

Abstract
The authors report a case involving a child with chronic respiratory symptoms, who did not respond to conventional treatment. Low serum immunoglobin levels and pathological findings on lung biopsy revealed an unusual diagnosis for his age group. A specific treatment led to clinical improvement.
Keywords: Child, Common variable immunodeficiency, Granulomatous lymphocytic interstitial lung disease, Hypogammaglobulinemia
Abstract
Les auteurs présentent le cas d’un enfant ayant des symptômes respiratoires chroniques, qui ne répondait pas au traitement classique. De faibles taux d’immunoglobine sérique et des observations pathologiques à la biopsie pulmonaire ont révélé un diagnostic inhabituel dans ce groupe d’âge. Un traitement spécifique a suscité une amélioration clinique.
Learning objectives
To recognize the usefulness of pathological diagnosis in a patient who may not be responding to therapy.
Pretest
When should an immune work-up be considered in a child with an unusual presentation of lung disease?
CASE PRESENTATION
A four-month-old boy was referred to the respiratory clinic with a history of cough, persistent wheeze, post-tussive emesis and grunting respirations since birth. Before his respiratory clinic assessment, he had presented to the emergency room five times, with a diagnosis of bronchiolitis on four of these occasions and wheeze of unknown etiology on one occasion. On one occasion, nasopharyngeal aspirate was positive for parainfluenza and positive for entero/rhinovirus on another. He had been admitted to the pediatric unit for four days at three months of age for respiratory syncytial virus bronchiolitis. He required supplemental oxygen during that hospital admission.
He was the product of an uncomplicated pregnancy at 35+4 weeks gestational age to nonconsanguineous parents. He was delivered vaginally, with a birth weight of 3.09 kg. His parents reported grunting respirations at birth that required no intervention and he was discharged home at 12 h of age. There was no family history of immune deficiency, although an older sister is being investigated for chronic cough.
At his respiratory consultation at four months of age, growth parameters showed weight between the 25th and 50th percentile, and height at the 75th percentile. He did not exhibit cyanosis; oxygen saturation was 96% on room air. He exhibited subcostal and intercostal retractions and grunting with deep breathing. On auscultation, inspiratory and expiratory large airway noise and expiratory wheeze were present.
Chest radiography showed hyperinflation of bilateral lung fields, right upper lobe infiltrate, bronchial wall thickening and diffuse interstitial infiltrates (Figure 1).
Figure 1).
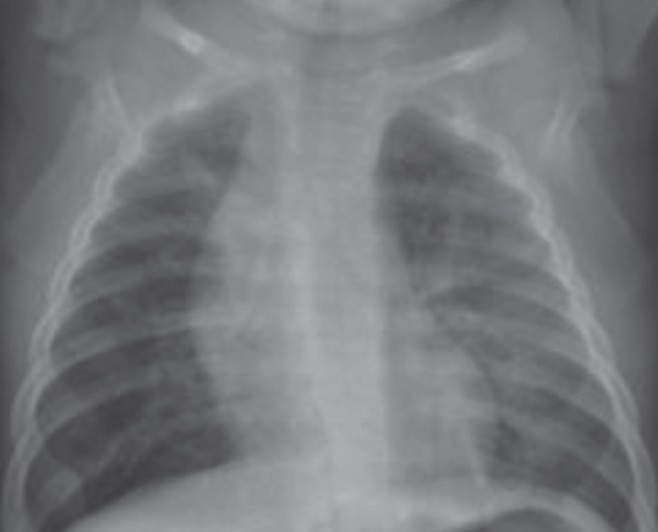
Chest x-ray showing hyperinflation and bilateral interstitial infiltrates
A single-contrast upper gastrointestinal study, performed to rule out large airway obstruction, was normal. A video fluoroscopic feeding study to investigate aspiration and 24 h pH probe to assess reflux showed no abnormalities. Echocardiogram was also normal. Computed tomography (CT) imaging revealed areas of atelectasis and mosaic attenuation (Figure 2).
Figure 2).
