). The reaction mixture was stirred at room temperature for 1 hour and then 1-fluoro-n-substituted benzene (2.4 mmol) was added. It was heated to 60°C for 2 hours then stirred for 16 hours at room temperature. The reaction mixture was diluted with ethyl acetate and washed with 10 mL of water (3 times) and brine. The organic layer was dried using sodium sulfate, filtered, and concentrated under vacuum. Then the product was purified using reversed phase column chromatography in gradient increase of methanol in 0.1% Formic acid solution31.
trans-4-(3-Fluorophenoxy)cyclohexylamine, 1
Using General Procedure A employing trans-4- aminocyclohexanol and 1,3-difluorobenzene to afford 1 as a white solid, 94% (394 mg) yield, mp. 176°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =9.966 min, purity of 99.2%; 1H NMR (399 MHz, DMSO-d6) δ 8.44 (s, 1H), 7.25 (q, J = 7.9 Hz, 1H), 6.75 (dddd, J = 31.0, 17.0, 8.8, 2.5 Hz, 2H), 4.63 (bs, 2H), 4.28 (tt, J = 9.3, 4.3 Hz, 1H), 3.07 – 2.78 (m, 1H), 1.99 (dd, J = 42.9, 11.4 Hz, 4H), 1.42 (dtd, J = 22.6, 12.9, 6.1 Hz, 4H).13C NMR (100 MHz, DMSO) δ 166.63, 159.40, 131.38, 131.28, 112.60, 112.58, 107.88, 107.67, 103.79, 103.55, 75.00, 48.85, 29.82, 29.44.19F NMR (376 MHz, DMSO) δ −112.16. LCMS(ESI) for C12H16FNO [M+H]+: m/z calcd; 210.12, found; 209.90.
trans-4-(2-Fluorophenoxy)cyclohexylamine, 2
Using General Procedure A employing trans-4- aminocyclohexanol and 1,2-difluorobenzene to afford 2 as a white solid, 90% (376 mg) yield, mp. 165°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =9.543 min, purity of 99.7%; 1H NMR (399 MHz, DMSO-d6) δ 8.44 (s, 1H), 7.25 – 7.11 (m, 1H), 7.07 (t, J = 7.8 Hz, 1H), 7.04 – 6.83 (m, 1H), 4.99 (s, 2H), 4.35 – 4.03 (m, 1H), 3.14 – 2.71 (m, 1H), 2.22 – 1.71 (m, 4H), 1.43 (h, J = 11.5, 10.3 Hz, 4H).13C NMR (100 MHz, DMSO) δ 166.73, 154.61, 125.43, 125.39, 122.24, 118.31, 116.99, 116.81, 76.59, 48.76, 29.93, 29.34.19F NMR (376 MHz, DMSO) δ −134.32. LCMS(ESI) for C12H16FNO [M+H]+: m/z calcd; 210.12, found; 209.84.
trans-4-(4-Chlorophenoxy)cyclohexylamine, 3
Using General Procedure A employing trans-4- aminocyclohexanol and 1-chloro-4-fluorobenzene to afford 3 as a white solid, 85% (383 mg) yield, mp. 198°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =19.302 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.40 (s, 1H), 7.39 – 7.15 (m, 2H), 7.12 – 6.80 (m, 2H), 4.22 (dd, J = 9.8, 4.9 Hz, 1H),3.63 (bs, 2H), 3.09 – 2.80 (m, 1H), 2.13 – 1.92 (m, 4H), 1.51 – 1.36 (m, 4H).13C NMR (100 MHz, DMSO) δ 156.78, 129.94, 124.67, 118.17, 75.08, 48.95, 29.84, 29.68. LCMS(ESI) for C12H16ClNO [M+H]+: m/z calcd; 226.09, found; 225.99.
trans-4-(4-(Trifluoromethyl)phenoxy)cyclohexylamine, 4
Using General Procedure A employing trans-4-aminocyclohexanol and 1-fluoro-4-(trifluoromethyl)benzene to afford 4 as a white solid, 95% (493 mg) yield, mp. 158°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =11.864 min, purity of 99.8%; 1H NMR (399 MHz, DMSO-d6) δ 8.44 (s, 1H), 7.58 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.5 Hz, 2H), 4.80 (s, 2H), 4.43 – 4.30 (m, 1H), 2.95 (dq, J = 11.1, 5.6, 5.1 Hz, 1H), 2.00 (dt, J = 47.0, 13.6 Hz, 4H), 1.43 (p, J = 16.1, 14.4 Hz, 4H).13C NMR (100 MHz, DMSO) δ 166.70, 160.88, 127.58, 127.57, 116.56, 74.95, 48.77, 29.70, 29.35.19F NMR (376 MHz, DMSO) δ −60.28. LCMS(ESI) for C13H16F3NO [M+H]+: m/z calcd; 260.12, found; 260.01.
trans-4-(4-(Trifluoromethoxy)phenoxy)cyclohexylamine, 5
Using General Procedure A employing trans-4-aminocyclohexanol and 1-fluoro-4-(trifluoromethoxy)benzene to afford 5 as a white solid, 93% (512 mg) yield, mp. 147°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =12.283 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.44 (s, 1H), 7.21 (d, J = 8.6 Hz, 2H), 7.01 (d, J = 9.1 Hz, 2H), 5.58 (s, 2H), 4.25 (tt, J = 8.7, 4.2 Hz, 1H), 2.97 (dq, J = 12.5, 5.8 Hz, 1H), 2.34 – 1.77 (m, 4H), 1.43 (dt, J = 22.4, 12.3 Hz, 4H).13C NMR (100 MHz, DMSO) δ 166.73, 156.76, 142.35, 123.09, 117.55, 75.14, 48.75, 29.76, 29.00.19F NMR (376 MHz, DMSO) δ −57.76. LCMS(ESI) for C13H16F3NO2 [M+H]+: m/z calcd; 276.11, found; 276.16.
