

Abstract
Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR) (MIM No.152950) is a rare autosomal dominant condition for which a causative gene has recently been identified. Mutations in the kinesin family member 11 (KIF11) gene have now been described in 16 families worldwide. This is a review of the condition based on the clinical features of 37 individuals from 22 families. This report includes nine previously unreported families and additional information for some of those reported previously. The condition arose de novo in 8/20 families (40%). The parental results were not available for two probands. The mutations were varied and include missense, nonsense, frameshift, and splice site and are distributed evenly throughout the KIF11 gene. In our cohort, 86% had microcephaly, 78% had an ocular abnormality consistent with the diagnosis, 46% had lymphoedema, 73% had mild-moderate learning difficulties, 8% had epilepsy, and 8% had a cardiac anomaly. We identified three individuals with KIF11 mutations but no clinical features of MCLMR demonstrating reduced penetrance. The variable expression of the phenotype and the presence of mildly affected individuals indicates that the prevalence may be higher than expected, and we would therefore recommend a low threshold for genetic testing.
Keywords: microcephaly, chorioretinal dysplasia, lymphoedema, KIF11, MCLMR
INTRODUCTION
Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR) (MIM No.152950) is a rare autosomal dominant condition characterised by variable expression of microcephaly, eye problems including chorioretinopathy, congenital lymphoedema of the lower limbs, and mild-to-moderate intellectual disability. It was originally described by Feingold and Bartoshesky1 who reported two unrelated patients in 1992.
It was recently reported that a significant proportion of cases of MCLMR are caused by mutations in the kinesin family member 11 (KIF11) gene.2 KIF11 encodes EG5, a homotetramer kinesin motor, likely to be important for the development and maintenance of retinal and lymphatic structures. Ostergaard et al2 in 2012 found that 15 of the 20 families tested had variants in KIF11 that were predicted to be deleterious, suggesting that a significant proportion of MCLMR cases are caused by KIF11 mutations but that the condition is genetically heterogeneous.
Herein we give a detailed report on the clinical features of 37 individuals from 22 families (9 previously unreported) with KIF11 mutations and review the literature, to further delineate this condition.
METHODS
Referral for genetic testing required a diagnosis of MCLMR by a clinical geneticist or chorioretinopathy by an ophthalmologist. Those with KIF11 mutations were selected; phenotypic data were collected by review of medical records, patient contact, and clinical photographs. The data were summarised by each collaborating clinician and forwarded to the author for review. Data on certain phenotypic characteristics, including level of intellectual disability, were not uniformly collected or standardised. Therefore the clinical judgement of the referring clinician was used.
Mutation analysis
Genomic DNA was extracted using standard protocols. Parental samples were analysed in all but two of the cases. Direct gene sequencing of all the 22 protein-coding exons and intron–exon boundaries of the KIF11 gene was performed using methods previously described.2 All old and new variants reported here are deposited in the LOVD KIF11 database http://databases.lovd.nl/shared/genes/KIF11 (using accession number NM_004523.3).
RESULTS
Twenty KIF11 variants were identified within the cohort (three unrelated families, XIII, XIV, and XV shared the same nonsense mutation) (Table 1). These 20 mutations included 4 missense, 6 nonsense, 6 frameshift, and 4 splice site mutations.
Table 1. Mutation spectrum and inheritance pattern.