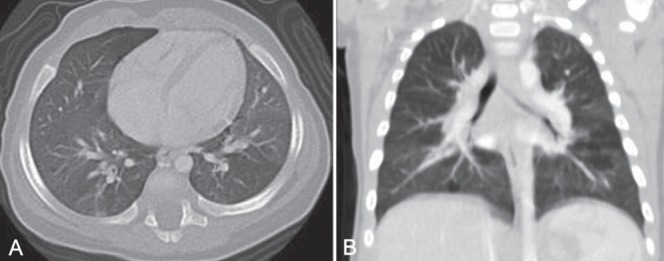
A and B. Computed tomography showing atelectasis and mosaic attenuation
Airway anatomy on flexible bronchoscopy was normal. Cytology from bronchoalveolar lavage showed neutrophilia (40%). Bronchoalveolar lavage culture grew Escherichia coli and Moraxella catarrhalis. Bordetella pertussis, HIV, Epstein-Barr virus (EBV) and human herpes virus 8 (HHV8) were not detected by polymerase chain reaction. Tuberculosis testing was negative. Investigations for cystic fibrosis and rheumatological work-up were negative. Angiotensinconverting enzyme analysis for sarcoidosis was negative.
Immunoglobulin (Ig) analysis revealed persistently low IgG levels (range 1.02 g/L to 1.87 g/L) between five months and 15 months of age (normal range for age 2.8 g/L to 16 g/L). IgA, IgM and IgE levels were normal on multiple occasions.
Vaccine titres could not be performed because the child had not received regular childhood immunizations. One dose of pneumococcal 13-valent conjugate vaccine was given to assess humoral responses at 11 months of age. Response was demonstrated in only three of nine serotypes tested (serotypes 3, 51 and 56, defined as a titre >1.4 μg/mL or fourfold increase). Isohemagglutinins were present at 19 months of age (blood type O, anti-A titre 128 (range 8 to 2048) and anti-B titre 64 (range 8 to 256).
Lymphocyte subsets were normal (CD3 3.92×109/L, CD4 2.968×109/L, CD8 0.616×109/L, CD19+ B cells 1.344×109/L and natural killer cells 0.336×109/L). CD4:CD8 ratio was elevated (4.6). Naive CD4+CD45RA+ and CD8+CD45RA+ T cells were normal (CD4+CD45RA+ 2.502×109/L, CD8+CD45RA+ 0.563×109/L). He had 2.2% IgD-CD27+ memory B cells. Mitogen stimulation assay was normal to phytohemagglutinin, concanavalin A and pokeweed mitogen. A neutrophil oxidative burst assay was normal, ruling out chronic granulomatous disease. Total hemolytic complement was normal.
Thoracoscopic wedge lung biopsy performed at nine months of age (five months following his initial respiratory assessment) revealed peribronchial lymphocytic inflammation and mild interstitial pneumonitis consisting mainly of CD3+ T cells (Figures 3 and 4). Small, poorly formed non-necrotizing granulomas were present within the interstitium, with some extending into the alveolar space (Figure 5). Mild pleural thickening was also present.
Figure 3).
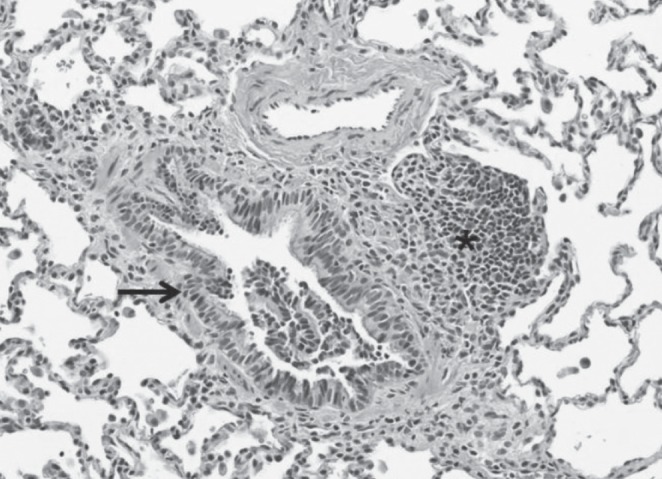
Peribronchial lymphocytic inflammation (asterisk). Bronchial epithelium (black arrow). Hematoxylin and eosin stain, original magnification ×200
Figure 4).