trans-4-(2-(Trifluoromethyl)phenoxy)cyclohexylamine, 6
Using General Procedure A employing trans-4-aminocyclohexanol and 1-fluoro-2-(trifluoromethyl)benzene to afford 6 as a white solid, 90% (467 mg) yield, mp. 140°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =11.595 min, purity of 99%; 1H NMR (399 MHz, DMSO-d6) δ 8.45 (s, 1H), 7.53 (d, J = 7.7 Hz, 2H), 7.31 (d, J = 8.6 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.56 (bs, 2H), 4.45 (ddt, J = 10.1, 7.9, 4.0 Hz, 1H), 3.04 (ddt, J = 10.5, 7.6, 3.8 Hz, 1H), 2.14 – 1.82 (m, 4H), 1.47 (dddd, J = 25.2, 15.9, 12.8, 6.5 Hz, 4H).13C NMR (100 MHz, DMSO) δ 166.90, 155.92, 134.62, 128.50, 127.42, 127.36, 125.79, 123.08, 120.82, 118.79, 118.49, 115.67, 75.43, 48.46, 29.38, 28.43.19F NMR (376 MHz, DMSO) δ −61.23. LCMS(ESI) for C13H16F3NO [M+H]+: m/z calcd; 260.12, found; 260.08.
trans-4-(3-(Trifluoromethyl)phenoxy)cyclohexylamine, 7
Using General Procedure A employing trans-4-aminocyclohexanol and 1-fluoro-3-(trifluoromethyl)benzene to afford 7 as a white solid, 92% (477 mg) yield, mp. 133°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =11.761 min, purity of 99%; 1H NMR (399 MHz, DMSO-d6) δ 8.44 (s, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.33 – 7.05 (m, 2H), 6.00 (s, 2H), 4.39 (td, J = 9.9, 5.0 Hz, 1H), 3.15 – 2.85 (m, 1H), 2.29 – 1.77 (m, 4H), 1.67 – 1.24 (m, 4H).13C NMR (100 MHz, DMSO) δ 166.72, 158.22, 131.38, 120.29, 117.66, 113.04, 74.94, 48.71, 29.67, 28.81.19F NMR (376 MHz, DMSO) δ −61.62. LCMS(ESI) for C13H16F3NO [M+H]+: m/z calcd; 260.12, found; 260.08.
trans-2-(4-(Trifluoromethyl)phenoxy)cyclohexylamine, 8
Using General Procedure A employing 2- aminocyclohexanol and 1-fluoro-4-(trifluoromethyl)benzene to afford 8 as a white solid, 47% (246 mg) yield, mp. 131°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =11.895 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.41 (s, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 6.09 (bs, 2H), 4.92–4.75 (m, 1H), 3.29 (d, J = 8.0 Hz, 1H), 2.03 – 1.89 (m, 1H), 1.88 – 1.59 (m, 3H), 1.52 (q, J = 7.0, 6.6 Hz, 1H), 1.44 – 1.24 (m, 3H). 13C NMR (100 MHz, DMSO) δ 166.57, 160.66, 127.54, 127.49, 117.33, 117.01, 74.25, 51.13, 27.00, 26.84, 23.26, 19.82.19F NMR (376 MHz, DMSO) δ −60.45. LCMS(ESI) for C13H16F3NO [M+H]+: m/z calcd; 260.12, found; 260.01.
trans-3-(4-(Trifluoromethyl)phenoxy)cyclohexylamine, 9
Using General Procedure A employing 3- aminocyclohexanol and 1-fluoro-4-(trifluoromethyl)benzene to afford 9 as a white solid, 38% (197 mg) yield, mp. 117°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =11.877 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.59 (d, J = 8.5 Hz, 2H), 7.11 (d, J = 8.5 Hz, 2H), 6.53 (s, 2H), 5.08 – 4.73 (m, 1H), 3.26 (td, J = 9.5, 8.1, 4.5 Hz, 1H), 2.35 – 1.42 (m, 8H).13C NMR (100 MHz, DMSO) δ 166.74, 160.45, 127.59, 127.45, 122.02, 121.71, 116.74, 72.04, 45.90, 34.21, 30.15, 28.59, 19.07.19F NMR (376 MHz, DMSO) δ −60.48. LCMS(ESI) for C13H16F3NO [M+H]+: m/z calcd; 260.12, found; 260.08.
trans-4-((4-Fluorobenzyl)oxy)cyclohexylamine, 10
2 mmol of trans-4-aminocyclohexanol was dissolved in 5 mL of dry Methanol, then 1.1 equiv of Boc2O were dissolved in 5 mL methanol and added drop wise at room temperature. The reaction was stirred until completion by TLC, then solvent was evapoarated to dryness. The produced crude was redissolved in 5 mL of dry THF, and cooled to 0°C. Then 2.2 mmol of 60% NaH in mineral oil were added. The reaction mixture was stirred at room temperature for 30 min and then 1.5 equiv of 4-fluorobenzylbromide was added. The reaction was stirred for 16 hours at room temperature. The reaction mixture was diluted with ethyl acetate and washed with 10 mL of water (3 times) and brine. The organic layer was dried using sodium sulfate, filtered, and concentrated under vacuum. Then the product was dissolved in 2 mL of DCM/TFA, 1:1, solvent mixture and stirred for 1 hr. The solvent was evaporated to dryness and the obtained solid was purified using reversed phase column chromatography in gradient increase of methanol in 0.1% Formic acid solution. White solid, 96% (429 mg) yield, mp. 166°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =9.528 min, purity of 97.8%; 1H NMR (399 MHz, DMSO-d6) δ 7.33 (dd, J = 8.1, 5.5 Hz, 2H), 7.13 (t, J = 8.6 Hz, 2H), 6.90 (bs, 2H), 4.46 (s, 2H), 4.28- 4.21 (m, 1H), 3.31–3.28 (m, 1H), 2.98 (dt, J = 10.9, 5.9 Hz, 1H), 1.97 (dd, J = 43.0, 12.0 Hz, 4H), 1.37 – 1.15 (m, 4H). 13C NMR (100 MHz, DMSO) δ 163.31, 135.33, 130.06, 129.98, 115.71, 115.50, 76.05, 69.07, 49.23, 30.07, 29.85, 28.84. 19F NMR (376 MHz, DMSO) δ −115.88. LCMS(ESI) for C13H18FNO [M+H]+: m/z calcd; 224.14, found; 223.81.