| Mutation | Exon | Protein | Inheritance | |
|---|---|---|---|---|
| Ia | c.139C>T | 2 | p.(Arg47*) | De novo |
| II | c.204dup | 2 | p.(Asp69*) | Maternal |
| III | c.385G>T | 4 | p.(Glu129*) | De novo |
| IV | c.387+1G>A | 4i | Splice site | Maternal |
| Vb | c.432T>G | 5 | p.(Phe144Leu) | Paternal |
| VIb | c.699-2A>G | 6i | Splice site | De novo |
| VIIb | c.700C>T | 7 | p.(Arg234Cys) | De novo |
| VIIIb | c.704C>G | 7 | p.(Ser235Cys) | Maternal |
| IX | c.775G>T | 7 | p.(Gly259*) | Maternal |
| X | c.757_758del | 7 | p.(Glu253Argfs*4) | Not known |
| XIb | c.1039_1040delCT | 9 | p.(Leu347Glufs*8) | Paternal |
| XII | c.1129-4_1133delinsTC | 9i | Splice site | Maternal |
| XIIIb | c.1159C>T | 10 | p.(Arg387*) | Paternal |
| XIVb | c.1159C>T | 10 | p.(Arg387*) | Maternal |
| XV | c.1159C>T | 10 | p.(Arg387*) | Maternal |
| XVIb | c.1804C>T | 14 | p.(Gln602*) | Not known |
| XVIIb | c.1963_1964dupAA | 15 | p.(His656Serfs*8) | Paternal |
| XVIII | c.2267+1G>A | 17i | Splice site | De novo |
| XIXb | c.2304_2305delCA | 18 | p.(His768Glnfs*7) | De novo |
| XX | c.2808_2813delinsCA | 20 | p.(Thr937Argfs*2) | De novo |
| XXIb,c | c.2830C>T | 20 | p.(Arg944Cys) | De novo |
| XXIIb | c.3016delA | 21 | p.(Ile1006Leufs*62) | Maternal |
Exons are numbered according to NM_004523.3.
Case description in Hazan et al.26
Case description in Ostergaard et al.2
Case description in Vasudevan et al.7
Parental samples were analysed in all the families except X and XVI (a sibling was available for testing in the latter). In eight cases, the mutation was confirmed to have arisen de novo (40%). The remaining 12 were inherited (60%), with 8 (67%) maternally and 4 (33%) paternally. In three families (IV, XVI, XVII), multiple siblings were affected. There was no significant gender disproportion in probands or total individuals (Table 2).
Table 2. Clinical features of all individuals with KIF11 mutations.
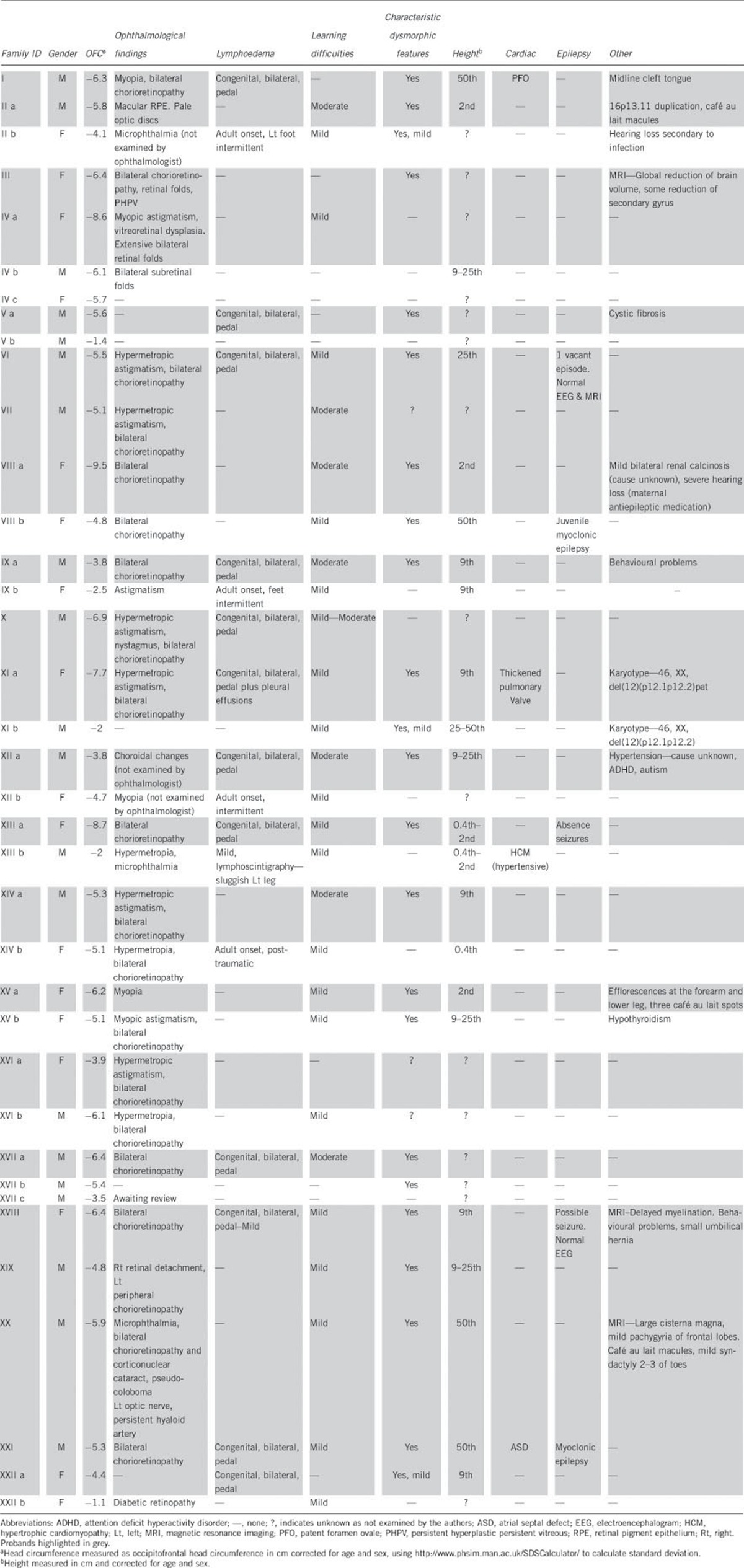
The predominant features in our cohort were microcephaly, lymphoedema, chorioretinopathy, and intellectual disability (Table 2). Of the 22 probands, all 22 (100%) had microcephaly (defined as <−3SD). The microcephaly was present at birth, with subsequent slow head growth and worsening head circumference in infancy. In the sibling/parental group, 10 