

Abstract
The quantification of gene expression at the single cell level uncovers novel regulatory mechanisms obscured in measurements performed at the population level. Two methods based on microscopy and flow cytometry are presented to demonstrate how such data can be acquired. The expression of a fluorescent reporter induced upon activation of the high osmolarity glycerol MAPK pathway in yeast is used as an example. The specific advantages of each method are highlighted. Flow cytometry measures a large number of cells (10,000) and provides a direct measure of the dynamics of protein expression independent of the slow maturation kinetics of the fluorescent protein. Imaging of living cells by microscopy is by contrast limited to the measurement of the matured form of the reporter in fewer cells. However, the data sets generated by this technique can be extremely rich thanks to the combinations of multiple reporters and to the spatial and temporal information obtained from individual cells. The combination of these two measurement methods can deliver new insights on the regulation of protein expression by signaling pathways.
Keywords: Cellular Biology, Issue 80, Yeasts, Flow Cytometry, Microscopy, Fluorescence, Signal Transduction, single cells, MAPK signaling, Saccharomyces cerevisiae
Introduction
Signaling via transduction cascades often culminates in the expression of proteins. The characterization of this expression profile is a key element in understanding the function of biological pathways. The identification of the spectrum of up-regulated proteins and the dynamics of their activation can be achieved by various techniques such as micro-arrays, northern blots or western blots1-3. However, these techniques average the response of an entire population of cells. To understand the fine regulation of the expression of proteins, it is desirable to gather measurements at the single cell level. Ideally, these measurements should also provide quantitative data amenable to develop mathematical models of the underlying pathway.
Microscopy and flow cytometry are two techniques, which are ideally suited to deliver such quantitative single cell measurements. The endogenous tagging of proteins with a fluorescent protein can be used to quantify their expression level 4. However, since the addition of a large fluorescent moiety at the terminus of the protein can render it non-functional, it is often more desirable to generate specific expression reporters based on a promoter driving the expression of a fluorescent construct. This reporter protein is exogenous to the cellular system and therefore does not influence the signaling events that are taking place in the cell.
Both microscopy and flow cytometry have been widely used in the yeast signaling field. As examples; Colman-Lerner and co-workers correlated, in single cells, the expression of mating specific and constitutively expressed reporters by microscopy to quantify the noise in yeast mating signal transduction cascade5, Acar and colleagues used flow cytometry to study the regulatory network controlling the expression of the GAL genes6. In a previous study7, we have used a combination of these two techniques to study the expression output from the high osmolarity glycerol (HOG) pathway in budding yeast. This mitogen activated protein kinase (MAPK) pathway is triggered by hyper-osmotic stress. It results in the activation of the MAPK Hog1, which translocates to the nucleus of the cell to induce a transcriptional program resulting in the expression of roughly 300 genes. To study this process, we had engineered an expression reporter based on the STL1 promoter (a gene induced specifically in response to Hog1 activity8) driving the expression of a quadruple Venus fluorescent protein (pSTL1-qV). Flow cytometry measurements uncovered the presence of two populations of cells at intermediate stress level (0.1 M NaCl) with only a fraction of the population expressing the fluorescent reporter. We used microscopy to further investigate this behavior and discovered that this noise in protein expression was governed by intrinsic factors9. We could further observe that cells with a similar level of Hog1 activity could display strikingly different expression outcomes. The combination of these two techniques allowed us to demonstrate how the slow remodeling of stress response genes influenced the expression outcome at the single cell level7.
In this paper, we use the expression of the pSTL1-qV reporter induced by hyper-osmotic shock as an example of the quantification of protein expression by microscopy and flow cytometry. The same strain subjected to 0.2 M NaCl stress was studied with both techniques. This will allow us to highlight some key differences in these two highly complementary techniques.
Protocol
1. Microscopy Measurements
Inoculate 5 ml of synthetic medium with yeast.
Grow cells at 30 °C overnight.
Measure the OD600 of the overnight culture the next morning.
Dilute overnight culture in 5 ml synthetic medium to OD600 0.05.
Grow the diluted culture at 30 °C for at least 4 hr.
Prepare 3-fold concentrated (0.6 M) NaCl solution in synthetic medium.
Prepare a 1 mg/ml solution of Concanavalin A (ConA) in PBS.
Filtrate ~200 μl of ConA solution into the well slide.
Let stand for 30 min.
Remove ConA solution.
Add 150 μl H2O to the well.