cis-Methyl 2-(3-(4-(4-fluorophenoxy)cyclohexyl)ureido)benzoate, I-11o
To a stirring solution of cis- 4-(4-fluorophenoxy)cyclohexylamine, 1b37, (20 mg, 0.09 mmol) in dichloromethane (2 mL) under nitrogen was added methyl 2-isocyanatobenzoate (15 mg, 0.08 mmol). The mixture was maintained at room temperature for 16 h and the reaction monitored for completion by TLC. Crude mixture was washed with brine and extracted using dichloromethane, dried under magnesium sulfate and column chromatographed using hexane and ethyl acetate gradient to obtain desired product as white amorphous solid (22 mg, 59% yield). 1H NMR (500 MHz, CDCl3) δ 10.33 (s, 1H), 8.53 (d, J = 8.5 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.49 (dd, J = 8.5 Hz, 7.5 Hz, 1H), 6.99 – 6.94 (m, 3H), 6.86 – 6.84 (m, 2H), 4.72 (d, J = 8.0 Hz, 1H), 4.40 (s, 1H), 3.90 (s, 3H), 3.79 – 3.75 (m, 1H), 2.04 – 2.00 (m, 2H), 1.86 – 1.84 (m, 2H), 1.74 – 1.66 (m, 4H); 13C NMR δ 169.46, 158.41, 156.51, 154.35, 153.62, 143.59, 134.84, 130.90, 120.75, 119.54, 117.60, 117.54, 116.16, 115.98, 113.73, 72.16, 52.35, 48.35, 28.56, 28.36, 28.04; 19F NMR (376 MHz, DMSO) δ −40.61. LCMS for C21H23FN2O4 [M+H]+: m/z calcd; 387.41, found: 387.20.
cis-2-(3-(4-(4-Fluorophenoxy)cyclohexyl)ureido)benzoic acid, I-12o
To a solution of urea I-11o (14 mg, 0.04 mmol) in 1 mL of acetonitrile was added lithium hydroxide (3 mg, 0.11 mmol) in water (0.3 mL) and reaction mixture was stirred at room temperature for 24 hours. Solvent was evaporated and the crude mixture was dissolved in methanol and filtered through a silica pipette to obtain pure product as white solid (10 mg, 74%). 1H NMR (500 MHz, CDCl3) δ 10.69 (s, 1H), 8.13 (d, J = 8.0 Hz, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.21 (t, J = 7.5 Hz, 1H), 7.14 (d, J = 8.5 Hz, 1H), 6.98 (d, J = 7.0 Hz, 4H), 6.04 (s, 1H), 5.56 (s, 1H), 5.03 (t, J = 12.5 Hz, 1H), 4.51 (s, 1H), 3.05 – 2.98 (m, 2H), 2.22 – 2.19 (m, 2H), 1.71 – 1.66 (m, 2H), 1.56 – 1.54 (m, 2H); 13C NMR δ 163.53, 158.44, 153.76, 152.56, 139.08, 135.03, 128.68, 123.38, 118.04, 117.98, 116.13, 115.95, 115.17, 114.99, 71.41, 53.18, 29.76, 23.09; 19F NMR (376 MHz, DMSO) δ −40.64. LCMS for C20H21FN2O4 [M+H]+: m/z calcd; 373.39, found; 373.15.
B. General Procedure for synthesis of the N-aryl,N′-cyclohexylureas, I-14-20 and III-1-5
To a stirred solution of 3-(trifluoromethyl)phenylisocyanate (1 mmol) in dry DMF (2 mL) and triethylamine (1 mmol), the desired amine (1 mmol) was added and the reaction was left at RT for 16 hrs. The reaction mixture was treated with DDW(5 mL) and the formed precipitate was collected, washed with water, and re-crystallized twice from methanol/water. In some cases further purification by reversed phase column chromatography employing a linear gradient of (0.1% formic acid/water) in methanol, (0%–100%) was needed to achieve purity >95%.
trans-1-(4-(2-Fluorophenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, I-14
Using General Procedure B employing trans-4-(2-fluorophenoxy)cyclohexylamine 2, to afford I-14 as a white solid, 56% (220 mg) yield, mp. 171°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR = 18.365 min, purity of 99%; 1H NMR (300 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.94 (s, 1H), 7.51-7.33 (m, 2H), 7.24 (dq, J = 20.4, 7.1, 6.4 Hz, 2H), 6.73 ((dt, J = 16.5, 11.0 Hz, 2H m, 3H), 6.24 (d, J = 5.5 Hz, 1H), 4.34 (q, J = 9.0, 8.4 Hz, 1H), 3.56-3.45 (m, 1H), 2.03-1.87(m, 4H), 1.49-1.31 (m, 4H). 13C NMR (100 MHz, DMSO) δ 155.08, 141.97, 130.38, 125.38, 122.11, 121.74, 118.26, 117.00, 116.82, 114.18, 76.74, 47.91, 30.43. 19F NMR (376 MHz, DMSO) δ −61.80, −134.26. HRMS(ESI) for C20H20F4N2O2 [M+H]+: m/z calcd; 397.15337, found; 397.15317.
