

Abstract
Much of the nutrient cycling and carbon processing in natural environments occurs through the activity of extracellular enzymes released by microorganisms. Thus, measurement of the activity of these extracellular enzymes can give insights into the rates of ecosystem level processes, such as organic matter decomposition or nitrogen and phosphorus mineralization. Assays of extracellular enzyme activity in environmental samples typically involve exposing the samples to artificial colorimetric or fluorometric substrates and tracking the rate of substrate hydrolysis. Here we describe microplate based methods for these procedures that allow the analysis of large numbers of samples within a short time frame. Samples are allowed to react with artificial substrates within 96-well microplates or deep well microplate blocks, and enzyme activity is subsequently determined by absorption or fluorescence of the resulting end product using a typical microplate reader or fluorometer. Such high throughput procedures not only facilitate comparisons between spatially separate sites or ecosystems, but also substantially reduce the cost of such assays by reducing overall reagent volumes needed per sample.
Keywords: Environmental Sciences, Issue 80, Environmental Monitoring, Ecological and Environmental Processes, Environmental Microbiology, Ecology, extracellular enzymes, freshwater microbiology, soil microbiology, microbial activity, enzyme activity
Introduction
Microorganisms such as bacteria and fungi obtain nutrients and carbon from complex organic compounds through the production of extracellular enzymes. These enzymes typically hydrolyze polymers into smaller subunits that can be taken into the cell. Therefore, at an ecological level, these microbial extracellular enzymes are responsible for much of the nutrient mineralization and organic matter decomposition that occurs in natural environments. Enzymes such as cellobiohydrolase (CBH) and β-glucosidase are important for cellulose degradation and work in unison to catalyze the hydrolysis of cellulose to glucose1,2, which provides a utilizable carbon substrate for microbial uptake and assimilation. The enzyme phosphatase releases soluble inorganic phosphate groups from organophosphates, essentially mineralizing phosphate and making it available for use by most organisms3. Other enzymes, such as N-acetylglucosaminidase (NAGase), are important in chitin degradation and can make both carbon and nitrogen available for microbial acquisition4.
One of the procedures for the assay of microbial extracellular enzyme activity in natural environments is the use of artificial p-nitrophenyl (pNP) linked substrates, an approach that was originally developed to detect soil phosphatase activity5. This approach relies on the detection of a colored end product, p-nitrophenol, which is released when the artificial substrate is hydrolyzed by the appropriate enzyme. The p-nitrophenol can be subsequently quantified colorimetrically by measuring its absorbance at around 400-410 nm. This method has since been applied to detect other enzymes such as NAGase6, and has been used in various studies looking at microbial extracellular enzyme activity in soils and sediments7-9.
An alternative approach that was originally developed to assess extracellular glucosidase activity in aquatic environments10,11 makes use of 4-methylumbelliferone (MUB) linked substrates. The end product released (4-methylumbelliferone) is highly fluorescent and can be detected using a fluorometer with an excitation/emission setting around 360/460 nm. A variety of MUB-linked artificial substrates are available, permitting the fluorometric measurement of the activity of at least as many enzymes (e.g. β-glucosidase, cellobiohydrolase, NAGase, phosphatase) as can be assayed using the pNP-substrate colorimetric procedure. Other microbial extracellular enzymes, such as the protein-degrading leucine aminopeptidase, can be assayed fluorometrically using 7-amino-4-methylcoumarin (COU) linked substrates. Both MUB- and COU-linked substrates have been used to determine enzyme activity in various terrestrial and aquatic samples12,13.
While previous studies have described fluorometric or colorimetric microplate approaches to determine extracellular enzyme activity14; there is a need for a clear presentation of how to conduct such assays. Here we demonstrate procedures for conducting high throughput microplate techniques for the analysis of extracellular enzyme activity in soils and sediments using the colorimetric pNP-linked substrates approach and in natural waters using the fluorescent MUB-linked substrates technique. We focus on the measurement of the activities of β-glucosidase, NAGase, and phosphatase as these enzymes can be tied to carbon, nitrogen, and phosphorus cycling, respectively. However, the procedures described here can be applied to the measurement of other extracellular enzymes using different artificial substrates.