out of 15 mutation-positive individuals had microcephaly, with the remaining 5 individuals at the lower end of the normal range (−1.1, −1.4, −2, −2, and −2.5 SD below the mean). Two of these had other features of MCLMR and, apart from mild learning difficulties in one case, the remaining three individuals did not have any features of the condition. Overall, 32/37 (86%) of individuals with KIF11 mutations had microcephaly (Table 3). There did not appear to be any significant correlation between degree of microcephaly in the probands and their affected parents.
Table 3. Percentage of patients in our cohort with each clinical feature.
| Clinical feature | Percentage of affected individuals |
|---|---|
| Microcephaly (<3 SD) | 86% |
| Consistent ocular abnormality | 78% |
| Chorioretinopathya | 59% |
| Hypermetropia/hypermetropic astigmatisma | 24% |
| Myopia/myopic astigmatisma | 14% |
| Retinal foldsa | 8% |
| Microphthalmiaa | 8% |
| Astigmatisma | 3% |
| Retinal detachmenta | 3% |
| PHPVa | 3% |
| Macular RPEa | 3% |
| Corticonuclear cataract, psuedocoloboma left optic nerve, persistent hyaloid arterya | 3% |
| Lymphoedema | 46% |
| Learning difficulties | 73% |
| Epilepsy | 8% |
| Cardiac anomaly | 8% |
Some individuals had overlapping ocular features.
Chorioretinopathy was also a significant feature and seen in 22/37 (59%) individuals. Four individuals had not been formally assessed by an ophthalmologist, but all had preliminary eye examinations. Other ophthalmological findings in this group included hypermetropia and hypermetropic astigmatism (nine individuals), myopia and myopic astigmatism (five individuals), bilateral retinal folds (three individuals), microphthalmia (three individuals), astigmatism (one individual), congenital unilateral retinal detachment (one individual), persistent hyperplastic primary vitreous (one individual), and macular retinal pigment epithelium (one individual), corticonuclear cataract, pseudocoloboma of the left optic nerve and persistent hyaloid artery (one individual); some individuals had more than one ocular feature (Table 2). Another individual with insulin-dependent diabetes mellitus was documented to have diabetic retinopathy. In total, 29/37 (78%) had ophthalmological features consistent with a diagnosis of MCLMR (Table 3).
