

Abstract
Hepatitis C virus (HCV) is a major cause of liver disease. Therapeutic options are limited and preventive strategies are absent. Entry is the first step of infection and requires the cooperative interaction of several host cell factors. Using a functional RNAi kinase screen we identified epidermal growth factor receptor and ephrin receptor A2 as host co-factors for HCV entry. Blocking of kinase function by approved inhibitors broadly inhibited HCV infection of all major HCV genotypes and viral escape variants in cell culture and an animal model in vivo. Receptor tyrosine kinases (RTKs) mediate HCV entry by regulating CD81-claudin-1 co-receptor associations and membrane fusion. These results identify RTKs as novel HCV entry co-factors and uncover that kinase inhibitors have significant antiviral activity. Inhibition of RTK function may constitute a novel approach for prevention and treatment of HCV infection.
Keywords: Animals; Antigens, CD; physiology; Antigens, CD81; Antiviral Agents; pharmacology; Base Sequence; Cell Line; Claudin-1; Hepacivirus; drug effects; physiology; Hepatitis C; physiopathology; prevention & control; therapy; virology; Host-Pathogen Interactions; physiology; Humans; Ligands; Membrane Proteins; physiology; Mice; Protein Kinase Inhibitors; pharmacology; Quinazolines; pharmacology; RNA Interference; RNA, Small Interfering; genetics; Receptor, EphA2; antagonists & inhibitors; genetics; physiology; Receptor, Epidermal Growth Factor; antagonists & inhibitors; genetics; physiology; Virus Internalization; drug effects
INTRODUCTION
Hepatitis C virus (HCV) is a major cause of liver cirrhosis and hepatocellular carcinoma. Current antiviral treatment is limited by resistance, toxicity and high costs [1]. Although the clinical development of novel antiviral substances targeting HCV protein processing has been shown to improve virological response, toxicity and resistance remain major challenges [2]. Thus, novel antiviral preventive and therapeutic strategies are urgently needed. Since HCV entry is required for initiation, dissemination and maintenance of viral infection, it is a promising target for antiviral therapy [3,4].
HCV entry is a multistep process involving several attachment and entry factors [5]. Attachment of the virus to the target cell is mediated through binding of HCV envelope glycoproteins to glycosaminoglycans [6]. HCV is internalized in a clathrin-dependent endocytic process requiring CD81 [7], scavenger receptor type B class I (SR-BI) [8], claudin-1 (CLDN1) [9] and occludin (OCLN) [10]. The impact of other host factors for HCV entry is poorly understood.
RESULTS
EGFR and EphA2 are host co-factors for HCV entry
Using a siRNA screen we identified a network of kinases with functional impact on HCV entry (Supplementary results, Supplementary Figs. 1 and 2). To study the relevance of the identified kinases on the HCV life cycle, we further validated and characterized the functional impact of epidermal growth factor receptor (EGFR) and ephrin receptor A2 (EphA2) on HCV entry. We focused on these kinases because they (i) are key components in the identified networks (Supplementary Fig. 2c), (ii) are highly expressed in human liver (Supplementary Table 2) and (iii) their kinase function is inhibited by clinically approved protein kinase inhibitors (PKIs) [11–13] allowing us to explore the potential of these molecules as therapeutic targets.