Protocol
Colorimetric Analysis of Extracellular Enzyme Activity in Soils and Sediments
1. Preparation of Substrate and Buffer Solutions for Colorimetric Analyses of Enzyme Activity
Prepare 50 mM acetate buffer (pH 5.0-5.5) by mixing 50 ml 0.1 M acetic acid (2.87 ml glacial acetic acid in 500 ml water), 150 ml 0.1 M sodium acetate, and 200 ml distilled H2O. Adjust pH to 5.0-5.5 with 0.1 M acetic acid if necessary.
Prepare a solution of 1 M sodium hydroxide (NaOH) in distilled H2O.
Prepare pNP-linked substrate solutions in 50 mM acetate buffer. To assay phosphatase prepare 5 mM pNP-phosphate in 50 mM acetate buffer; to assay β-glucosidase prepare 5 mM pNP-β-glucopyranoside; to assay NAGase prepare 2 mM pNP-β-N-acetylglucosaminide. Prepare all substrate solutions in sterile 15 ml or 50 ml centrifuge tubes. Solutions can be stored at 4 °C for 2-3 weeks.
2. Determination of a Standard to Convert Absorbance to pNP Concentration
Prepare standard solutions of p-nitrophenol in 50 mM acetate buffer. Concentrations should range from 0.025-1 mM.
Transfer three replicates of 100 μl of each concentration to a clear 96-well microplate. Add 10 μl 1 M NaOH and 190 μl distilled H2O to each well.
Record absorbance at 410 nm using a microplate reader.
Multiply concentrations in the standard curve by 0.3 to get μmoles pNP per 300 μl reaction volume. Plot a curve of absorbance vs. μmole of pNP. The slope of the curve serves as the conversion factor (C) that will relate absorbance to μmole of pNP in each enzyme reaction.
3. Conducting the Enzyme Assay
For soil, prepare a slurry of each sample to be assayed in a sterile 15 ml centrifuge tube at a concentration of approximately 1 g/ml-1 using 50 mM acetate buffer. For sediments, add enough acetate buffer to make the slurry easily pipettable. The exact volume of slurry needed will vary according to the number of enzymes assayed, but a minimum of 5 ml is recommended. Vortex each slurry until all clumps of soil or sediment have dispersed and note the final volume.
Re-vortex each slurry and immediately pipette 150 μl into each of six wells on a 96-well deepwell block (Figure 1). It is important to vortex thoroughly and frequently in order to keep soil particles in suspension. Leave at least two wells per block empty to serve as a substrate control. Note: clip the end of pipette tips with scissors prior to pipetting as soil slurries tend to clog tips. Prepare one 96-well block for each enzyme to be assayed.
Pour approximately 5 ml of acetate buffer into a pipette reservoir and use an 8-channel pipettor to add 150 μl of the buffer to the last two wells of each sample (these will be sample buffer controls) and the two substrate control wells (Figure 1)
Pour approximately 10 ml of the appropriate pNP-substrate solution into a pipette reservoir and use an 8-channel pipettor to add 150 μl of the substrate solution to the first four wells of each sample and the two substrate control wells (Figure 1). Note the time as the reaction begins as soon as the substrate solution is added.
Incubate plates at RT (22 °C) for 0.5-4 hr. Exact incubation time will vary depending on activity level in samples and the enzyme to be assayed. For most soils and sediments, phosphatase and β-glucosidase require incubation times of 0.5-1.5 hr, whereas NAGase and other enzymes require incubation times of >2 hr.
While assays are incubating prepare clear 96-well microplates to read absorbance. Prepare one microplate for each deepwell block (i.e. for each enzyme assayed). Pipette 10 μl 1 M NaOH and 190 μl of distilled water into each well of the microplate. Note: NaOH slows the enzymatic reaction and raises the pH which enhances the color of the pNP released during the reaction.
After incubation, centrifuge the 96-well blocks at 2,000-5,000 x g for 5 min to pellet soil particles.
Use a multichannel pipette to withdraw 100 μl from each well, being careful to avoid the pellet, and transfer it to the corresponding well on prepared clear 96-well microplate.