Lymphoedema was present in 17 individuals. In all the probands (n=12), this was congenital, bilateral, and only affected the lower limbs (Table 2). The type of swelling was highly reminiscent of that seen in Milroy disease,3 with small, dysplastic nails, deep interphalangeal creases of the toes and swelling, particularly of the dorsum of the feet and toes. Congenital lymphoedema was not reported in the sibling/parental group; however, five individuals had adult onset oedema. In general, this was intermittent, particularly towards the end of the day. However, one individual did have residual unilateral leg swelling following a femoral fracture. Two individuals in the cohort had lymphoscintigraphy, proband XIIIa and her father XIIIb. Lymphoscintigraphy is the imaging of the lymphatic system by injecting radioactive isotope into the web spaces between the toes and quantification of uptake into the inguinal lymph nodes after 2 h. In the proband who presented with congenital lymphoedema, there was no isotope uptake after 2 h. In her father who had mild clinical signs of lymphoedema, lymphoscintigraphy showed slow uptake in one leg. Overall, 17/37 (46%) of those with KIF11 mutations had lymphoedema (Table 3).
Learning difficulties within the mild–moderate range of impairment were present in 27/37 (73%) of the cohort, this included 17 probands and 10 individuals from the sibling/parental group (Table 2).
Details of dysmorphic features or clinical photographs were available for 34/37 (92%) individuals; and the characteristic facial phenotype of upslanting palpebral fissures, broad nose with rounded tip, long philtrum with thin upper lip, and prominent, large ears were seen in the majority of individuals (Figure 1). Probands IIa and XIa had an additional chromosome abnormality (paternally inherited 16p13.11 duplication and paternally inherited 12p12.1 microdeletion, respectively), which may have contributed to the facial phenotype. The parental group did not have the same dysmorphic features seen in their children; suggesting that any facial dysmorphism may become less obvious with age. Unfortunately, we do not have childhood photographs of the parental group for comparison.
Figure 1.

(a) Clinical photographs showing facial features of the probands with upslanting palpebral fissures, broad nose with rounded tip, long philtrum with thin upper lip, prominent chin, and prominent ears. Pedigree no. (Left—Right) Row 1—I, II, III, V, VI, VIII; Row 2—IX, XI, XII, XIII, XIV, XV; and Row 3—XVII, XVIII, XIX, XXI, XXII. (b) Parents with less obvious dysmorphism. Pedigree no. (Left—Right) Row 1—II, IX, XII, XIV, XV, XVII.
Cardiac abnormalities were documented in three individuals (3/37, 8%) and included congenital thickened pulmonary valve, atrial septal defect, and patent foramen ovale. Another individual from the parental group had an acquired hypertrophic cardiomyopathy, possibly secondary to hypertension. Epilepsy was diagnosed in three individuals (3/37, 8%), two with myoclonic epilepsy, and an individual with absence of seizures. A magnetic resonance imaging (MRI) report was only available in one of these patients (XIII), and this was normal. Two more probands were reported to have possible seizure activity but with normal electroencephalograms. Within the cohort, eight additional probands underwent MRI to investigate the microcephaly (pedigrees I, III, VI, VIII, X, XVIII, XIX, XX). In five cases, there was normal brain parenchyma (three of these had reduced cerebral volumes); one child had delayed myelination; one had some reduction in secondary gyrus; and the remaining had a large cisterna magna and possible mild frontal pachygyria.
In all individuals in whom height had been documented, this fell within the normal range. One individual also suffered from cystic fibrosis, another individual was found to have a midline cleft tongue. Other features seen include mild, bilateral renal calcinosis of unknown cause, severe hearing loss thought to be secondary to antenatal exposure to anti-epileptic medication in one individual (VIIIa) whose mother had epilepsy and a KIF11 mutation, hearing loss in another individual thought to be secondary to infection, and umbilical hernia and hypospadias in two more individuals. Three individuals had behavioural problems.