trans-1-(4-(3-Fluorophenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, I-15
Using General Procedure B employing trans-4-(3-fluorophenoxy)cyclohexylamine 1, to afford I-15 as a white solid, 66% (260 mg) yield, mp. 193°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =18.857 min, purity of 100%; 1H NMR (300 MHz, DMSO-d6) δ 8.64 (s, 1H), 7.94 (s, 1H), 7.58 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 5.5 Hz, 2H), 7.18 (d, J = 5.9 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.24 (d, J = 7.5 Hz, 1H), 4.57- 4.33 (m, 1H), 3.55-3.46 (m, 1H), 2.13-1.75 (m, 4H), 1.41 (dp, J= 23.6, 12.0 Hz, 4H). 13C NMR (100 MHz, DMSO) δ 155.06, 141.96, 131.38, 131.26, 130.39, 121.74, 117.83, 114.14, 112.59, 107.58, 103.49, 75.16, 47.96, 30.54, 30.29. 19F NMR (376 MHz, DMSO) δ −61.78, −112.16. HRMS(ESI) for C20H20F4N2O2 [M+H]+: m/z calcd; 397.15337, found; 397.15429.
trans-1-(4-(4-Chlorophenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, I-16
Using General Procedure B employing trans-4-(4-chlorophenoxy)cyclohexylamine 3, to afford I-16 as a white solid, 50% (206 mg) yield, mp. 244°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =19.617 min, purity of 100%; 1H NMR (300 MHz, DMSO-d6): δ 8.65 (s, 1H), 7.95 (s, 1H), 7.42 (d, J = 7.9 Hz, 2H), 7.27 (dd, J = 9.0, 3.3 Hz, 2H), 7.19 (d, J = 6.8 Hz, 1H), 6.95 (dd, J = 8.7, 3.5 Hz, 2H), 6.25 (d, J = 4.0 Hz, 1H), 4.32 – 4.27 (m, 1H), 3.55 – 3.48 (m, 1H), 2.50 – 1.90 (m, 4H), 1.48-1.29 (m, 4H). 13C NMR (100 MHz, DMSO) δ 156.84, 155.06, 141.96, 130.40, 129.92, 124.70, 121.73, 118.08, 117.85, 114.14, 75.13, 47.96, 30.57, 30.30. 19F NMR (376 MHz, DMSO) δ −61.79. HRMS(ESI) for C20H20ClF3N2O2 [M+H]+: m/z calcd; 413.12382, found; 413.12507.
trans-1-(4-(4-(trifluoromethoxy)phenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, I-17
Using General Procedure B employing trans-4-(4-(trifluoromethoxy)phenoxy)cyclohexylamine 5, to afford I-17 as a white solid, 54% (251 mg) yield, mp. 190°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR= 20.231 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.66 (s, 1H), 7.96 (s, 1H), 7.46 – 7.41 (m, 2H), 7.39-7.04 (m, 3H), 7.01 (d, J = 8.5 Hz, 2H), 6.25 (d, J = 7.6 Hz, 1H), 4.35-4.30 (m, 1H), 3.54-3.49 (dd, J = 13.8, 7.3 Hz, 1H), 2.04 – 1.90 (m, 4H), 1.47-1.33 (m, 4H). 13C NMR (100 MHz, DMSO) δ 156.88, 155.07, 141.96, 130.36, 123.14, 121.71, 117.47, 114.14, 75.32, 47.96, 30.56, 30.30. 19F NMR (376 MHz, DMSO) δ −57.78, −61.87. LCMS(ESI) for C21H20F6N2O3 [M+H]+: m/z calcd; 463.13, found; 463.11.
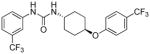
trans-1-(4-(4-(Trifluoromethyl)phenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, I-18
Using General Procedure B employing trans-4-(4-(trifluoromethyl)phenoxy)cyclohexylamine 4, to afford I-18 as a white solid, 43% (194 mg) yield, mp. 176°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =19.617 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.64 (s, 1H), 7.94 (s, 1H), 7.58 (d, J = 8.1 Hz, 2H), 7.41 (d, 2H), 7.25-7.15 (m, 1H), 7.09 (t, J = 9.3 Hz, 2H), 6.26 (d, J = 7.5 Hz, 1H), 4.58-4.31 (m, 1H), 3.72 – 3.39 (m, 1H), 2.25 – 1.73 (m, 4H), 1.53-1.29 (m, 4H). 13C NMR (100 MHz, DMSO) δ 160.97, 155.06, 141.94, 138.79, 130.44, 127.64, 121.76, 116.00, 114.13, 75.08, 47.91, 30.53, 30.22. 19F NMR (376 MHz, DMSO) δ −60.21, −61.78. HRMS(ESI) for C21H20F6N2O2 [M+H]+: m/z calcd; 447.15017, found; 447.15126.