Turn on the microplate reader and set up any necessary software. Record absorbance at 410 nm. If the absorbance of a particular well is above the linear detection limit of the plate reader, dilute that well 1:1 with water and re-measure. If absorbance is still too high, the assay should be repeated with a shorter incubation time.
4. Determination of Dry Mass of Samples
Pipette 1 ml of each sample slurry into a preweighed aluminum pan.
Dry in a 75 °C drying oven for 48 hr and weigh. Subtract the weight of the pan from this value to obtain the dry mass of soil or sediment in 1 ml of the slurry. Multiply by a factor of 0.15 to determine the dry mass of sample in the 150 μl added to each well in the enzyme assay.
5. Calculation of Enzyme Activity per Dry Mass of Soil or Sediment
Calculate final absorbance of each sample by subtracting the sample control absorbance from the sample assay absorbance. If substrate controls have high absorbance (roughly >0.060) then subtract those also.
Calculate enzyme activity in μmoles hr-1 g dry mass-1 from the equation:
Enzyme activity = Final absorbance / (C x incubation time x sample dry mass)
Fluorescent Analysis of Extracellular Enzyme Activity in Natural Waters
1. Preparation of Substrate, Standard, and Buffer Solutions for Fluorometric Analyses of Enzyme Activity
Prepare 200 μM solutions of MUB-linked substrates (e.g. 4-MUB-β-glucopyranoside, 4-MUB-phosphate, 4-MUB-N-acetyl-β-D-glucosaminide) by dissolving the appropriate substrate in sterile (autoclaved) distilled H2O in sterile 15 ml or 50 ml centrifuge tubes. Wrap tubes in aluminum foil to exclude light and store in refrigerator prior to use. Substrates should be stable for at least 1 week if stored in this way.
Prepare a MUB standard by making a stock solution of 100 μM 4-methylumbelliferone in sterile distilled H2O. Store refrigerated in amber or foil-wrapped bottle. Immediately prior to use, dilute the 100 μM stock solution by 1/10 into sterile H2O to make a working solution of 10 μM for enzyme assays.
Prepare a stock solution of 100 mM bicarbonate buffer by dissolving 8.4 g of NaHCO3 into 1 L H2O and autoclaving. Dilute this stock solution 1/20 into sterile H2O as needed to make a working solution of 5 mM for enzyme assays.
2. Organizing Water Samples on a 96-Well Black Microplate
Organize a microplate for each enzyme following the example shown in Figure 2. Note that allowing for adequate replication, standards, and controls, this procedure can assay the activity of a single enzyme for up to nine water samples on one 96-well black microplate.
Pour approximately 5 ml of the first sample into a pipette reservoir and use an 8-channel pipettor to pipette200 μl into all of the wells in column 1 of the microplate(s). Discard used pipette tips and repeat as needed for each water sample to fill columns 1-9.
3. Setting up Sample, Standard, Quench, and Substrate Controls
Set up controls to account for samples, standards, substrate and quenching on the same black microplate as the samples (Figure 2).
Sample controls contain sample water and bicarbonate buffer, and are not used in the activity calculations but will demonstrate reading consistency throughout the course of the experiment. The quench controls consist of sample water and a standard amount of the fluorescent tag and are used to measure the diffraction of fluorescence in sample water. Substrate and standard controls are made up of either substrate-linked or the standard fluorescent tag, respectively, and bicarbonate buffer.
Pour approximately 5 ml of 5 mM bicarbonate buffer into a clean pipette reservoir. Pipette 50 μl of buffer into microplate wells 1 through 9 in Rows D and E to form two replicate wells of sample controls per sample. Change pipette tips then transfer 200 μl of bicarbonate buffer to wells 10 through 12 in Rows A and H.
Reduce ambient lighting by dimming or turning off lights as the fluorescent standard is light sensitive.
Pour approximately 5 ml of 10 μM 4-methylumbelliferone into a clean pipette reservoir. Pipette 50 μl into microplate wells 1 through 12 in Row H, and into wells 1 through 9 into Rows G and F to form three replicates of quench controls per sample and overall standard controls. Either place the microplate in the dark or cover with an opaque lid to reduce light degradation of MUB.
Turn on the fluorometer and set-up any necessary software to be ready to read before adding the substrate. Note: some fluorometer bulbs may require a warm-up time of 3 min or more.