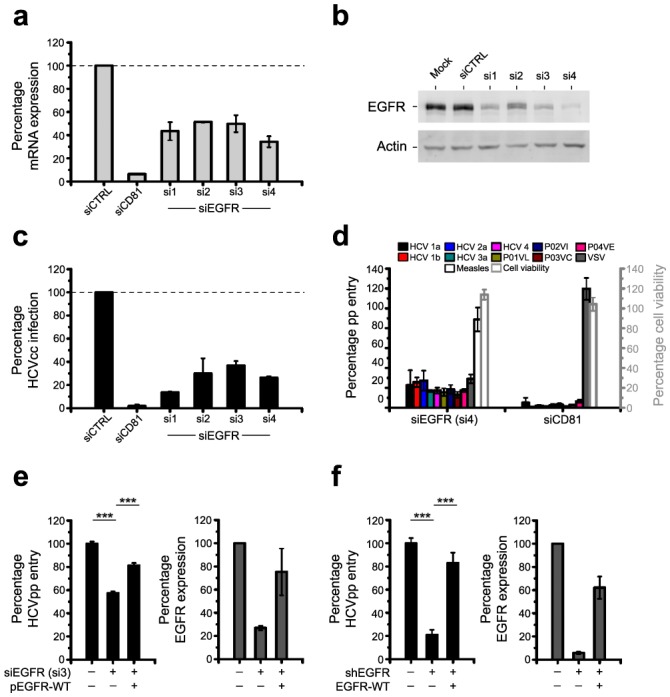
Using individual siRNAs we first confirmed that silencing of mRNAs reduced EGFR and EphA2 mRNA and protein expression (Fig. 1a,b and Supplementary Fig. 3a,b). Inhibition of infection with cell culture-derived HCV (HCVcc) in silenced cells demonstrated that both EGFR and EphA2 are important for initiation of a productive infection (Fig. 1c and Supplementary Fig. 3c). Silencing of kinase expression inhibited the entry of HCV pseudoparticles (HCVpp) derived from major genotypes including highly diverse HCV strains [14] (Fig. 1d and Supplementary Fig. 3d). Effects of silencing of endogenous EGFR or EphA2 on HCV infection were rescued by RNAi-resistant ectopic expression of wild-type EGFR or EphA2 (Fig. 1e,f and Supplementary Fig. 3e,f). These results largely excluded the possibility of off-target effects causing the observed phenotype. Furthermore, silencing and rescue experiments using well characterized lentiviral vectors expressing EGFR-specific shRNA unambiguously and specifically demonstrated a key role for EGFR in HCV entry into primary human hepatocytes (PHH) (Fig. 1f). The functional impact of EGFR as a co-factor for HCV entry was further confirmed by expressing human EGFR in mouse hepatoma cell lines engineered to express the four human entry factors CD81, SR-BI, CLDN1 and OCLN (AML12 4R, Supplementary Fig. 4). Cell surface expression of human EGFR in AML12 4R cells markedly increased HCVpp entry into mouse cells (Supplementary Fig. 4).
Fig. 1. EGFR is a co-factor for HCV entry.
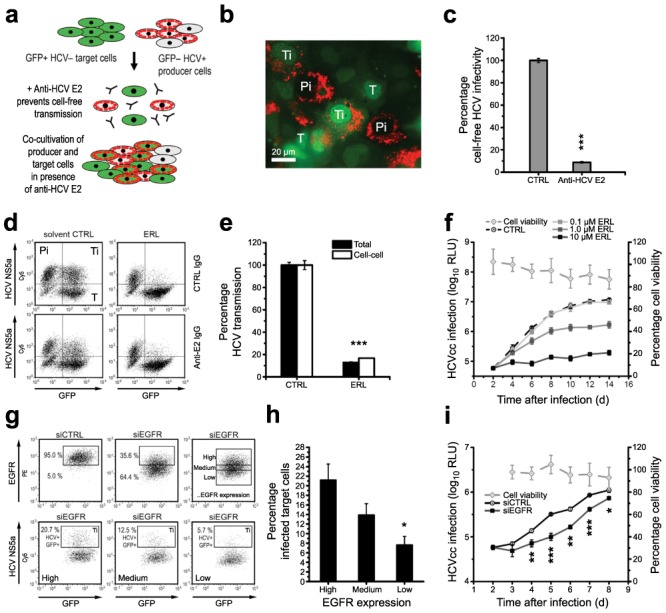
(a,b) Silencing EGFR expression in HCV-permissive cells. (a) EGFR mRNA (qRT-PCR analysis) and (b) protein expression (Western blot) in Huh7.5.1 cells transfected with EGFR-specific individual siRNAs (si1–4). Silencing of CD81 mRNA expression by CD81-specific siRNA served as control. EGFR mRNA (relative to GAPDH mRNA) and protein expression compared to cells transfected with control siRNA (siCTRL) is shown. (c,d) Inhibition of HCV infection and entry in cells with silenced EGFR expression. (c) HCVcc infection in Huh7.5.1 cells transfected with individual siRNAs shown in panels a,b. siCTRL and CD81-specific siRNA served as internal controls. Data are expressed as percentage HCVcc infection relative to siCTRL-transfected cells. (d) Entry of HCVpp containing envelope glycoproteins of various isolates14],39] in Huh7.5.1 cells transfected with si4. VSV and measles virus pp entry or cells transfected with CD81 siRNA served as controls. Data are expressed as percentage pp entry relative to siCTRL-transfected cells. (e,f) Rescue of HCV entry in cells with silenced EGFR expression by exogenous EGFR. (e) HCVpp entry and EGFR protein expression in Huh7.5.1 cells co-transfected with EGFR-specific individual si3 and a cDNA encoding for RNAi-resistant EGFR (pEGFR-WT)40]. (f) HCVpp entry and EGFR protein expression in PHH co-transduced with lentiviruses expressing shEGFR and wild-type EGFR cDNA (EGFR-WT)40]. Protein expression was quantified using Image Quant analysis of Western blots. Data are expressed as percentage HCVpp entry relative to CTRL cells or as percentage EGFR expression normalized for β-actin expression. *** P<0.0005.