trans-1-(4-(2-(Trifluoromethyl)phenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, I-19
Using General Procedure B employing trans-4-(2-(trifluoromethyl)phenoxy)cyclohexylamine 6, to afford I-19 as a white solid, 69 % (308 mg) yield, mp. 121°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =19.868 min, purity of 99%;1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.96 (s, 1H), 7.59 (d, J = 7.7 Hz, 2H), 7.45 – 7.41 (m, 2H), 7.31 (d, J = 8.6 Hz, 1H), 7.20 (d, J = 7.7 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 6.36 (d, J = 7.8 Hz, 1H), 4.56 (td, J = 9.4, 4.8 Hz, 1H), 3.56 (dtt, J = 15.2, 9.9, 4.9 Hz, 1H), 2.05-1.87 (m, 4H), 1.54-1.32 (m, 4H). 13C NMR (100 MHz, DMSO) δ 154.19,153.28, 147.42, 146.89, 140.15, 132.83, 128.55, 128.14, 125.66, 119.89, 118.88, 116.01, 113.71, 112.30, 73.64, 45.71, 28.09, 27.88. 19F NMR (376 MHz, DMSO) δ −62.10, −62.71. HRMS(ESI) for C21H20F6N2O2 [M+H]+: m/z calcd; 447.15017, found; 447.15147.
trans-1-(4-(3-(Trifluoromethyl)phenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, I-20
Using General Procedure B employing trans-4-(3-(trifluoromethyl)phenoxy)cyclohexylamine 7, to afford I-20 as a white solid, 59 % (264 mg) yield, mp. 160°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR = 20.083 min, purity of 99%; 1H NMR (300 MHz, DMSO-d6) δ 8.67 (s, 1H), 7.94 (s, 1H), 7.64 – 7.31 (m, 3H), 7.35 – 7.07 (m, 4H), 6.26 (d, J = 7.5 Hz, 1H), 4.45 (tt, J = 9.5, 4.0 Hz, 1H), 3.72 – 3.38 (m, 1H), 2.01 – 1.76 (m, 4H), 1.63 – 1.21 (m, 4H). 13C NMR (100 MHz, DMSO) δ 155.10, 142.00, 131.37, 130.40, 121.75, 120.37, 117.73, 114.23, 112.97, 75.24, 47.93, 30.48, 30.24. 19F NMR (376 MHz, DMSO) δ −61.50, −61.77. HRMS(ESI) for C21H20F6N2O2 [M+H]+: m/z calcd; 447.15017, found; 447.15145.
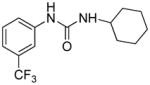
1-Cyclohexyl-3-(3-(trifluoromethyl)phenyl)urea, III-1
Using General Procedure B employing, cyclohexylamine to afford III-1 as a white solid, 63% (180 mg) yield, mp. 180°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR = 16.943 min, purity of 98%; 1H NMR (399 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.95 (s, 1H), 7.42 (t, J = 7.4 Hz, 2H), 7.16 (d, J = 7.4 Hz, 1H), 6.14 (d, J = 8.3 Hz, 1H), 3.47 (m, 1H), 1.78 (d, J = 12.0 Hz, 2H), 1.62 (d, J = 13.2 Hz, 3H), 1.50 (d, J = 13.4 Hz, 1H), 1.21 (dp, J = 34.6, 12.4, 11.8 Hz, 5H). 13C NMR (100 MHz, DMSO) δ 154.92, 142.04, 130.30, 121.63, 117.73, 114.11, 48.43, 33.49, 25.85, 25.01. 19F NMR (376 MHz, DMSO) δ −61.91. HRMS(ESI) for C14H17F3N2O [M+H]+: m/z calcd; 287.13657, found; 287.13707.
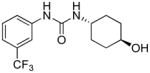
trans-1-(4-Hydroxycyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, III-2
Using General Procedure B employing trans-4-aminocyclohexanol, to afford III-2 as a white solid, 59 % (178 mg) yield, mp. 249°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR = 12.467 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.94 (s, 1H), 7.41 (td, J = 10.8, 7.2 Hz, 2H), 7.17 (d, J = 7.1 Hz, 1H), 6.09 (d, J = 8.3 Hz, 1H), 4.52-4.47 (m, 1H), 3.39-3.20 (m, 2H), 1.85 – 1.78 (m, 4H), 1.24 – 1.13 (m, 4H). 13C NMR (100 MHz, DMSO) δ 155.07, 142.01, 130.31, 130.00, 121.71, 117.79, 114.19, 68.68, 48.43, 34.45, 31.32. 19F NMR (376 MHz, DMSO) δ −61.88. HRMS(ESI) for C14H17F3N2O2 [M+H]+: m/z calcd; 303.13149, found; 303.13204.
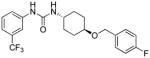
trans-1-(4-((4-Fluorobenzyl)oxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, III-3
Using General Procedure B employing trans-4-((4-fluorobenzyl)oxy)cyclohexylamine 10, to afford III-3 as a white solid, 33% (136 mg) yield, mp. 162°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR =18.230 min, purity of 95%; 1H NMR (300 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.93 (s, 1H), 7.41-7.39 (m, 2H), 7.34 – 7.30 (m, 1H), 7.18 – 7.09 (m, 3H), 6.14 (d, J = 7.7 Hz, 1H), 4.44 (s, 2H), 3.42 (dd, J= 12.3, 5.4 Hz, 1H), 3.49-3.36 (m, 1H), 3.35-3.33 (m, 1H), 2.09 – 1.71 (m, 4H), 1.24 (dq, J= 21.0, 11.0, 4H). 13C NMR (100 MHz, DMSO) δ 155.12, 142.09, 136.18, 130.36, 130.03, 129.95, 121.68, 117.71, 115.70, 115.49, 76.42, 68.94, 48.21, 30.90, 30.85. 19F NMR (376 MHz, DMSO) δ −61.78, −116.01. LCMS(ESI) for C21H22F4N2O2 [M+H]+: m/z calcd; 411.16, found;411.12.