Pour approximately 5 ml of the appropriate MUB-linked substrate (e.g. 4-MUB-phosphate) into a clean pipette reservoir. Use a 12-channel pipettor to pipette 50 μl into microplate wells 1 through 12 in Row A, and into wells 1 through 9 in Rows B and C to form three replicate assays for each sample and three substrate controls. Immediately proceed to step 4.1, recording fluorescence.
4. Recording Fluorescence
Read the initial fluorescence immediately after substrate addition to the microplate. After reading fluorescence, incubate the microplate at RT (22 °C) either in the dark or covered with an opaque lid to reduce light degradation of MUB.
The incubation time required to measure the maximum potential enzyme activity in a water sample will depend on the enzyme concentration within the sample. Since this is unknown before the assay is completed, the microplate will have to be read at multiple time steps. Typically, reading at intervals of 10-15 min over the course of 1 hr is acceptable for many enzymes, although samples with very high activity for certain enzymes may peak before 10 min.
Continue reading fluorescence in the microplate at your designated intervals for at least 1 hr. Be sure to keep the microplate covered or in the dark between readings.
5. Calculation of Enzyme Activity per Volume of Water
For each sample at each time interval calculate the: mean initial sample fluorescence (wells D and E), the mean final sample fluorescence (wells A-C), the mean standard fluorescence (wells H10-11), and the mean quench control fluorescence (wells F-G).
For each time interval, calculate enzyme activity in nmoles hr-1 ml-1 from the equation:
Enzyme activity = (mean sample fluorescence - mean initial sample fluorescence) / ((mean standard fluorescence / 0.5 mol) x (mean quench control fluorescence / mean standard fluorescence) x (0.2 ml) x (time in hr))
Examine the activity values calculated for each time step. Determine final potential activity from the time step with the highest activity. If activity values continue to increase then later time steps may be required; if activity values fall throughout the course of the run, then run again with shorter time steps. Final activity is in nmoles of substrate consumed hr-1 ml-1 but can be scaled up to express as μmoles hr-1 L-1.
Representative Results
Soils and aquatic sediments typically have appreciable levels of extracellular enzyme activity as a result of attached microbial communities (biofilms) growing on the surface of particles. Figure 3 shows how this activity changes depending on the size of particles obtained from the surface sediment of a third order stream in northern Mississippi, USA. A previous study has shown that the bacterial communities on sediment particles from this stream can be separated into three distinct groups based on molecular analysis of their community structure: those on 0.063 mm particles, those on 0.125, 0.25, and 0.5 mm particles, and those on 1 mm particles15. Analysis of patterns in extracellular enzyme activity supports this conclusion; with phosphatase (Figure 3A) being similar on 0.125, 0.25, and 0.5 mm particles but much higher on the 1 and 0.063 mm fractions when measured using the pNP-linked substrate technique. Other enzymes such as β-glucosidase (Figure 3B), and NAGase (Figure 3C) show similar peaks on the finest particles, but are not elevated on 1 mm particles, highlighting the fact that different enzymes may show different environmental distributions and these can be elucidated using this assay. The relatively low error bars show that colorimetric assays of enzyme activity on sediments are reproducible and thus amenable to statistical analysis when comparing different environmental samples.
Natural waters tend to have lower extracellular enzyme activity per ml than soils or sediments do per g. As such, they should be assayed fluorometrically using MUB-linked substrates. Figure 4 shows how the activity of the enzymes phosphatase (Figure 4A), β-glucosidase (Figure 4B), and NAGase (Figure 4C) varies with depth in a shallow lake in northern Mississippi, USA. The lake (Boondoggle Lake) is known to be nutrient poor16, which is also suggested by the relatively high activity of phosphatase (Figure 4A), an enzyme that microorganisms produce in order to acquire phosphate from organic compounds. For all three of the enzymes assayed, samples collected at the water surface (0 cm) and 50 cm depth showed similar activity, whereas activity was elevated in the 100 cm sample. This sample was essentially taken from the water-sediment interface, and the presence of sediment particles in the sample likely accounts for the higher activity seen in this sample, especially for β-glucosidase and NAGase. As with the pNP substrates, the low error bars show that even with just three replicate readings, the reproducibility of the fluorometric assays using MUB substrates is high.