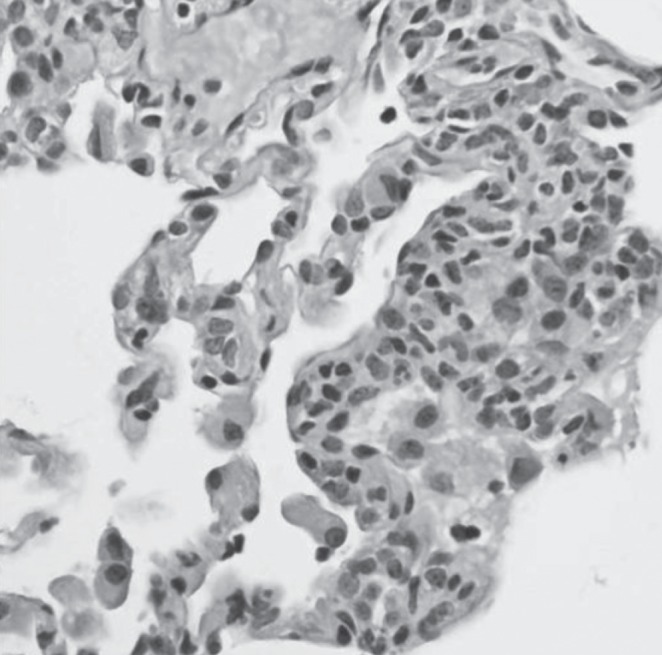
Mild lymphoplasmacytic interstitial pneumonitis. Hematoxylin and eosin stain, original magnification ×400
Figure 5).
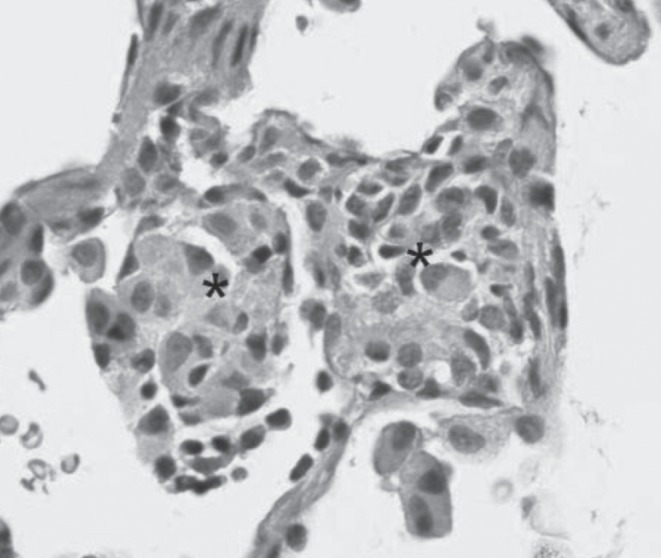
Poorly formed non-necrotizing granulomas (asterisks). Hematoxylin and eosin stain, original magnification ×400
Special stains for microorganisms (including fungi, mycobacteria and bacteria) and immunohistochemistry for HHV8 and EBV were negative. There was no evidence of pulmonary neuroendocrine cell hyperplasia, pulmonary interstitial glycogenosis or aspiration.
DISCUSSION
The finding of persistent low IgG levels led to suspicion of an immune deficient process. Histology consistent with a diagnosis of granulomatous lymphocytic interstitial lung disease (GLILD) was unexpected. The term ‘GLILD’ was coined by Bates et al (1) who reported this unique histological pattern in a subgroup of patients with common variable immunodeficiency (CVID), who also had chronic respiratory symptoms and diffuse radiographic abnormalities. These patients were reported to have a worse prognosis and increased prevalence of lymphoproliferative disorders. Most recently De Dios et al (2) decribed an 18-year-old woman with Kabuki syndrome, immunoglobulin deficiency and GLILD.
Recurrent pneumonias are common in patients with CVID and represent a significant cause of morbidity. Pulmonary fibrosis and bronchiectasis may also be present (3). The most common pulmonary CT findings include airway disease, ground-glass attenuation, nodules and parenchymal opacification (4). In children, air trapping on expiratory CT scans is the most frequent abnormality (5).