trans-1-(2-(4-(Trifluoromethyl)phenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, III-4
Using General Procedure B employing trans-2-(4-(trifluoromethyl)phenoxy)cyclohexylamine 8, to afford III-4 as a white solid, 79 % (354 mg) yield, mp. 58°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR = 20.047 min, purity of 97%; 1H NMR (500 MHz, DMSO-d6) δ 8.84 (s, 1H), 7.92 (s, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.45 – 7.31 (m, 2H), 7.18 (d, J = 8.4 Hz, 3H), 6.38 (d, J = 8.0 Hz, 1H), 4.70 (dt, J = 5.1, 2.58 Hz, 1H), 3.96-3.77 (m, 1H), 1.99 – 1.84 (m, 1H), 1.67 (q, J= 4.5, 4.0 Hz, 3H), 1.65 (dd, J = 14.5, 7.8 Hz, 1H), 1.46-1.31 (m, 3H). 13C NMR (100 MHz, DMSO) δ 16.77, 154.70, 147.32, 141.57, 130.21, 127.36, 121.33, 116.72, 75.26, 49.83, 28.15, 27.56, 23.83, 19.91. 19F NMR (376 MHz, DMSO) δ −59.88, −61.42. HRMS(ESI) for C21H20F6N2O2 [M+H]+: m/z calcd; 447.15017, found; 447.15122.
trans-1-(3-(4-(Trifluoromethyl)phenoxy)cyclohexyl)-3-(3-(trifluoromethyl)phenyl)urea, III-5
Using General Procedure B employing trans-3-(4-(trifluoromethyl)phenoxy)cyclohexylamine 9, to afford III-5 as a white solid, 69% (306 mg) yield, mp. 62°C. RP-HPLC (C18): 0 to 100% (ACN/Water/0.1%TFA) in 25 min, tR = 20.183 min, purity of 100%; 1H NMR (399 MHz, DMSO-d6) δ 8.74 (s, 1H), 7.94 (s, 1H), 7.60 (d, J = 8.6 Hz, 2H), 7.42 (dt, J = 15.5, 8.2 Hz, 2H), 7.17 (d, J = 7.7 Hz, 1H), 7.10 (d, J = 8.5 Hz, 2H), 6.35 (d, J = 8.1 Hz, 1H), 4.80 (s, 1H), 3.90 (dp, J = 14.0, 5.4, 4.6 Hz, 1H), 2.01-1.91 (m, 1H), 1.87-1.69 (m, 2H), 1.68-1.50 (m, 4H), 1.44-1.21(m, 1H). 13C NMR (100 MHz, DMSO) δ 160.76, 155.01, 142.00, 130.33, 127.66, 127.75, 117.82, 116.61, 114.19, 73.09, 44.70, 36.55, 32.45, 29.33, 19.90. 19F NMR (376 MHz, DMSO) δ −60.34, −61.86. HRMS(ESI) for C21H20F6N2O2 [M+H]+: m/z calcd; 447.15017, found; 447.15159.
Plasmids and Ternary complex assay
The dual luciferase expression vector and other plasmids used for these studies are described in32. The ternary complex assay, surrogate of eIF2α phosphorylation, has been described elsewhere25. Briefly, the pBISA vector, which contains seven copies of the tetracycline-regulated transactivator response element (TRE), is flanked on both sides by human cytomegalovirus (CMV) minimal promoters allowing bi-directional transcription and two MCSs. Firely and renilla luciferases were subcloned into MCS-I and MCS-II, respectively. This plasmid, designated pBISA-DL, transcribes two mRNAs that contain the 90 nucleotide plasmid derived 5′UTR (same sequence in both mRNAs), and the ORF encoding either firefly or renilla luciferase followed by a polyadenylation sequence. This plasmid was further modified by inserting the 5′UTR of ATF-4 mRNA into MCS-I in front of the firefly luciferase mRNA. Transcription from this unit generates an mRNA that contains the firefly luciferase ORF preceded by a 5′UTR composed of 90 nucleotides derived from the plasmid and 267 nucleotides derived from the 5′UTR of ATF-4 mRNA. Transcription from the other unit generates an mRNA that contains the renilla luciferase ORF proceeded only by the 90-nucleotide plasmid-derived sequence in the 5′UTR (pBISA-DL(ATF-4))25.
Dual luciferase (DLR) assay
Cells expressing firefly and renilla luciferases were assayed with a dual glow luciferase assay kit, per manufacturer’s instruction (Promega Inc., Madison, WI). The data calculations were carried out as the ratio of firefly to renilla luciferase signal. Dose-response curves were obtained and triplicate data points were fitted to the logistical sigmoidal model using nonlinear least-squares regression performed in GraphPad Prism 6, where Emax is the maximal stimulatory effect of F/R Ratio, and C3X is the concentration at which F/R ratio=3 on the curve.
Stable and transient transfection
Stable cell lines utilized in this study are generated as described elsewhere22. Briefly, cells were seeded at the density of 105 in 60-mm dish (stable transfection) or 104 cells per well of 96-well plate (transient transfection) and transfected one day later using the Lipofectamine 2000 (Invitrogen). For selection of stable cell lines, transfected cells were transferred to 100-mm plates and selected with appropriate antibiotics22.
Western blotting
Cells cultured under recommended media conditions, were plated and maintained in serum-containing media without antibiotics in 14-cm plates (Nunc) until reaching 70% confluence. Cells were then treated with compounds for 6 hours, washed with cold PBS once, and lysed with M-PER Mammalian Protein Extraction Reagent (Pierce) for 30 minutes on ice. The cell lysates were centrifuged at 12,000 RPM for 15 min and the supernatants were transferred to fresh tubes and the concentrations were determined by BCA (Pierce). Equal amount of proteins were mixed with Laemmli Sample Buffer, heated at 100°C for 5 min and separated by SDS-PAGE and probed with anti-phosphoserine-51-eIF2α (Phos-eIF2α), anti-total eIF2α-specific antibodies (Total-eIF2α) (Biosource International, Hopkinton, MA), anti-CHOP, anti-cyclin D1 or anti-actin (Santa Cruz Biotechnology, CA) essentially as described38.