Note that units for Figure 4 are in nmoles of substrate consumed whereas those for Figure 3 are in μmoles of substrate consumed, even though the per unit size is comparable (either per ml or per g). This highlights the fact that soils and sediments tend to have much higher extracellular activity than natural waters (the activities of each specific enzyme in Figure 3 are approximately 10-100x higher than the activities of the equivalent enzyme in Figure 4). It also demonstrates the increased sensitivity of the MUB-linked substrate technique, and the necessity of using this technique for assaying extracellular enzyme activity in water samples.
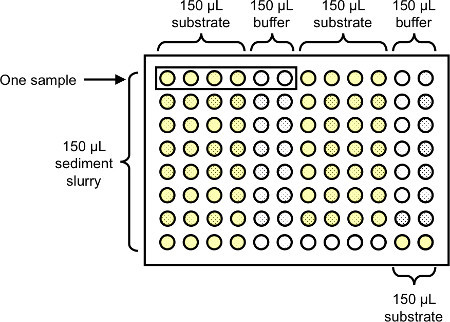 Figure 1. Suggested layout of 96-well deep well blocks for the assay of extracellular enzyme activity in soils or sediments using the colorimetric pNP-linked substrate technique. Each well should receive a total of 300 μl, made up of sample slurry, substrate solution, or acetate buffer depending upon whether it is a sample run, sample control, or substrate control.
Figure 1. Suggested layout of 96-well deep well blocks for the assay of extracellular enzyme activity in soils or sediments using the colorimetric pNP-linked substrate technique. Each well should receive a total of 300 μl, made up of sample slurry, substrate solution, or acetate buffer depending upon whether it is a sample run, sample control, or substrate control.
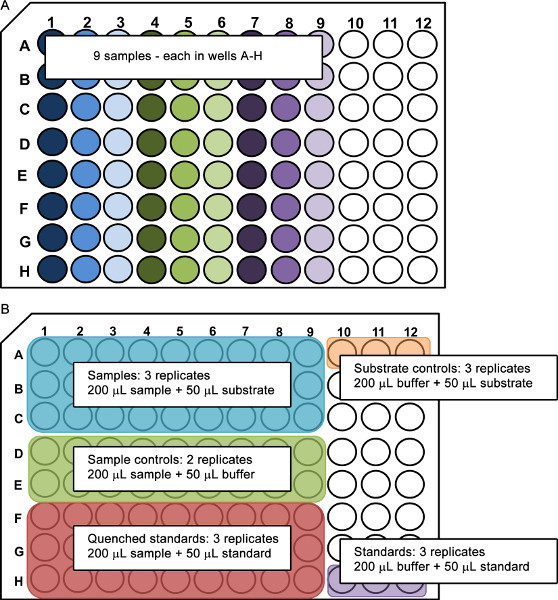 Figure 2. Suggested layout of 96-well black microplates for the assay of extracellular enzyme activity in water samples using the fluorometric MUB-linked substrate technique. Nine samples can be arranged vertically on the plate (A) each occupying eight wells. These eight wells are used for sample runs, sample controls, and quench controls while remaining wells are used for substrate controls and MUB standards (B).
Figure 2. Suggested layout of 96-well black microplates for the assay of extracellular enzyme activity in water samples using the fluorometric MUB-linked substrate technique. Nine samples can be arranged vertically on the plate (A) each occupying eight wells. These eight wells are used for sample runs, sample controls, and quench controls while remaining wells are used for substrate controls and MUB standards (B).
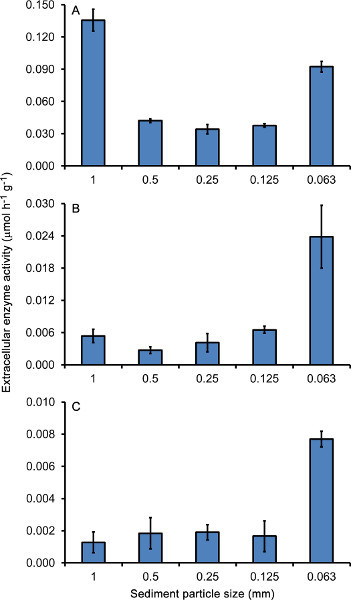 Figure 3. Extracellular enzyme activity on different sizes of surface sediment particles collected from a small stream in north Mississippi, USA. Each particle size fraction was assayed for the activity of phosphatase (A), β-glucosidase (B), and NAGase (C) following the pNP-substrate colorimetric procedure. Activity is reported in μmoles substrate consumed hr-1 g dry mass of sediment-1 and is the mean (+ S.E.) of three replicate readings per particle size fraction.