RTK kinase function is important for HCV entry
The functional relevance of the identified kinases and their kinase function for HCV entry and infection was further studied using PKIs. Erlotinib (an EGFR inhibitor) and Dasatinib (an EphA2 inhibitor) inhibited HCV infection in a dose-dependent manner whilst having no detectable effect on replication of the corresponding subgenomic replicon (Fig. 2a–c and Supplementary Fig. 5a–c). The formally calculated IC50 values for Erlotinib and Dasatinib to inhibit HCVpp entry (Erlotinib 0.45 ± 0.09 μM, Dasatinib 0.53 ± 0.02 μM) and HCVcc infection (Erlotinib 0.53 ± 0.08 μM, Dasatinib 0.50 ± 0.30 μM) of Huh7.5.1 cells were comparable (Fig. 2a and Supplementary Fig. 5a,b). These data suggest that RTKs inhibited by Erlotinib and Dasatinib predominantly play a role during the HCV entry process.
Fig. 2. Inhibition of EGFR activation by kinase inhibitors reduces HCV entry and infection.
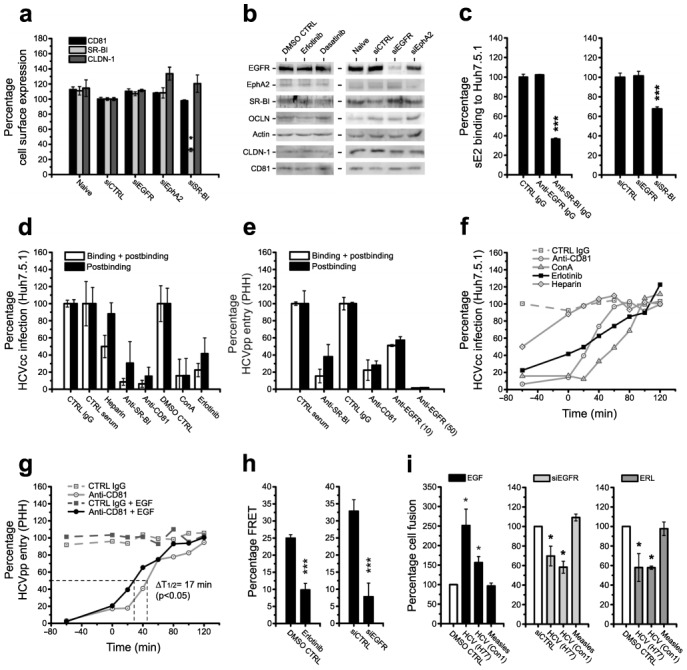
(a) Effect of Erlotinib on HCV entry and infection in Huh7.5.1 cells. HCVcc (Luc-Jc1; J6-JFH1) infection and HCVpp (J6) entry in Huh7.5.1 cells pre-incubated with indicated concentrations of Erlotinib are shown. Data are expressed as percentage HCVcc infection or HCVpp entry relative to solvent-treated control cells (means ± SEM). (b) Effect of Erlotinib on HCV replication. Northern blot analysis of HCV RNA and GAPDH mRNA in Huh7.5 cells electroporated with RNA from subgenomic HCV JFH1 replicon and incubated with solvent CTRL, HCV protease inhibitor BILN-2061 or Erlotinib (ERL) is shown. Analysis of HCV RNA in cells transfected with replication incompetent HCV RNA (GND, Δ) served as negative control. (c) Effect of Erlotinib on HCVpp and MLVpp entry in HepG2-CD81 cells. Pseudovirus entry into non-polarized and polarized HepG2-CD81 cells (generated as described15]) pre-incubated with Erlotinib (10 μM) is shown. (d) Effect of Erlotinib on HCVpp entry into PHH. HCVpp entry in PHH pre-incubated with Erlotinib is shown relative to entry into solvent-treated control cells. IC50 value is expressed as median of three independent experiments ± standard error of the median. (e,f) Effect of PKIs on HCV entry and infection in PHH and Huh7.5.1 cells. (e) HCVpp entry into PHH and (f) HCVcc infection in Huh7.5.1 pre-incubated with 1 μM Erlotinib (ERL), Gefitinib (GEF), Lapatinib (LAP), Blebbistatin (BLEB) or Wortmannin (WORT) is shown. Cell viability was assessed using MTT assay.