Remove H2O.
Measure OD600 of cell culture.
Prepare 300 μl of cell culture at OD600 = 0.02 in a 1.5 ml microtube.
Sonicate the cells in a water bath for 45 sec.
Vortex briefly the microtube.
Sonicate the cells in a water bath for 45 sec.
Vortex briefly the microtube.
Add 200 μl of cell culture to the well.
Wait 30 min to let the cells settle to the bottom of the well.
Place the well slide on the microscope.
Select the illumination settings for the fluorescent channel to be recorded.
Select the illumination settings for two bright field images (one slightly out of focus: 3 μm below the focal plane to allow for a proper segmentation of the cells).
Select the time intervals for the time-lapse measurement
Select the fields of view to image.
Start the acquisition of a few frames.
Pause the acquisition to add the 100 μl of 0.6 M NaCl medium.
Resume imaging.
2. Flow Cytometry Measurements
Inoculate 5 ml of synthetic medium with yeast.
Grow at 30 °C overnight.
Measure the OD600 of the overnight culture the next morning.
Dilute overnight culture in 5 ml synthetic medium to OD600 0.1.
Grow at 30 °C for at least 4 hr to reach an OD600 of 0.2-0.4.
Prepare cycloheximide solution 1 mg/ml in H2O.
Prepare 3-fold concentrated (0.6 M) NaCl solution in synthetic medium.
Prepare one 1.5 ml microtube with 100 μl of 0.6 M NaCl medium for each time point to be measured.
At time 0, add 200 μl of cell culture to each microtube.
Incubate with shaking at 30 °C.
At time T, add 30 μl cycloheximide to a microtube.
Repeat step 2.11 for all the desired time points.
Incubate all microtubes for at least 45 min and up to 3 hr at 30 °C with shaking.
Prepare FACS tubes with 400 μl PBS.
Sonicate the cells in water bath for 1 min.
Vortex briefly the microtubes.
Sonicate the cells in water bath for 1 min.
Vortex briefly the microtubes.
Add 100 μl cell culture from each microtube to its corresponding FACS tube.
Select the 488 nm excitation laser and 530/30 nm bandpass filter for detection.
Test non-expressing and fully expressing sample to verify if they fall in the detector sensitivity range.
Measure 10,000 cells for each sample acquired.
Representative Results
Microscopy
Yeast cells bearing the expression reporter pSTL1-qV (ySP97) were attached to the bottom of the well-slide and placed under the microscope. The cells were stimulated by the addition of stress medium directly into the well during the course of the imaging session. This allows us to acquire a few images of the cells before pathway induction and follow their fate after the stimulation. In the present case, the cells were followed for ~2 hr with a time interval of 10 min. Figure 1A is an image of non-fluorescent cells before the induction and Figure 1B displays the same cells 2 hr after stress when the expression reporter has been induced and has become fluorescent.
To extract the quantitative information present in these images, a complex image analysis process has to be performed. It includes the segmentation of the cells to define objects in the image, the tracking of these objects from one frame to the next and the measurements of the features of these objects (shape and intensity properties). We have developed our own platform named YeastQuant to perform this analysis10. A few other non-commercial softwares are available to perform these tasks: CellProfiler 11, CellID 12 or CellX13. Figure 1C displays the results of an analysis performed with YeastQuant. Each red trace represents the temporal evolution of the average fluorescence intensity of a single cell. The blue line displays the median of more than 500 cells which were segmented and tracked throughout the 20 frames of five time-lapse movies acquired from five different fields of views from the same well. The light blue area represents the 25th and 75th percentile of all intensities measured at a given time point.
Flow Cytometry
For the flow cytometry measurements, the cell culture was spilt in microtubes containing the stress medium. To study the temporal evolution of the pSTL1-qV expression, the translation was inhibited at different time points (5 to 10 min intervals) with cycloheximide (Figure 2). This drug blocks the synthesis of new proteins, but the maturation of the fluorescent protein already expressed is not perturbed. Therefore, after 45 min to one hour of incubation, the cells can be measured in the flow cytometer allowing quantification of the amount of protein produced at the time of cycloheximide addition. This strategy circumvents the problem of fluorescent protein maturation and thereby quantifies the true dynamics of fluorescent protein synthesis.