Figure 3. Extracellular enzyme activity on different sizes of surface sediment particles collected from a small stream in north Mississippi, USA. Each particle size fraction was assayed for the activity of phosphatase (A), β-glucosidase (B), and NAGase (C) following the pNP-substrate colorimetric procedure. Activity is reported in μmoles substrate consumed hr-1 g dry mass of sediment-1 and is the mean (+ S.E.) of three replicate readings per particle size fraction.
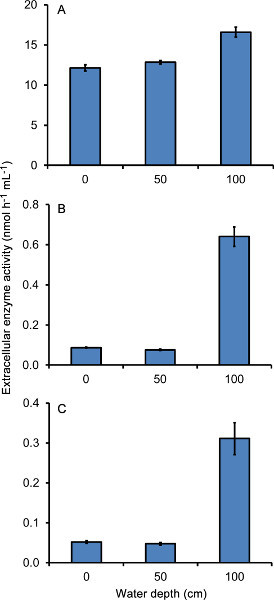 Figure 4. Extracellular enzyme activity in water taken at three different depths (0, 50, and 100 cm) from a shallow lake in north Mississippi, USA. Water was assayed for the activity of phosphatase (A), β-glucosidase (B), and NAGase (C) following the MUB-substrate fluorometric procedure. Activity is reported in nmoles substrate consumed h-1 ml of water-1 and is the mean (+ S.E.) of three replicate readings per sample.
Figure 4. Extracellular enzyme activity in water taken at three different depths (0, 50, and 100 cm) from a shallow lake in north Mississippi, USA. Water was assayed for the activity of phosphatase (A), β-glucosidase (B), and NAGase (C) following the MUB-substrate fluorometric procedure. Activity is reported in nmoles substrate consumed h-1 ml of water-1 and is the mean (+ S.E.) of three replicate readings per sample.
Discussion
Determining the activity of a variety of microbial extracellular enzymes in soils and sediments can provide useful insights into rates of nutrient mineralization and organic matter processing17. However, soils can vary in their moisture levels, so it is important to standardize activity to soil dry weight. This requires an additional drying step (typically of two days) beyond simply measuring enzyme activity. Thus, in contrast to assays of enzyme activity in water samples that provide near instantaneous results, reliable assays of enzyme activity in soil and sediments take a few days. For some soils, it may even be more appropriate to express activity per gram of organic matter or ash free dry mass, requiring an additional ashing step (typically 2 hr at 500 °C) beyond the drying procedure. Regardless, the most critical step in the colorimetric assay of soil or sediment enzyme activity is the withdrawal of the supernatant from the deep-well block following centrifugation at the end of the incubation period. Because this procedure relies on measuring absorbance, even the presence of a few stray soil particles in the final microplate can lead to spurious results. Higher centrifugation speeds (>5,000 x g) might help mitigate this problem, but in our experience centrifuge rotors that accept microplates usually have rotational limits below this threshold, and the blocks themselves may not tolerate much higher speeds. Essentially, this particular pipetting step requires a combination of care and speed to use a multi-channel pipettor to quickly transfer the required amount of supernatant from the deep-well block to the microplate.