To confirm that the inhibitors reduced HCV entry into cells more closely resembling HCV target cells in vivo, we investigated their effect(s) on HCVpp entry into polarized HepG2-CD81 cells15] and PHH. PKIs markedly and significantly (P<0.005) inhibited HCVpp entry into polarized HepG2-CD81 cells (Fig. 2c and Supplementary Fig. 5d) and PHH (Fig. 2d and Supplementary Fig. 5e). Similar results were obtained for infection of PHH with HCVcc and serum-derived HCV (Fig. 3h,i and Supplementary Fig. 5f) confirming the relevance of the kinases as auxiliary host cell co-factors in models which most closely mimic in vivo infection.
Fig. 3. Modulation of HCV entry by EGFR ligands and an EGFR-specific antibody.
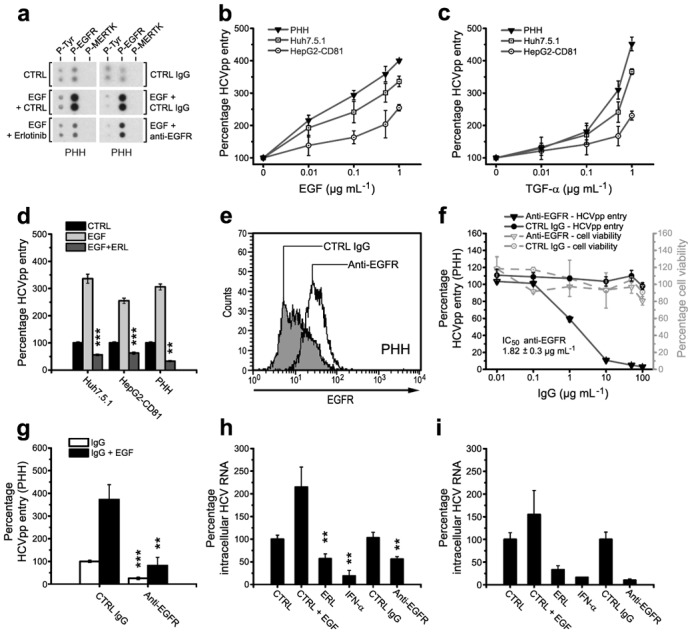
(a) Modulation of EGFR phosphorylation by EGF, Erlotinib and EGFR-specific antibody. EGFR activation was assessed in PHH incubated with the indicated compounds using the Human Phospho-RTK Array Kit. Phospho-tyrosine (P-Tyr) and phosphorylation of an unrelated kinase (MERTK) served as internal positive and negative controls. (b,c) Effect of EGFR ligands on HCVpp entry. HCVpp entry (HCV-J) into serum-starved Huh7.5.1, polarized HepG2-CD81 and PHH in the presence of EGF (b) and TGF-α (c) is shown. (d) Reversion of EGF mediated-enhancement of HCVpp entry by Erlotinib. HCVpp entry into Huh7.5.1, polarized HepG2-CD81 and PHH incubated with EGF or EGF and Erlotinib is shown. (e) Flow cytometric analysis of non-permeabilized PHH binding EGFR-specific or control monoclonal antibody (mAb). (f) Inhibition of HCV entry by EGFR-specific mAb. HCVpp entry into PHH pre-incubated with EGFR-specific or control mAb is shown. Viability of cells was assessed using MTT assay. IC50 value is expressed as median of three independent experiments ± standard error of the median. (g) Reversion of EGF-induced enhancement of HCV entry by an EGFR-specific antibody. HCVpp entry into PHH pre-incubated with EGF and EGFR-specific mAb. (h,i) Effect of EGF, EGFR-specific mAb and Erlotinib on HCV infection in PHH. Intracellular HCV RNA in PHH infected with (h) HCVcc or (i) serum-derived HCV (one representative experiment) was measured by qRT-PCR. **, P<0.005; ***, P<0.0005. Unless otherwise indicated: EGFR-specific and control mAbs: 10 μg mL−1, EGF: 1 μg mL−1, ERL= Erlotinib: 10 μM.
