

Abstract
Glutamate excitotoxicity contributes to the neuronal injury and death associated with many neurodegenerative diseases. The glutamate transporter EAAT2, which is primarily localized on astrocytic processes, facilitates glutamate clearance from synapses, thus preventing neuronal damage. In this issue of the JCI, Kong et al. characterize a compound that upregulates EAAT2 translation, thereby increasing glutamate uptake by glial cells. Furthermore, this strategy for alleviating excitotoxicity was found to be beneficial in mouse models of both amyotrophic lateral sclerosis (ALS) and epilepsy, suggesting that future development in this chemical series may lead to much-needed treatments for these disorders.
Current therapies for neurodegenerative diseases offer limited relief
Current treatments for many neurodegenerative diseases provide only very modest effects on disease progression and symptomology. While neurodegenerative disorders such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS) affect an ever-growing number of individuals, our ability to treat these fatal illnesses is very limited. For example, ALS leads to the degeneration and death of motor neurons, causing muscle wasting, paralysis, and eventual death of affected individuals; however, the lone FDA-approved treatment currently available for this disorder only extends lifespan by a matter of months (1).
In this issue, Kong et al. propose a therapeutic strategy for the treatment of neurodegenerative disorders that targets the neuronal death and dysfunction caused by glutamate excitotoxicity (2). Damage to neurons caused by excess glutamate contributes to ALS and is a feature of many neurological conditions, including epilepsy, which is a group of disorders characterized by epileptic seizures resulting from aberrant cortical neuronal activity (3). Kong and colleagues characterize a compound, LDN/OSU-0212320, that upregulates the translation of the glutamate transporter EAAT2 in astrocytes, thus increasing EAAT2 on glial cell surfaces and enhancing glutamate uptake (2). Previous work by this group entailed a small-molecule screen in PA-EAAT2 cells, an EAAT2-expressing primary astrocyte line, for compounds that induce translational upregulation of EAAT2 (4). This led to a structure-activity relationship (SAR) study performed on the pyridazine-based series of small molecules identified in the aforementioned screen (5). The characterization of LDN/OSU-0212320 is the culmination of this work, demonstrating the efficacy of a representative compound from the pyridazine series in animal models of neuronal disorders.
Kong et al. demonstrate herein that within 2 hours of treatment, LDN/OSU-0212320 enhanced the localization of EAAT2 mRNA within polyribosome cell fractions, enhanced EAAT2 expression on the plasma membrane, and increased glutamate uptake in cultured astrocytes (2). Furthermore, in primary neuron-astrocyte cocultures, LDN/OSU-0212320 substantially protected neurons from glutamate-induced toxicity. LDN/OSU-0212320 demonstrated favorable pharmacokinetics (PKs) and localized to the brain of treated animals in appreciable concentrations following i.p. administration. These results, combined with the compound’s good in vivo potency and low toxicity, open the possibility that LDN/OSU-0212320 is a reasonable therapeutic option for the treatment of diseases that display glutamate-induced neurodegeneration.
Enhanced glutamate transport in murine models of neurodegenerative disease
Kong and colleagues tested LDN/OSU-0212320 in two murine models of neurodegenerative disease: a model of chronic excitotoxicity and an acute excitotoxicity model (2). ALS is a disease characterized by loss of EAAT2 expression and chronic glutamate toxicity. SOD1(G93A) mice, an extensively used rodent model of ALS, were treated with LDN/OSU-021230 following symptom onset. LDN/OSU-0212320 treatment restored EAAT2 expression in the brain and resulted in both an extension of lifespan and an enhancement of motor function. Additionally, LDN/OSU-0212320 reduced mortality and spontaneous recurrent seizures in pilocarpine-induced status epilepticus mice, indicating that translational upregulation of EAAT2 may also be a viable treatment for epilepsy, which is also associated with acute glutamate toxicity.
The comprehensive study by Kong et al. on the potential for a pyridazine-based compound to increase EAAT2 translation and provide therapeutic efficacy in rodent models of neurodegenerative disease has compelling implications for ALS and epilepsy treatment (2). However, further studies will need to be performed to fully understand how these compounds may advance neurodegenerative therapies. While LDN/OSU-021320–treated SOD1(G93A) mice exhibited pronounced symptom reduction and increased lifespan, it is important to note that many compounds that have demonstrated efficacy in this ALS model have had no significant effects in human trials. A pertinent example of such a compound is the β-lactam antibiotic ceftriaxone, which restores EAAT2 expression in SOD1(G93A) mice through increased Eaat2 transcription (6). Despite positive effects in SOD1(G93A) animals, phase III clinical trials of ceftriaxone in ALS patients were halted because the drug did not meet the predetermined efficacy criteria. Although SOD1(G93A) mice have historically been the most widely used murine ALS model, researchers are relying less and less on these animals as predictors of human outcomes.