Assays of extracellular enzyme activity in water samples have neither the need for a drying period nor the centrifugation step, the latter because both the enzyme-substrate reaction and measurement of fluorescence of the end product occur in the same microplate. As such, these assays are typically faster and may initially appear easier to run. However, they have their own limitations in that, whenever possible, the MUB standard and MUB-linked substrates should not be exposed to light. In our experience, dimming the lights as much a possible during pipetting and incubating the microplates in the dark (or covered with an opaque lid) is a necessity. Because these assays also require the plates to be read at multiple time points, this often results in the need for very rapid switching between plates when assaying multiple enzymes at the same time. For example, in order to assay nine water samples for the activity of six enzymes simultaneously (i.e. using six different microplates), it becomes necessary to read a plate every 2 min for 1 hr in order to ensure that each individual plate is read every 10-15 min (in this case, every 12 min). Care must be taken to keep track of the particular plate being read, as well as to ensure that no liquid is spilled from the plate as it is transferred into and out of the microplate fluorometer. When assaying multiple enzymes at once, it is also important to keep in mind the time that it takes for the microplate reader to actually read the plate and to stagger reading intervals accordingly. A final concern with the fluorescent assay of extracellular enzyme activity in water samples is when working with samples that are turbid. Water samples that contain suspended particles should be shaken before initially pouring into the pipette reservoir and mixed again by withdrawing and ejecting with the pipettor prior to being loaded onto the microplate. Higher numbers of particles in a sample also typically results in greater quenching of the fluorescent signal, and the importance of quench controls for each sample cannot be stressed enough.
While MUB- and pNP-linked substrates typically show similar Vmax values for environmental enzymes, the Km values can differ, and enzymes such as β-glucosidases and phosphatases may have higher affinity for MUB-linked substrates than their pNP-linked analogs14. Therefore, MUB-linked substrates are likely to be more sensitive than those linked to pNP, which suggests that they would be a better choice to use when measuring enzyme activity in soils and sediments. However the fluorogenic end product that is measured during MUB assays is subject to potential quenching in some soil extracts, and fluorescence readings can be unstable over time12. Soils and sediments also tend to have higher extracellular enzyme activity than natural waters, so the increased sensitivity of the fluorometric MUB-linked assays may offer little advantage over the colorimetric pNP methods given the potential problems with quenching when analyzing certain soils. As a side note, the pNP-linked substrates and the pNP standard itself are generally more affordable than their MUB counterparts; an additional reason for their use if budgetary restraints apply.
Perhaps the most important aspect of being able to assay microbial extracellular enzyme activity in aquatic and terrestrial environments is that while these techniques measure microbial physiological processes, these processes have a direct influence on ecosystem level transformations of carbon and nutrients. Rates of organic matter decomposition have been directly linked to the activity of extracellular cellulases in sediments18,19, so that rapid measurements of enzyme activity potentially facilitate the determination of instantaneous in situ decomposition rates in environmental samples. Using high throughput microplate approaches allows for the simultaneous measurement of enzyme activity in larger numbers of samples than more typical single tube approaches, so that variation in enzyme activity (and by extension in ecosystem level processes) in response to factors such as depth20 or environmental perturbations9,21 can be examined. Similarly, assays of enzymes involved in nutrient cycling, such as phosphatase and NAGase, can provide insights into nutrient limitation in specific environments, for instance, the relative importance of organic phosphorus to organic nitrogen during soil development8.
Because enzyme activities have been measured in environmental samples for over thirty years, comparisons between studies using advanced meta-analyses are now becoming possible. Such studies suggest that, at the global scale, enzyme activity, and therefore rates of organic matter decomposition and nutrient mineralization, are tied to pH, substrate availability, and nutrient stoichiometry17. At the same time, the use of a microplate-based protocol has begun to permit the analysis of fine scale patterns in enzyme activity using geostatistical mapping techniques22. Such techniques can examine patterns in spatial variability, and future developments could include the use of 384-well based microplates to permit the instantaneous analysis of even more samples and the finer resolution of spatial patterns.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
Funding for aspects of this work was provided by various sources including the United States Department of Agriculture Specific Cooperative Agreement 58-6408-1-595 and the National Science Foundation (award 1049911).