Comparison of the two techniques
Figure 3A compares the dynamics of the expression reporter measured by microscopy and flow cytometry. While the fluorescent signal starts to rise 30-40 min after stress with microscopy, the flow cytometry measurements clearly prove that the proteins are already produced between 10 to 15 min after the stimulus. This half-hour delay is due to the slow maturation of the fluorescent proteins14. Although both methods quantify the fluorescent intensity of the cells, one has to bear in mind that in this study, they don't measure the same biological process. While, microscopy follows the apparition of the fluorescent signal, the flow cytometry actually quantifies the synthesis of the protein. It is essential to take into account this maturation step when performing live-cell imaging. In a situation where an endogenous protein is tagged with a fluorescent protein, the endogenous protein will be expressed and active before the fluorescence of its tag can be detected.
As indicated above, both techniques deliver complementary information. Flow cytometry can measure a large number of cells very rapidly (10,000 or more). It can therefore deliver statistically rich data sets where two overlapping populations can be identified or a few rare events can be observed. Microscopy provides information on a limited number of cells (on the order of 100). However, because this data set contains temporal and spatial information and allows the correlation of multiple measurements in the same cell, it can offer highly valuable insights into the investigated biological process.
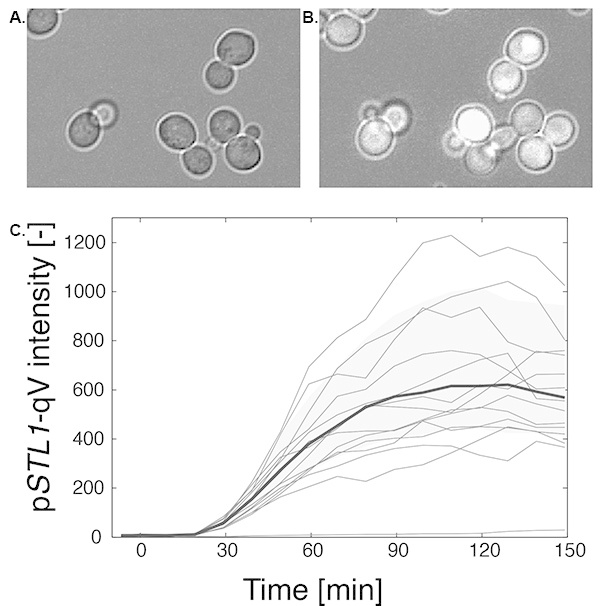 Figure 1. Measurement of reporter expression by microscopy. A and B. Images of cells bearing the fluorescent expression reporter pSTL1-qV before (A) and 2 hr after stimulation with 0.2 M NaCl (B). C. Time course of fluorescence apparition. The red curves represent the average cellular fluorescence intensity of a few representative single cell traces. The blue curve represents the median fluorescence of 550 single cell traces. The light blue area represents the 25th and 75th percentile of the fluorescent expression of the cell population at each time point. Click here to view larger image.
Figure 1. Measurement of reporter expression by microscopy. A and B. Images of cells bearing the fluorescent expression reporter pSTL1-qV before (A) and 2 hr after stimulation with 0.2 M NaCl (B). C. Time course of fluorescence apparition. The red curves represent the average cellular fluorescence intensity of a few representative single cell traces. The blue curve represents the median fluorescence of 550 single cell traces. The light blue area represents the 25th and 75th percentile of the fluorescent expression of the cell population at each time point. Click here to view larger image.
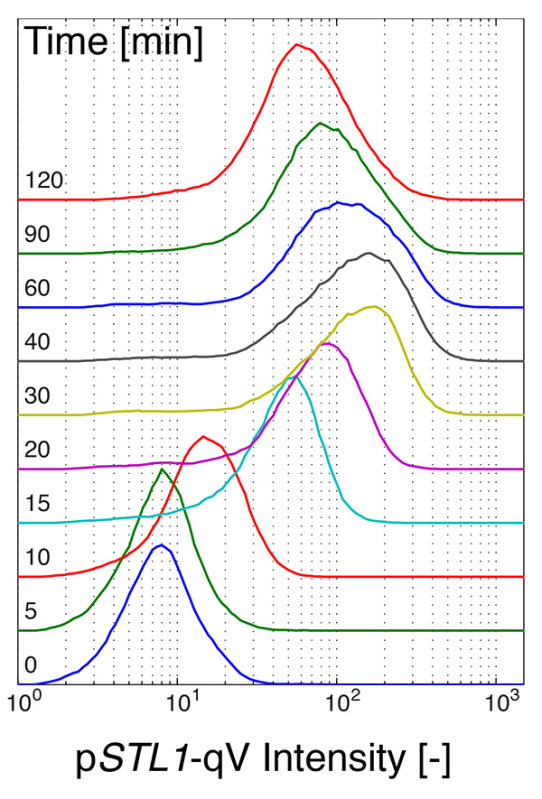 Figure 2. Measurement of reporter expression by flow cytometry. Histograms of the cellular fluorescence of cells bearing the fluorescent expression reporter pSTL1-qV treated with 0.2 M NaCl and inhibited with cycloheximide at different time points. Click here to view larger image.