Currently, the only FDA-approved treatment for ALS is riluzole, a compound that also reduces glutamate toxicity in vivo; however, riluzole confers few advantages to patients and only increases lifespan by several months (7). The minuscule benefits of riluzole, combined with the ineffectiveness of ceftriaxone in clinical trials, raises the question of whether a reduction in glutamate toxicity alone is an adequate treatment for ALS. As neurons in affected individuals suffer from a variety of biological insults, perhaps a reduction of excitotoxicity alone is not a realistic mechanism for therapeutic development. However, it will be imperative to determine whether drugs that reduce glutamate toxicity could be efficacious in ALS patients when used in combination with other treatments.
Additionally, Kong et al. provide data suggesting that translational enhancers of EAAT2 are advantageous for the treatment of epilepsy (2). Current anticonvulsants, predominantly Na+ channel blockers, do not meet patients’ needs, and approximately 30% of epilepsy disorders are reported to be drug resistant. Furthermore, many anticonvulsants have undesirable side effects, including sedation, ataxia, and weight loss or gain (8). Upregulation of glutamate transporters on astrocytes may be an effective treatment strategy, because excessive glutamatergic neurotransmission is believed to be one of the major pathological mechanisms of epilepsy. Notably, transgenic mice that lack EAAT2 have spontaneous epileptic activity (9). In addition, astrocytes contribute to epilepsy by releasing glutamate during seizure activity, implying that astrocytes induce irregular neuronal activity (10). These data suggest that a drug that enhances glutamate uptake by astrocytes may be a powerful tool for epilepsy treatment.
While LDN/OSU-0212320 did not have any measurable effect on acute seizure activity, Kong et al. demonstrated that treatment with this compound reduces neuronal death and mortality rates following seizure activity in mice (2). Importantly, LDN/OSU-0212320 substantially reduced recurrent seizures; therefore, this compound may attenuate the progression of this disease. Together, these data suggest that upregulation of EAAT2 expression and function may be an appropriate mechanism for the development of new therapeutics for patients with epilepsy. Kong and colleagues have provided evidence that initiation of EAAT2 translation is a drugable target that may allow for the treatment of drug-resistant forms of epilepsy. Additionally, LDN/OSU-0212320 appears to have a low side-effect profile, indicating that drugs developed around this target may have fewer adverse side effects than current anticonvulsants. A treatment strategy centered on astrocytes rather than neurons has exciting potential for epilepsy management.
LDN/OSU-0212320 promotes EAAT2 translation via a YB-1–dependent mechanism
Kong et al. also attempted to decipher the mechanism of action of LDN/OSU-0212320 and demonstrated that this compound increases the association of EAAT2 mRNA with Y-box–binding protein 1 (YB-1), a nucleic acid–binding protein that is important for translational regulation (2) (Figure 1). Knockdown of YB-1 with siRNA reduced the ability of LDN/OSU-0212320 to upregulate EAAT2 expression, indicating that YB-1 is required for enhanced EAAT2-mediated glutamate uptake. Additionally, LDN/OSU-0212320 increased the phosphorylation of both YB-1 and the signaling effector protein kinase C (PKC). Further studies indicated that PKC activity is required, but not sufficient, for LDN/OSU-0212320 activation of EAAT2 translation. While these data provide some insight into the mechanism of action, the molecular target of LDN/OSU-0212320 is still unknown. Due to the efficiency of biological systems, it seems unlikely that there is a molecular target with the sole function of regulating the translation of EAAT2 mRNA. While upregulation of actin mRNAs in polyribosomal fractions was not observed after LDN/OSU-0212320 treatment, the possibility that a series of other related mRNAs are recruited to polyribosomes following drug treatment cannot be eliminated. Additional investigations into the mechanism of action and molecular targets of this compound are necessary to predict its effects in human trials and to facilitate drug discovery efforts. Furthermore, future studies that include a screen to analyze protein upregulation following chronic compound treatment should be considered.
Figure 1. LDN/OSU-021230 upregulates EAAT2 expression in astrocytes.