Cell growth assay
Cells were seeded in 96-well plates and maintained for 5 days in the presence of 0.5 to 20 μM of individual compound, and cell proliferation was measured by the sulforhodamine B (SRB) assay as described33: briefly, at the end of a 5-day treatment, cells were fixed in 10% cold trichloroacetic acid and cell numbers were estimated by measuring the remaining bound dye of sulforhodamine B after washing. The percentage of growth was calculated by using the equation: 100× [(T−T0)/(C−T0)], where T and C represent the absorbance in treated and control cultures at Day 5, and T0 at time zero, respectively. If T is less than T0, cell death has occurred and can be calculated from 100× [(T−T0)/T0].
Supplementary Material
Acknowledgments
This work was supported by NCI grant #1RO1CA152312 to B.H. Aktas, as well as NIEHS grant ES02710 and NIEHS Superfund Basic Research Program grant P42 ES04699 to B. D. Hammock.
Abbreviations used
- HRI
Heme-regulated inhibitor kinase
- eIF2α
eukaryotic translation initiation factor 2 alpha
- Met-tRNAi
methionine transfer RNA
- Pi
inorganic phosphate
- PKR
protein kinase R
- PERK
PKR-like endoplasmic reticulum kinase
- GCN2
general control non-derepressible-2
- AUDA
12-(3-adamantane-1-ylureido)dodecanoic acid
- sEH
soluble epoxide hydrolase
- CHOP
CCAAT/enhancer-binding protein homologous protein
- SRB
sulforhodamine B
- RP-HPLC
high performance liquid chromatography
- DLR
dual luciferase
- uORF
upstream open reading frame
- UTR
untranslated region
- F
firely
- R
renilla
- ATF-4
activating transcription factor 4
- πx
high lipophilicity parameters
- P-eIF2α
phosphorylated eIF2α
- T-eIF2α
total eIF2α
- TLC
thin layer chromatography
- TFA
trifluoracetic acid
- δ
chemical shifts
- TRE
transactivator response element
- CMV
minimal human cytomegalovirus
- MCS
multiple cloning sites
Footnotes
“Supporting Information Available: The screened compound library showing compounds’ effect on F/R ratio in the surrogate eIF2α phosphorylation assay, activity of N-phenyl-N′-(4-((2,6-difluorobenzyl)oxy)cyclohexyl)ureas in the surrogate eIF2α phosphorylation and cell proliferation assay, time response studies of selected compounds in the surrogate eIF2α phosphorylation assay, and HRMS data of the synthesized compounds. This material is available free of charge via the internet at http://pubs.acs.org.”
References
- 1.Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol. 2001;21:5018–5030. doi: 10.1128/MCB.21.15.5018-5030.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kimball SR, Fabian JR, Pavitt GD, Hinnebusch AG, Jefferson LS. Regulation of guanine nucleotide exchange through phosphorylation of eukaryotic initiation factor eIF2alpha. Role of the alpha-and delta-subunits of eiF2b. J Biol Chem. 1998;273:12841–1285. doi: 10.1074/jbc.273.21.12841. [DOI] [PubMed] [Google Scholar]
- 3.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 4.Raven JF, Koromilas AE. PERK and PKR: old kinases learn new tricks. Cell Cycle. 2008;7:1146–1150. doi: 10.4161/cc.7.9.5811. [DOI] [PubMed] [Google Scholar]
- 5.Baltzis D, Qu LK, Papadopoulou S, Blais JD, Bell JC, Sonenberg N, Koromilas AE. Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J Virol. 2004;78:12747–12761. doi: 10.1128/JVI.78.23.12747-12761.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 7.Takada Y, Ichikawa H, Pataer A, Swisher S, Aggarwal BB. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene. 2007;26:1201–1212. doi: 10.1038/sj.onc.1209906. [DOI] [PubMed] [Google Scholar]
- 8.Cabanski M, Steinmuller M, Marsh LM, Surdziel E, Seeger W, Lohmeyer J. PKR regulates TLR2/TLR4-dependent signaling in murine alveolar macrophages. Am J Respir Cell Mol Biol. 2008;38:26–31. doi: 10.1165/rcmb.2007-0010OC. [DOI] [PubMed] [Google Scholar]
- 9.Raven JF, Baltzis D, Wang S, Mounir Z, Papadakis AI, Gao HQ, Koromilas AE. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J Biol Chem. 2008;283:3097–3108. doi: 10.1074/jbc.M709677200. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Taylor SI, Tan SL, Sonenberg N. When translation meets metabolism: multiple links to diabetes. Endocr Rev. 2003;24:91–101. doi: 10.1210/er.2002-0018. [DOI] [PubMed] [Google Scholar]
- 11.Biason-Lauber A, Lang-Muritano M, Vaccaro T, Schoenle EJ. Loss of kinase activity in a patient with Wolcott-Rallison syndrome caused by a novel mutation in the EIF2AK3 gene. Diabetes. 2002;51:2301–2305. doi: 10.2337/diabetes.51.7.2301. [DOI] [PubMed] [Google Scholar]
- 12.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109:2693–2699. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen JJ. Heme-regulated eIF2α Kinase. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational control of gene expression. Cold spring harbor laboratory press; New York: 2000. pp. 529–546. [Google Scholar]
- 14.Han AP, Fleming MD, Chen JJ. Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia. J Clin Invest. 2005;115:1562–1570. doi: 10.1172/JCI24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couturier J, Page G, Morel M, Gontier C, Claude J, Pontcharraud R, Fauconneau B, Paccalin M. Inhibition of double-stranded RNA-dependent protein kinase strongly decreases cytokine production and release in peripheral blood mononuclear cells from patients with Alzheimer’s disease. J Alzheimers Dis. 2010;21:1217–1231. doi: 10.3233/jad-2010-100258. [DOI] [PubMed] [Google Scholar]