DISCUSSION
MCLMR presents with a variable spectrum of central nervous system, lymphatic and ocular developmental anomalies. Phenotypic abnormalities are described in 37 individuals with mutations in KIF11. Three of these individuals (8%) were found to carry disease-causing mutations but were clinically unaffected. Of the 34 clinically affected individuals with KIF11 mutations, microcephaly, chorioretinopathy, and learning difficulties were the most consistent findings, although the presence of lymphoedema tended to alert the clinician to the diagnosis at an earlier age.
Microcephaly is usually defined as head circumference of 3 SDs below the mean when adjusted for age and sex (<−3SD).4 Primary microcephaly is present at birth and is a static developmental anomaly, whereas secondary microcephaly develops postnatally and indicates a progressive neurodegenerative condition.5 Pathogenesis is heterogeneous and both may have genetic or environmental aetiology.6 The microcephaly seen in MCLMR is primary,7 and although the majority of individuals (86%) with KIF11 mutations have microcephaly, the clinical spectrum is extremely variable, ranging from −9.5 SDs below the mean to some individuals with head circumference in the normal range (−1.1 SD).
Microcephaly is strongly associated with intellectual disability.8 Those with learning difficulties in our cohort were in the mild-to-moderate range. Although individuals with microcephaly, chorioretinal dysplasia, and severe learning difficulties have been reported,9 we would suggest that this is not typical in those with KIF11 mutations. It has been suggested that abnormal findings of brain anatomy, including cerebral atrophy, cortical dysplasia, myelination delay, and white matter hypoplasia, are more significantly correlated with poor developmental performance than the severity of microcephaly.10 Brain abnormalities, including pachymicrogyria11 and lissencephaly,12, 13 have been reported in association with MCLMR. It is thought that the developmental anomalies in the retina could be analogous to central nervous system anomalies. One individual (XX), was reported to have mild pachygyria of the frontal lobes. Interestingly, this individual had a similar ocular phenotype to the case previously reported,11 although our patient only had mild learning difficulties.
There is also an autosomal recessive form of microcephaly and chorioretinopathy with intellectual disability, which is caused by homozygous or biallelic mutations in the TUBGCP6 gene.14 The features are similar to MCLMR; however, this condition does not appear to be associated with lymphoedema (Dr Puffenberger, pers. comm.), the intellectual disability is more severe and polymicrogyria is seen on the brain MRI.
Epilepsy is a common feature in individuals with microcephaly of all causes.15 In the KIF11 cohort, 3 (8%) of individuals had epilepsy, and two more individuals had a history suggestive of possible seizure activity. Therefore epilepsy; in particular myoclonic epilepsy, could be a minor feature of this condition.
The ocular features of MCLMR have been described as choroidal atrophy and dysplasia, which are thought to be non-progressive. The typical fundus features are of focal areas of lacunar atrophy of the choroid and retina.11 Other previously reported features include microphthalmia, myopic and hypermetropic astigmatism, and persistent hyperplastic primary vitreous,9, 16, 17 concordant with our observations.
In 1981, Jarmas et al,18 reported a family with microcephaly, microphthalmia, bilateral falciform retinal folds, and blindness, and subsequently, retinal folds were described in another family with microcephaly, lymphoedema, and microphthalmia.19 Bilateral retinal folds were seen in two families within our cohort, as was microphthalmia, suggesting that some cases of Jarmas syndrome could be allelic but that there is likely to be genetic heterogeneity of this condition.
A characteristic facial phenotype with upslanting palpebral fissures, broad nose with rounded tip, long philtrum with thin upper lip, and prominent ears has been well documented,7, 20 and this was consistent with the majority of our probands but became less prominent with age. Lymphoedema is not seen in all individuals with KIF11 mutations. It is generally congenital, bilateral, and confined to the dorsa of the feet (Figure 2a) and resembles the lymphoedema seen in Milroy disease.3, 21 It can be seen on antenatal ultrasound in the third trimester (Figure 2b), and this could be a diagnostic clue in those with a family history. However, there is likely to be underlying lymphatic insufficiency in those who do not present with congenital lymphoedema, as a proportion of individuals had adult onset, intermittent lymphoedema.