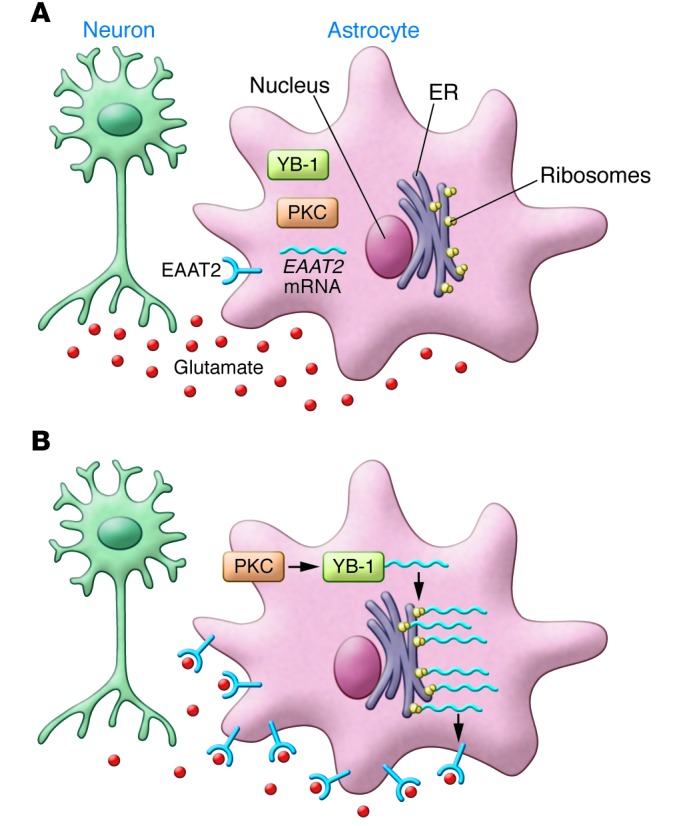
(A) In many neurodegenerative diseases, excess glutamate (red circles) causes damage to neurons (aqua), which can be mitigated by glutamate uptake by astrocytes (pink). (B) LDN/OSU-021230 increases glutamate uptake through the upregulation of EAAT2 receptors on astrocytic processes. Following LDN/OSU-021230 treatment, PKC becomes activated. This activation is required for phosphorylation of the translational regulator YB-1. LDN/OSU-021230 enhances the interaction of YB-1 with EAAT2 mRNA and upregulates the translation of EAAT2 protein. EAAT2 localizes to the plasma membrane, where it facilitates glutamate uptake by the cell, thus reducing glutamate excitotoxicity.
Conclusions and future directions
In conclusion, the demonstrated efficacy of an EAAT2 translational enhancer has enticing implications for the treatment of neurodegenerative diseases characterized by both chronic and acute glutamate toxicity. As a treatment for ALS, the study by Kong and colleagues (2) should be viewed with cautious optimism, because the preponderance of evidence suggests that due to the complex symptomology of ALS, a reduction of excess glutamate alone will be of little benefit to patients. However, elimination of excitotoxicity in combination with other treatments will likely improve patient outcomes. The development of compounds targeting EAAT2 translation for ALS therapy should be viewed in the context of a combinatorial treatment option, with discovery efforts focused on the development of compounds with drug-like properties including PK profiles that favor use with other therapeutic agents.
In this study, Kong et al. also demonstrate that upregulation of glutamate transporters on astrocytes has pronounced benefits in mouse models of temporal lobe epilepsy (2). The fact that LDN/OSU-0212320 targets translation rather than transcription of EAAT2 suggests that upregulation of this protein can be achieved quickly, providing protection from the acute excitotoxicity seen following status eptilepticus. While no mutations involving glutamatergic signaling have been identified in epilepsy patients (11), deficiencies in glutamate transport function have been documented (12); therefore, enhancing EAAT2 expression may be beneficial, and reduction of astrocytic glutamate may enhance the outcomes of conventional epilepsy treatments.
Finally, despite the substantial data included in this study, further investigation of the mechanism of action of these compounds is required. Understanding how these drugs directly target EAAT2 translation is important, especially for the prediction of drug interactions and side effects of this compound in patients.
Acknowledgments
This work was supported in part by the Department of Defense Amyotrophic Lateral Sclerosis Research Program under award number W81XWH-12-1-0373 (to N.D.P. Cosford). The views and opinions of and endorsements by the authors do not reflect those of the U.S. Army or the Department of Defense.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(3):964–967. doi:10.1172/JCI74608.
See the related article beginning on page 1255.
References
- 1.Limpert AS, Mattmann ME, Cosford ND. Recent progress in the discovery of small molecules for the treatment of amyotrophic lateral sclerosis (ALS). Beilstein J Org Chem. 2013;9:717–732. doi: 10.3762/bjoc.9.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kong Q, et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest. 2014;124(3):1255–1267. doi: 10.1172/JCI66163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher RS, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005;46(4):470–472. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- 4.Colton CK, et al. Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen. 2010;15(6):653–662. doi: 10.1177/1087057110370998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing X, et al. Structure-activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg Med Chem Lett. 2011;21(19):5774–5777. doi: 10.1016/j.bmcl.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berry JD, et al. Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS One. 2013;8(4):e61177. doi: 10.1371/journal.pone.0061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glicksman MA. The preclinical discovery of amyotrophic lateral sclerosis drugs. Expert Opin Drug Discov. 2011;6(11):1127–1138. doi: 10.1517/17460441.2011.628654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moldrich RX, Chapman AG, De Sarro G, Meldrum BS. Glutamate metabotropic receptors as targets for drug therapy in epilepsy. Eur J Pharmacol. 2003;476(1–2):3–16. doi: 10.1016/S0014-2999(03)02149-6. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276(5319):1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 10.Tian GF, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11(9):973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman AG. Glutamate and epilepsy. J Nutr. 2000;130(4S Suppl):1043S–1045S. doi: 10.1093/jn/130.4.1043S. [DOI] [PubMed] [Google Scholar]
- 12.Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis. 2012;47(2):145–154. doi: 10.1016/j.nbd.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]