References
- Ljungdahl LG, Eriksson K-E. Ecology of microbial cellulose degradation. Advances in microbial ecology. 1985;8:237–299. [Google Scholar]
- Sinsabaugh RL, Antibus RK, Linkins AE, Mclaugherty CA, Rayburn L, Repert D, Weiland T. Wood decomposition over a first-order watershed: mass loss as a function of lignocellulase activity. Soil biology and biochemistry. 1992;24:743–749. [Google Scholar]
- Dalal RC. Soil organic phosphorus. Advances in agronomy. 1977;29:83–113. [Google Scholar]
- Sinsabaugh RL, Moorhead DL. Resource allocation to extracellular enzyme production: a model for nitrogen and phosphorus control of litter decomposition. Soil biology and biochemistry. 1995;26:1305–1311. [Google Scholar]
- Tabatabai MA, Bremner JM. Use of p-nitrophenyl phosphate for assay of soil phosphatase activity. Soil biology and biochemistry. 1969;1:301–307. [Google Scholar]
- Parham JA, Deng SP. Detection, quantification and characterization of β-glucosaminidase activity in soil. Soil biology and biochemistry. 2000;32:1183–1190. [Google Scholar]
- Kuperman RG, Carreiro MM. Soil heavy metal concentrations, microbial biomass and enzyme activities in a contaminated grassland ecosystem. Soil biology and biochemistry. 1997;29:179–190. [Google Scholar]
- Olander LP, Vitousek PM. Regulation of soil phosphatase and chitinase activity by N and P availability. Biogeochemistry. 2000;49:175–190. [Google Scholar]
- Jackson CR, Vallaire SC. Effects of salinity and nutrient enrichment on microbial assemblages in Louisiana wetland sediments. Wetlands. 2009;29:277–287. [Google Scholar]
- Hoppe H-G. Significance of exoenzymatic activities in the ecology of brackish water: measurements by means of methylumbelliferyl-substrates. Marine ecology progress series. 1983;11:299–308. [Google Scholar]
- Somville M. Measurement and study of substrate specificity of exoglucosidase activity in eutrophic water. Applied and environmental microbiology. 1984;48:1181–1185. doi: 10.1128/aem.48.6.1181-1185.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman C, Liska G, Ostle NJ, Jones SE, Lock MA. The use of fluorogenic substrates for measuring enzyme activity in peatlands. Plant and soil. 1995;175:147–152. [Google Scholar]
- Sinsabaugh RL, Findlay S, Franchini P, Fischer D. Enzymatic analysis of riverine bacterioplankton production. Limnology and oceanography. 1997;42:29–38. [Google Scholar]
- Marx M-C, Wood M, Jarvis SC. A microplate fluorometric assay for the study of enzyme diversity in soils. Soil biology and biochemistry. 2001;33:1633–1640. [Google Scholar]
- Jackson CR, Weeks AQ. Influence of particle size on bacterial community structure in aquatic sediments as revealed by 16S rRNA gene sequence analysis. Applied and environmental microbiology. 2008;74:5237–5240. doi: 10.1128/AEM.00923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canion AK, Ochs C. The population dynamics of freshwater armored dinoflagellates in a small lake in Mississippi. Journal of freshwater ecology. 2005;20:617–626. [Google Scholar]
- Sinsabaugh RL, Lauber CL, et al. Stoichiometry of soil enzyme activity at global scale. Ecology letters. 2008;11:1252–1264. doi: 10.1111/j.1461-0248.2008.01245.x. [DOI] [PubMed] [Google Scholar]
- Jackson CR, Foreman CM, Sinsabaugh RL. Microbial enzyme activities as indicators of organic matter processing rates in a Lake Erie coastal wetland. Freshwater biology. 1995;34:329–342. [Google Scholar]
- Jackson CR, Vallaire SC. Microbial activity and decomposition of fine particulate organic matter in a Louisiana cypress swamp. Journal of the north american benthological society. 2007;26:743–753. [Google Scholar]
- Jackson CR, Liew KC, Yule CM. Structural and functional changes with depth in microbial communities in a tropical Malaysian peat swamp forest. Microbial ecology. 2009;57:402–412. doi: 10.1007/s00248-008-9409-4. [DOI] [PubMed] [Google Scholar]
- Rietl AJ, Jackson CR. Effects of the ecological restoration practices of prescribed burning and mechanical thinning on soil microbial enzyme activities and leaf litter decomposition. Soil biology and biochemistry. 2012;50:47–57. [Google Scholar]
- Smart KA, Jackson CR. Fine scale patterns in microbial extracellular enzyme activity during leaf litter decomposition in a stream and its floodplain. Microbial ecology. 2009;58:591–598. doi: 10.1007/s00248-009-9512-1. [DOI] [PubMed] [Google Scholar]