Figure 2.

(a) Lymphoedema is typically congenital, bilateral, and confined to the dorsa of the feet. (b) Lymphoedema of the feet detected by antenatal ultrasound scan. (c) Fundal images demonstrating characteristic changes of chorioretinal dysplasia.
Cardiac defects have been reported in individuals with MCLMR,22 and were seen in a small proportion of our cohort. However, cardiac anomalies are a relatively common congenital abnormality within the general population,23 therefore it would be difficult to conclude definitively the association with MCLMR. Short stature has also been reported in association with MCLMR;24, 25 however, this was not a feature in any of our patients, and we believe that this is not related to this condition. Recently a midline cleft tongue was reported26 and this individual (I) has been included in our cohort, although this feature was not present in any other individuals. Midline cleft tongue is a rare anomaly; it is generally associated with an underlying syndromic diagnosis, most commonly orofaciodigital syndrome type 1 (OFD1), which is a disorder of the cilia. OFD1 is characterised by malformations of the face, oral cavity, and digits.27 KIF11 encodes a kinesin which is not a ciliary protein, and therefore we would not expect this feature to be associated with MCLMR. It may be that this patient had more than one diagnosis, particularly in view of the parental consanguinity.
The combination of microcephaly and lymphoedema has been reported in a variety of conditions, including various chromosomal microdeletions (19p13.3, 3q21.1-q21.3, 5q14.3, 22q13 and 8q24), carbohydrate-deficient glycoprotein syndrome type 1a, progressive encephalopathy–oedema–hypsarrhythmia–optic atrophy, Aicardi–Goutieres, and some other rare genetic disorders.28 However, the phenotype in MCLMR is quite specific and, in general, these other conditions have other distinctive features.
There appears to be very little genotype–phenotype correlation. Three unrelated families had the same mutation (c.1159C>T, p.(Arg387*)) in exon 10 but with evidence of inter- and intra-familial variation. We observed no significant phenotypic differences between individuals grouped by mutation type, although both cases with myoclonic epilepsy had missense mutations (c.704C>G, p.(Ser235Cys) and c.2830C>T, p.(Arg944Cys)), the sample size is too small for accurate interpretation. There were two families with bilateral retinal folds in the cohort (III, IV). In both the families, the mutation was at the terminal end of exon 4 separated only by three nucleotides (c.385G>T, p.(Glu129*) and c.387+1G>A, splice mutation), which could suggest that this feature is particular to mutations in this region.
There was no significant correlation of clinical features in the three families with the same mutation to indicate genotype–phenotype association; larger studies would be required to determine this conclusively. There was some intra-familial correlation in the presence/absence of the main clinical features (microcephaly, lymphoedema, chorioretinal dysplasia, and intellectual disability), in particular pedigrees IV, VIII, XIV, XV, and XVI. Interestingly, pedigree XXII (c.3016delA, p.(Ile1006Leufs*62)) appeared to have a milder phenotype, in both the proband and her affected mother. This frameshift mutation is a single-base deletion in the second-to-last exon and is predicted to result in substitution of the terminal 50 residues of the 1056 amino-acid wild-type protein and extension of the reading frame by a further 12 residues.2 Furthermore, the parents in pedigrees V and XI, who both carry a pathogenic KIF11 mutation did not show any clinical features of MCLMR, demonstrating reduced penetrance. This suggests that the prevalence of this condition is likely to be higher than expected. We would therefore recommend a low threshold for consideration of genetic testing. Genetic testing should be considered in individuals with isolated microcephaly (particularly if clearly dominant), congenital lymphoedema but with no mutation in VEGFR3 (the gene associated with Milroy disease), or chorioretinopathy. Given the incomplete penetrance, it would also be prudent to perform genetic testing in the parents of apparently sporadic or isolated cases.