Figure 2. Measurement of reporter expression by flow cytometry. Histograms of the cellular fluorescence of cells bearing the fluorescent expression reporter pSTL1-qV treated with 0.2 M NaCl and inhibited with cycloheximide at different time points. Click here to view larger image.
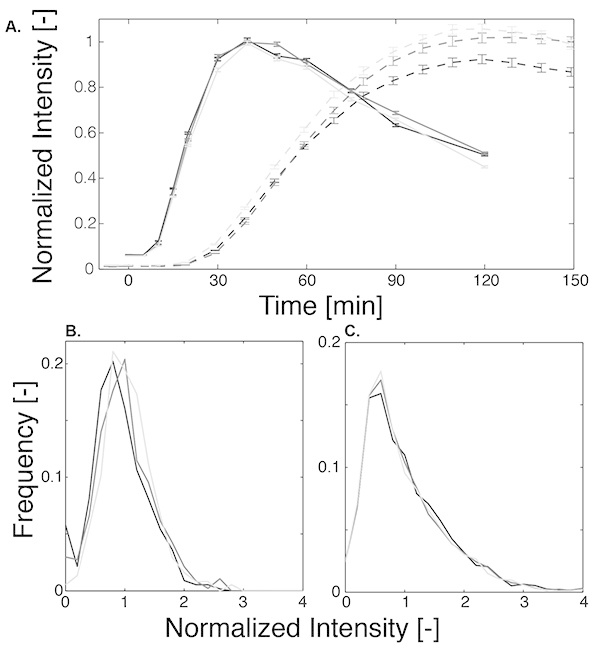 Figure 3. Comparison of the expression dynamics measured by microscopy and flow cytometry. A. Mean of fluorescence intensity measured by flow cytometry (solid line) and microscopy (dashed line) as function of time for three biological replicates. The error bars represent the standard error of the mean. B and C. Histograms of the normalized fluorescence intensity at 120 min after stress for microscopy (B) and 60 min after stress for flow cytometry (C) for the three replicates. Click here to view larger image.
Figure 3. Comparison of the expression dynamics measured by microscopy and flow cytometry. A. Mean of fluorescence intensity measured by flow cytometry (solid line) and microscopy (dashed line) as function of time for three biological replicates. The error bars represent the standard error of the mean. B and C. Histograms of the normalized fluorescence intensity at 120 min after stress for microscopy (B) and 60 min after stress for flow cytometry (C) for the three replicates. Click here to view larger image.
Discussion
Microscopy
The treatment of the well with ConA is an essential step to ensure a proper imaging of the cells. Because ConA has low solubility in PBS (5 mg/ml), the filtration process allows the removal of large aggregates that are present in the unfiltered solution and interfere with the imaging. Cells attach relatively strongly to the treated surface and the addition of the inducing solution should not disturb the localization of the cells, allowing for a continuous tracking before and after the stimulus. This treatment can also be applied to flow channels or microfluidic devices were the cells are subjected to a constant flow7,15. If the well is not properly rinsed with water after the treatment, the cells will not adhere properly to the glass, presumably because the ConA in solution binds directly to the cells and prevents them to form a strong bond at the glass surface. After the washing step, the well can remain dry for 3-4 hr before the addition of the cells without affecting noticeably the binding of the cells to the glass.
The selection of the exposure time for the fluorescent channel is a critical setting for the success of the experiment. Since the fluorescence of the cell will increase throughout the time-lapse, it is important to test beforehand with an induced sample what is the maximal exposure time that can be used to avoid saturation of the images during the acquisition. Another parameter to take into consideration when selecting the exposure time is the bleaching of the fluorescent signal. For each sample, one has to find a trade-off between brightness of the image and degradation of the signal due to bleaching during the time-lapse. The time interval between each acquisition also influences strongly the bleaching of the fluorophore. Since protein expression is a relatively slow process, we typically use 10-15 min. intervals. If other faster processes need to be followed in parallel to the protein expression, it might be advisable to use different time steps across the whole time-lapse movie (1-2 min at the beginning and 5-15 min for later acquisitions).