- 16.Donze O, Jagus R, Koromilas AE, Hershey JW, Sonenberg N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. Embo J. 1995;14:3828–3834. doi: 10.1002/j.1460-2075.1995.tb00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berns A. A tRNA with oncogenic capacity. Cell. 2008;133:29–30. doi: 10.1016/j.cell.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 18.Marshall L, Kenneth NS, White RJ. Elevated tRNA(iMet) synthesis can drive cell proliferation and oncogenic transformation. Cell. 2008;133:78–89. doi: 10.1016/j.cell.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 19.Dever TE, Yang W, Astrom S, Bystrom AS, Hinnebusch AG. Modulation of tRNA(iMet), eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2.GTP. Met-tRNA(iMet) ternary complexes. Mol Cell Biol. 1995;15:6351–6363. doi: 10.1128/mcb.15.11.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aktas H, Fluckiger R, Acosta JA, Savage JM, Palakurthi SS, Halperin JA. Depletion of intracellular Ca2+ stores, phosphorylation of eIF2alpha, and sustained inhibition of translation initiation mediate the anticancer effects of clotrimazole. Proc Natl Acad Sci U S A. 1998;95:8280–8285. doi: 10.1073/pnas.95.14.8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aktas BH, Qiao Y, Ozdelen E, Schubert R, Sevinc S, Harbinski F, Grubissich L, Singer S, Halperin JA. Small-Molecule targeting of translation initiation for cancer therapy. Oncotarget. 2013;4:1606–1617. doi: 10.18632/oncotarget.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai H, Chen T, Ming J, Sun H, Cao P, Fusco DN, Chung RT, Chorev M, Jin Q, Aktas BH. Dual activators of protein kinase R (PKR) and protein kinase R-like kinase PERK identify common and divergent catalytic targets. Chembiochem. 2013;14:1255–1262. doi: 10.1002/cbic.201300177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denoyelle S, Chen T, Yang H, Chen L, Zhang Y, Halperin JA, Aktas BH, Chorev M. Synthesis and SAR study of novel 3,3-diphenyl-1,3-dihydroindol-2-one derivatives as potent eIF2.GTP. Met-tRNA ternary complex inhibitors. Eur J Med Chem. 2013;69C:537–553. doi: 10.1016/j.ejmech.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 24.Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, Supko JG, Chorev M, Wagner G, Halperin JA. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget. 2012;3:869–881. doi: 10.18632/oncotarget.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen T, Ozel D, Qiao Y, Harbinski F, Chen L, Denoyelle S, He X, Zvereva N, Supko JG, Chorev M, Halperin JA, Aktas BH. Chemical genetics identify eIF2alpha kinase heme-regulated inhibitor as an anticancer target. Nat Chem Biol. 2011;7:610–616. doi: 10.1038/nchembio.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denoyelle S, Chen T, Chen L, Wang Y, Klosi E, Halperin JA, Aktas BH, Chorev M. In vitro inhibition of translation initiation by N,N′-diarylureas--potential anti-cancer agents. Bioorg Med Chem Lett. 2012;22:402–409. doi: 10.1016/j.bmcl.2011.10.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen HC, Hammock BD. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem. 2012;55:1789–1808. doi: 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scherman MS, North EJ, Jones V, Hess TN, Grzegorzewicz AE, Kasagami T, Kim IH, Merzlikin O, Lenaerts AJ, Lee RE, Jackson M, Morisseau C, McNeil MR. Screening a library of 1600 adamantyl ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability. Bioorg Med Chem. 2012;20:3255–3262. doi: 10.1016/j.bmc.2012.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang SH, Morisseau C, Do Z, Hammock BD. Solid-phase combinatorial approach for the optimization of soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2006;16:5773–5777. doi: 10.1016/j.bmcl.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madar DJ, Djuric SW, Michmerhuizen MJ, Kopecka HA, Li X, Longenecker KL, Pei Z, Pireh D, Sham HL, Stewart KD, Szczepankiewicz BG, Wiedeman PE, Yong H. Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) 20050215784 A1 US Patent. 2005
- 32.Ziegeler G, Ming J, Koseki JC, Sevinc S, Chen T, Ergun S, Qin X, Aktas BH. Embryonic lethal abnormal vision-like HuR-dependent mRNA stability regulates post-transcriptional expression of cyclin-dependent kinase inhibitor p27Kip1. J Biol Chem. 2010;285:15408–15419. doi: 10.1074/jbc.M110.113365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palakurthi SS, Fluckiger R, Aktas H, Changolkar AK, Shahsafaei A, Harneit S, Kilic E, Halperin JA. Inhibition of translation initiation mediates the anticancer effect of the n-3 polyunsaturated fatty acid eicosapentaenoic acid. Cancer Res. 2000;60:2919–2925. [PubMed] [Google Scholar]
- 34.Sheppard CW. The Effect of Fluorine Substitution on the Electronic Properties of Alkoxy, Alkylthio and Alkylsulfonyl Groups. J Am Chem Soc. 1963;85:1314–1318. [Google Scholar]
- 35.AH . The Element Displacement Principle: A new Guide in p-Block Element Chemistry. Vol. 28. Academic Press Inc; Orlando, FL: 1884. pp. 167–202. [Google Scholar]
- 36.O’Hagan D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem Soc Rev. 37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 37.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol Cell Biol. 1997;17:3850–3857. doi: 10.1128/mcb.17.7.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.