Our patient presented with cough, persistent wheeze, post-tussive emesis, grunting respirations, interstitial changes on chest imaging and hypogammaglobulinemia (IgG line only). While histology was consistent with a diagnosis of GLILD, he did not meet the full criteria for CVID. Diagnostic criteria for CVID include: four years of age or older; low serum IgG levels (<2.5th percentile for age) and levels below normal for age of IgA or IgM; lack of antibody response to protein antigens following immunization; and exclusion of other known causes of failure of immunoglobulin production (3).
The histopathological patterns observed in GLILD patients described by Bates et al (1) include non-necrotizing granulomas, and a spectrum of lymphocytic inflammation including lymphocytic interstitial pneumonia (LIP), follicular bronchiolitis and lymphoid hyperplasia. Our patient showed only mild interstitial lymphocytic inflammation and mild follicular bronchiolitis with occasional small non-necrotizing granulomas.
LIP has been associated with several autoimmune disorders suggesting an underlying immunological pathogenesis. EBV, HHV8 and HIV have been suggested to play a role in the pathogenesis of LIP. Based on these interactions, in their review, Park et al (6) propose a possible role for HHV8 and GLILD in CVID.
A review of the literature identified no other childhood conditions with hypogammaglobulinemia and the pathological findings of GLILD (7). Classifications of pediatric interstitial lung disease (ILD) have recently been revised (8). Causes of early ILD diagnosed in infancy include developmental disorders related to defects in alveolar formation, surfactant dysfunction disorders, neuroendocrine cell hyperplasia of infancy and pulmonary interstitial glycogenosis. CT and histological data from our patient, however, did not support these diagnoses. LIP is a rare paediatric ILD classified under idiopathic interstitial pneumonias that present in older children. It has, however, been reported in association with hypogammaglobulinemia in children (9). More commonly, when apparent in children, it is a harbinger for AIDS (10). HIV testing was also negative in our patient.
The decision to perform lung biopsy was a difficult choice given the patient’s age. The decision to proceed with lung biopsy, however, was based on persistent symptoms, lack of response to treatment and parental consent. Despite treatment with inhaled corticosteroids, oral steroids, ventolin and azithromycin, the patient continued to experience symptoms of cough, wheeze and post-tussive vomiting. Following the finding of persistent low IgG levels and histological features consistent with GLILD, a trial of intravenous immunoglobulin (IVIG) therapy was initiated. To date, he has responded well to this treatment and his symptoms have improved significantly including resolved grunting, good feeding with no post-tussive emesis and maintaining appropriate weight for height.
In addition, repeat CT imaging at five and 10 months (ie, at 20 and 25 months of age, respectively) following initiation of IVIG treatment showed improvement. There remain areas of air trapping in the lower lobes (Figure 6).
Figure 6).
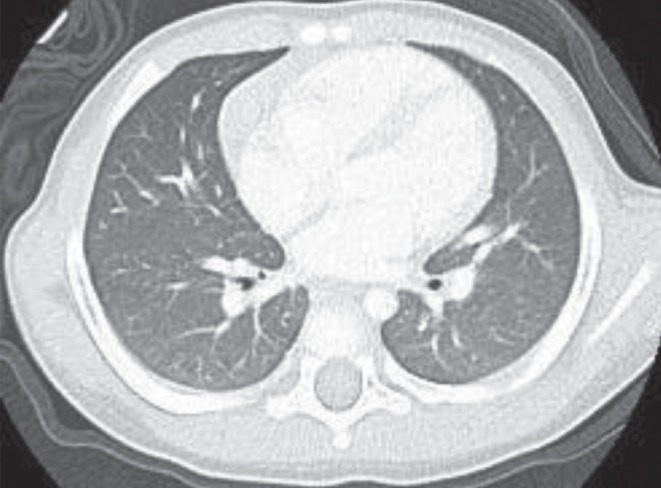
Areas of gas trapping in the lower lobes
The main treatment for hypogammaglobulinemia is IVIG. In their study investigating the efficacy of IVIG in children with CVID, Baris et al (11) noted fewer respiratory tract infections, fewer and shorter hospital stays, and reduced antibiotic usage in a group of 29 children with CVID.