The control of the cell density is a crucial parameter to take into account. If the density is too low, only a few cells will be present in each field of view resulting in poor statistics for the data set. However, if the density is too high, it might hinder the segmentation, tracking and quantification of the cells, making it difficult to extract the desired information from the images. To determine the seeding density, one additional parameter to take into account is the duration of the experiment. If an experiment lasts for multiple division cycles, it is advisable to start with a sparse field of view that will gradually fill during the time course of the imaging due to division of the cells and settling down of cells in solution. In the protocol, we suggest using an OD600 of 0.02, depending on the type of experiment we perform we use OD600 from 0.05 for short time-lapse movies to 0.005 for longer ones.
The number of cells analyzed is clearly a key parameter for the validity of the experiment. Sometimes as few as 20-30 cells are combined to generate one dataset. However, to start having statistically significant datasets, a hundred cells or more are probably necessary. The magnification of the objective determines the size of the imaged area and thereby the number of imaged cells. For protein expression measurements, where the aim is to measure the average fluorescence of the cell, a high numerical aperture 40X immersion objective is the best solution providing both good signal to noise ratio and a large field of view. If sub-cellular structures have to be analyzed, higher magnifications (60X or 100X objectives) are often necessary, resulting in the measurement of fewer cells. The advent of sCMOS cameras with a detector size almost four times larger than the one of conventional CCD is clearly an advantage to obtain more cells per field of view.
With a motorized XY-stage, it is possible to image in parallel multiple fields of view to increase the number of cells in the final dataset. However, there is a trade-off between the number of XY positions visited and the speed of the image acquisition. Depending on the speed of the stage and the number of illuminations recorded, it is typically feasible to image ten positions in less than a minute. To quantify fast signaling events, this can become a limiting factor. For protein expression measurements, where fluorescence changes happen on a slower time scale, it is usually not a limitation. Alternatively, because these expression experiments last for a few hours and because of the slow temporal resolution required, it becomes advantageous to image multiple samples in parallel. Multiple neighboring wells can be seeded with wild type and mutant cells, different stimulus can be applied to each well or biological replicates can be measured in parallel. Scanning through all wells from a 96-well plate is routinely achieved with air objective for microscopy screens16. Due to the low abundance of endogenous proteins in yeast, their quantification requires an oil objective with high numerical aperture. This limits severely the range that can be travelled with the objective. Nonetheless we routinely image four neighboring wells by staying close to their common corner. It is however more challenging to image a larger number of wells. Water objectives with automatic dispensers might circumvent this problem at the expense of a minor loss in brightness.
Flow cytometry
Collecting a sufficient number of cells is hardly a problem with flow cytometers. Typically 10,000-100,000 cells can be acquired in a minute. However, because no image of the cell is acquired, it is important to select properly the cell population to be analyzed. Gating of the cells based on the measured forward and side scatters can allow to select individual cells rather than clusters of cells and to remove cell debris from the analysis. In Figure 2, each histogram represents a population of roughly 5,000 cells, which were selected based on their scattering properties from the 10,000 events measured.
By breaking clusters of cells into individual cells, sonication of the cell culture is an additional way to improve the quality of the data acquired. This holds true for microscopy where clusters of cells can be very difficult to segment properly. The duration of this step has to be tested experimentally. We usually perform two rounds of sonication in a water bath of 30 sec to a minute followed by vortexing of the cell suspension. Prolonged sonication can result in the lysis of a few cells. However, we did not observe an up-regulation of stress responsive genes due to this experimental step. Note that the aggregation of the cells is a background dependent phenotype, for instance W303 cells display a stronger tendency to aggregate than S288C cells.
Variability in single cell measurements
In Figure 3A, the mean and standard error for three independent experiments performed by flow cytometry and microscopy is plotted. The minor differences between the three curves arise from the fact that the experimental errors are relatively small for these simple measurements and that many cells are measured (more than 5,000 for flow cytometry, between 370-550 for microscopy). The temporal evolution of the microscopy curves is smoother than the one of the flow cytometry data. One probable reason for this behavior is the sample preparation process. For microscopy, all time points in an experiment result from the same stress event, while for the FACS, the sample for each time point is stressed individually. Small variations in the volume of the stress and culture solutions can result in a small difference in the gene expression output. One alternative strategy would be to stress 5 ml culture of cells and remove aliquots at the desired times to inhibit them with cycloheximide. Both methods work well, we use the microtube preparation method because it offers more flexibility and is also well-suited for the measurements of dose-response curves where multiple NaCl concentrations have to be measured in parallel.