In conclusion, we have explored the relationship between KIF11 genotype and some of the major phenotypic characteristics of MCLMR. We have shown that there is reduced penetrance and variable expression of the phenotype. Our sample size was relatively small; therefore analysis of specific genotype–phenotype relationships using a larger set of MCLMR cases, and to compare the features with those without KIF11 mutations, would be valuable to further our knowledge of this condition.
Acknowledgments
We thank the families for agreeing to the publication of their clinical details and photographs. We also thank the British Heart Foundation (PG/10/58/28477 to PO) and (FS/11/40/28739 to KG), Sheffield Children's Hospital Charity, and the NIHR Biomedical Research Centre at the Moorfields Eye Hospital and the Institute of Ophthalmology for supporting the research.
The authors declare no conflict of interest.
Footnotes
ETHICS
The Ethical approval for this work was obtained from the South West London Research Ethics Committee (REC Ref: 05/Q0803/257) and the North Sheffield Research Ethics Committee (REC Ref: 05/Q2308/156).
References
- Feingold M, Bartoshesky Λ. Microcephaly, lymphoedema, and chorioretinal dysplasia: a distinct syndrome. Am J Med Genet. 1992;43:1030–1031. doi: 10.1002/ajmg.1320430623. [DOI] [PubMed] [Google Scholar]
- Ostergaard P, Simpson MA, Mendola A, et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphoedema and chorioretinopathy. Am J Hum Genet. 2012;90:56–62. doi: 10.1016/j.ajhg.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell FC, Gordon K, Brice G, et al. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clin Genet. 2013;84:303–314. doi: 10.1111/cge.12173. [DOI] [PubMed] [Google Scholar]
- Baraitser M.Microcephaly; in The genetics of Neurological Disorders2nd edn.Oxford: Oxford Medical Publications; 1990vol 1826–33. [Google Scholar]
- Woods CG, Parker A. Investigating microcephaly. Arch Dis Child. 2013;0:1–7. doi: 10.1136/archdischild-2012-302882. [DOI] [PubMed] [Google Scholar]
- Abuelo D. Microcephaly syndromes. Semin Pediatr Neurol. 2007;14:118–127. doi: 10.1016/j.spen.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Vasudevan PC, Garcia-Minaur S, Botella MP, Perez-Aytes A, Shannon NL, Quarrell OWJ. Microcephaly-lymphoedema-chorioretinal dysplasia: three cases to delineate the facial phenotype and review of the literature. Clin Dysmorphol. 2005;14:109–116. [PubMed] [Google Scholar]
- Dolk H. The predictive value of microcephaly during the first year of life for mental retardation at seven years. Dev Med Child Neurol. 1991;33:974–983. doi: 10.1111/j.1469-8749.1991.tb14813.x. [DOI] [PubMed] [Google Scholar]
- Trzupek KM, Falk RE, Demer JL, Weleber RG. Microcephaly with chorioretinopathy in a brother–sister pair: evidence for germ line mosacism and further delineation of the ocular phenotype. Am J Med Genet A. 2007;143A:1218–1222. doi: 10.1002/ajmg.a.31717. [DOI] [PubMed] [Google Scholar]
- Custer DA, Vezina G, Vaught DR, et al. Neurodevelopmental and neuroimaging correlates in nonsyndromal microcephalic children. J Dev Behav Pediatr. 2000;21:12–18. doi: 10.1097/00004703-200002000-00003. [DOI] [PubMed] [Google Scholar]
- Pastora N, Peralta J, Canal-Fontcuberta I, et al. Microcephaly-lymphedema-chorioretinal dysplasia associated with pachymicrogyria and atrophy of the cerebellar vermis: an integration of brain-ocular migration disorders. Ophthalmic Genet. 2012;33:116–118. doi: 10.3109/13816810.2011.626012. [DOI] [PubMed] [Google Scholar]