The final diagnosis for this child still remains unclear. Differential diagnoses include transient hypogammaglobulinemia of infancy, nonspecific hypogammaglobulinemia, a mild combined immune deficiency or ILD. This patient does not meet full criteria for a diagnosis of CVID due to age, and only one Ig isotype being decreased. Also, we have been unable to adequately assess response to vaccines, though it is possible that he will progress to develop CVID. Currently we plan to stop IVIG after two years of age and better assess his humoral immune response with a nonconjugated pneumococcal vaccine challenge. Abnormal response or clinical worsening off IVIG will warrant further immune investigation. The clinical response to IVIG and subsequent improvement in his follow-up CT scan supports the diagnosis of hypogammaglobulinemia in this case. Thus, a diagnosis of GLILD presenting at <1 year of age is favoured. The present article describes the first case of this constellation of symptoms and signs in a child <1 year of age.
Post-test
The presence of a persistently low Ig level should always prompt consideration of immunological work-up. The present case highlights the spectrum of presentation of children with hypogammaglobulinemia. The pathological diagnosis gave credence to the diagnosis of an immunodeficiency and justification for use of IVIG.
REFERENCES
- 1.Bates C, Ellison M, Lynch D, et al. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114:415–21. doi: 10.1016/j.jaci.2004.05.057. [DOI] [PubMed] [Google Scholar]
- 2.De Dios JA, Javaid AA, Ballesteros E, Metesky ML. An 18-year-old woman with Kabuki syndrome, immunoglobulin deficiency and granulomatous lymphocytic interstitial lung disease. Conn Med. 2012;76:15–8. [PubMed] [Google Scholar]
- 3.Chapel H, Cunningham-Rundles C. Updates in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haem. 2009;145:709–27. doi: 10.1111/j.1365-2141.2009.07669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanaka N, Kim J, Bates C, et al. Lung diseases in patients with common variable immunodeficiency: Chest radiographic and computed tomographic findings. J Comput Assist Tomogr. 2006;30:828–38. doi: 10.1097/01.rct.0000228163.08968.26. [DOI] [PubMed] [Google Scholar]
- 5.van de Ven A, van Montfrans J, Terheggen-Largo S, et al. A CT scan score for the assessment of lung disease in children with common variable immunodeficiency disorders. Chest. 2010;138:371–9. doi: 10.1378/chest.09-2398. [DOI] [PubMed] [Google Scholar]
- 6.Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID) Clin Immunol. 2010;134:97–103. doi: 10.1016/j.clim.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Kinane TB, Shailam R, Mark EJ. Case 37-2011: A 9-month-old boy with recurrent tachypnea and respiratory distress. N Engl J Med. 2011;365:2221–8. doi: 10.1056/NEJMcpc1103560. [DOI] [PubMed] [Google Scholar]
- 8.Das S, Langston C, Fan LL. Interstitial lung disease in children. Curr Opin Pediatr. 2011;23:325–31. doi: 10.1097/MOP.0b013e3283464a37. [DOI] [PubMed] [Google Scholar]
- 9.Church JA, Isaacs H, Saxon A, Keens TG, Richards W. Lymphoid interstitial pneumonitis and hypogammaglobulinemia in children. Am Rev Respir Dis. 1981;124:491–6. doi: 10.1164/arrd.1981.124.4.491. [DOI] [PubMed] [Google Scholar]
- 10.Joshi VV, Oleske JM, Minnefor AB, et al. Pathologic pulmonary findings in children with the acquired immunodeficiency syndrome: A study of ten cases. Hum Pathol. 1985;16:241–6. doi: 10.1016/s0046-8177(85)80009-5. [DOI] [PubMed] [Google Scholar]
- 11.Baris S, Ercan H, Hasret Cagan H, et al. Efficacy of intravenous immunoglobulin treatment in children with common variable immunodeficiency. J Investig Allergol Clin Immunol. 2011;21:514–21. [PubMed] [Google Scholar]