The low noise in these averaged curves however does not reflect the full range of variability present at the single cell level. In Figure 3B and 3C, the histograms of the expression measured by microscopy and flow cytometry at a single time point are plotted. These two panels illustrate well the fact that the technical variations between replicates are relatively small, especially in comparison to the large variability observed in the expression of individual cells. The difference in the shapes of the distribution between the microscopy and the flow cytometry measurements arises from the fact that with flow cytometry the total cellular intensity is quantified while in the microscopy images the average cellular intensity was calculated. This latter measurement averages out variations in cell size and therefore results in narrower distributions.
The two techniques presented here allow to study quantitatively the behavior of single cells. This feature is usually hidden in traditional biological measurements performed at the population level. Understanding how the variability in the response of individual cells emerges can offer important insights into the complex regulation of biological processes.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors thank Matthias Peter and his group at the Institute of Biochemistry at the ETH in Zürich where these methods have been developed. This work has been supported by the Swiss National Science Foundation.
References
- Gasch AP, Spellman PT, et al. Genomic expression programs in the response of yeast cells to environmental changes. Molecular biology of the cell. 2000;11(12):4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nadal E, Zapater M, Alepuz PM, Sumoy L, Mas G, Posas F. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature. 2004;427(6972):370–374. doi: 10.1038/nature02258. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh W-K, et al. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Wu J-Q, Pollard TD. Counting cytokinesis proteins globally and locally in fission yeast. Science. 2005;310(5746):310–314. doi: 10.1126/science.1113230. [DOI] [PubMed] [Google Scholar]
- Colman-Lerner A, Gordon A, et al. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437(7059):699–706. doi: 10.1038/nature03998. [DOI] [PubMed] [Google Scholar]
- Acar M, Becskei A, van Oudenaarden A. Enhancement of cellular memory by reducing stochastic transitions. Nature. 2005;435(7039):228–232. doi: 10.1038/nature03524. [DOI] [PubMed] [Google Scholar]
- Pelet S, Rudolf F, Nadal-Ribelles M, de Nadal E, Posas F, Peter M. Transient activation of the HOG MAPK pathway regulates bimodal gene expression. Science. 2011;332(6030):732–735. doi: 10.1126/science.1198851. [DOI] [PubMed] [Google Scholar]
- de Nadal E, Casadome L, Posas F. Targeting the MEF2-like transcription factor Smp1 by the stress-activated Hog1 mitogen-activated protein kinase. Molecular and Cellular Biology. 2003;23(1):229–237. doi: 10.1128/MCB.23.1.229-237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297(5584):1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Pelet S, Dechant R, Lee SS, van Drogen F, Peter M. An integrated image analysis platform to quantify signal transduction in single cells. Integrative biology : quantitative biosciences from nano to macro. 2012;4(10):1274–1282. doi: 10.1039/c2ib20139a. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome biology. 2006;7(10):R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomoretz A, Bush A, Yu R, Gordon A, Colman-Lerner A. Using Cell-ID 1.4 with R for microscope-based cytometry. Current protocols in molecular biology. 2008;Chapter 14:Unit 14.18. doi: 10.1002/0471142727.mb1418s84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimopoulos S, Mayer C, Rudolf F, Stelling J. Automatic Single Cell Segmentation in a Variety of Microscopy Images. Current protocols in molecular biology. 2012.
- Miyawaki A, Nagai T, Mizuno H. Mechanisms of protein fluorophore formation and engineering. Current Opinion in Chemical Biology. 2003;7(5):557–562. doi: 10.1016/s1367-5931(03)00097-8. [DOI] [PubMed] [Google Scholar]
- Hersen P, McClean MN, Mahadevan L, Ramanathan S. Signal processing by the HOG MAP kinase pathway. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(20):7165–7170. doi: 10.1073/pnas.0710770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperkok R, Ellenberg J. High-throughput fluorescence microscopy for systems biology. Nature reviews Molecular cell biology. 2006;7(9):690–696. doi: 10.1038/nrm1979. [DOI] [PubMed] [Google Scholar]