- Lee BJ, Kim JH, Yu YS. Lissencephaly and mild cerebellar vermis hypoplasia in a case of microcephaly and chorioretinal dysplasia. Ophthalmic Genet. 2010;31:89–93. doi: 10.3109/13816811003620509. [DOI] [PubMed] [Google Scholar]
- Casteels I, Devriendt K, Leys A, et al. Autosomal dominant microcephaly-lymphoedema-chorioretinal dysplasia syndrome. Br J Ophthalmol. 2001;85:496. doi: 10.1136/bjo.85.4.496d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenberger EG, Jinks RN, Sougnez C, et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Salam GM, Halasz AA, Czeizel AE. Association of epilepsy with different groups of microcephaly. Dev Med Child Neurol. 2000;42:760–767. doi: 10.1017/s0012162200001419. [DOI] [PubMed] [Google Scholar]
- Hordijk R, van de Logt F, Houtman WA, van Essen AJ. Chorioretinal dysplasia-microcephaly-mental retardation syndrome: another family with autosomal dominant inheritance. Genet Couns. 1996;7:113–122. [PubMed] [Google Scholar]
- Atchaneeyasakul LO, Linck L, Weleber RG. Microcephaly with chorioretinal degeneration. Ophthalmic Genet. 1998;19:39–48. doi: 10.1076/opge.19.1.39.2178. [DOI] [PubMed] [Google Scholar]
- Jarmas AL, Weaver DD, Ellis FD, Davis A. Microcephaly, microphthalmia, falciform retinal folds, and blindness. A new syndrome. Am J Dis Child. 1981;135:930–933. doi: 10.1001/archpedi.1981.02130340036013. [DOI] [PubMed] [Google Scholar]
- Young ID, Fielder AR, Simpson K. Microcephaly, microphthalmos and retinal folds: report of a family. J Med Genet. 1987;24:172–174. doi: 10.1136/jmg.24.3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limwongse C, Wyszynski RE, Dickerman LH, Robin NH. Microcephaly-lymphoedema-chorioretinal dysplasia: a unique genetic syndrome with variable expression and possible characteristic facial appearance. Am J Med Genet. 1999;86:215–218. [PubMed] [Google Scholar]
- Milroy WF. An undescribed variety of heritable oedema. NY Med J. 1892;56:505–508. [Google Scholar]
- Eventov-Friedman S, Singer A, Shinwell ES. Microcephaly, lymphoedema, chorioretinopathy and atrial septal defect: a case report and review of the literature. Acta Paediatr. 2009;98:758–759. doi: 10.1111/j.1651-2227.2008.01161.x. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Ma X, Jia B, Huang G. Prevalence of congenital heart disease at live birth: an accurate assessment by echocardiographic screening. Acta Paediatr. 2013;102:397–402. doi: 10.1111/apa.12170. [DOI] [PubMed] [Google Scholar]
- McKusick VA, Stauffer M, Knox DL, Clark DB. Chorioretinopathy with hereditary microcephaly. Arch Ophthal. 1966;75:597–600. doi: 10.1001/archopht.1966.00970050599003. [DOI] [PubMed] [Google Scholar]
- Strenge S, Froster UG. Microcephaly-lymphoedema syndrome: report of a family with short stature as an additional feature. Am J Med Genet. 1998;80:506–509. doi: 10.1002/(sici)1096-8628(19981228)80:5<506::aid-ajmg13>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Hazan F, Ostergaard P, Ozturk T, et al. A novel KIF11 mutation in a Turkish patient with microcephaly, lymphoedema, and chorioretinal dysplasia from a consanguineous family. Am J Med Genet A. 2012;158A:1686–1689. doi: 10.1002/ajmg.a.35371. [DOI] [PubMed] [Google Scholar]
- Prattichizzo C, Macca M, Novelli V, et al. Mutational spectrum of the oral-facial-digital type I syndrome: a study on a large collection of patients. Hum. Mutat. 2008;29:1237–1246. doi: 10.1002/humu.20792. [DOI] [PubMed] [Google Scholar]
- Winter RM, Baraitser M. London Medical Databases: Winter Baraitser Dysmorphology Database Version 1.0.24. Oxford: Oxford University Press; 2007. [Google Scholar]