A specific effect of Erlotinib on EGFR-mediated HCV entry was further confirmed by the investigation of additional EGFR inhibitors: EGFR-inhibitors Gefitinib and Lapatinib markedly inhibited HCVpp entry and HCVcc infection in PHH and Huh7.5.1 similar to Erlotinib (Fig. 2e,f). The specific action of PKIs on RTKs as HCV entry factors was further corroborated by absent effects on MLV and measles virus (Fig. 2c and Supplementary Fig. 10) and silencing/rescue experiments: PKIs specifically reversed rescue of HCV entry when added to silenced Huh7.5.1 cells expressing EGFR and EphA2 in trans (data not shown). Taken together, these results suggest that the RTK kinase function is important for HCV entry.
RTK-specific ligands and antibodies modulate HCV entry
To investigate the functional role of RTK ligand binding domains for viral entry, we assessed virus entry in the presence of RTK-specific ligands and antibodies. Epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α) are well characterized EGFR ligands, where ligand binding promotes receptor homodimerization and subsequent phosphorylation of the intracytoplasmic kinase domain16]. To confirm the biological activity of EGFR-specific reagents in the target cells of our HCV model systems, we first studied their effect on EGFR phosphorylation. Pre-incubation of PHH with EGF markedly increased basal levels of EGFR phosphorylation (Fig. 3a, upper and middle panel). In contrast, EGF had no effect on the phosphorylation of an unrelated kinase. EGF-induced enhancement of basal EGFR phosphorylation was markedly inhibited by Erlotinib and an EGFR-specific antibody (Fig. 3a, lower panel) demonstrating their specific effect(s) on EGFR phosphorylation and activation.
To elucidate the role of the EGFR ligand domain, we assessed the effect of EGFR ligands on HCV entry. Binding of EGF and TGF-α markedly enhanced entry of HCVpp into serum-starved Huh7.5.1, HepG2-CD81 and PHH (Fig. 3b,c) whereas TGF-β had no effect (data not shown). These data suggest that direct interaction of EGF or TGF-α with the EGFR ligand-binding domain modulates HCV entry. The higher affinity of EGF for EGFR on hepatocytes17] may explain the differences between EGF and TGF-α in enhancing HCVpp entry. Erlotinib at doses used in HCV entry inhibition experiments reversed the enhancing effect(s) of EGF (Fig. 3d) and TGF-α (data not shown) on HCV entry. These data confirm that Erlotinib inhibits HCV entry by modulating EGFR activity.
We screened a large panel of EGFR-specific antibodies and identified a monoclonal human EGFR-specific antibody that bound to PHH (Fig. 3e) and dose-dependently inhibited HCV entry (Fig. 3f) with an IC50 value of 1.82 ± 0.3 μg mL−1. The antibody inhibited EGFR phosphorylation (Fig. 3a) and reversed the EGF-induced enhancement of HCV entry (Fig. 3g). Ligand-induced enhancement and EGFR-specific antibody-mediated inhibition of HCV entry were also observed for infection of PHH with HCVcc (Fig. 3h) and serum-derived HCV (Fig. 3i). Taken together, these results suggest that the EGFR ligand domain plays a crucial role in HCV entry. Similarly, EphA2 ligands and EphA2-specific antibodies modulated HCV entry confirming a functional relevance of the EphA2 ligand-binding domain for HCV entry (Supplementary results and Supplementary Fig. 6).
RTKs promote CD81-CLDN1 associations and membrane fusion
To understand the mechanistic role of EGFR and EphA2 in HCV entry, we first investigated whether the RTKs regulate HCV entry factor expression. Silencing RTK expression with specific siRNAs or inhibiting RTK function with PKIs had no significant effect on HCV entry factor expression (Fig. 4a,b) suggesting that RTKs do not mediate entry by modulating expression levels of the known human HCV entry factors.
Fig. 4. EGFR mediates HCV entry at postbinding steps by promoting CD81-CLDN1 co-receptor interactions and membrane fusion.
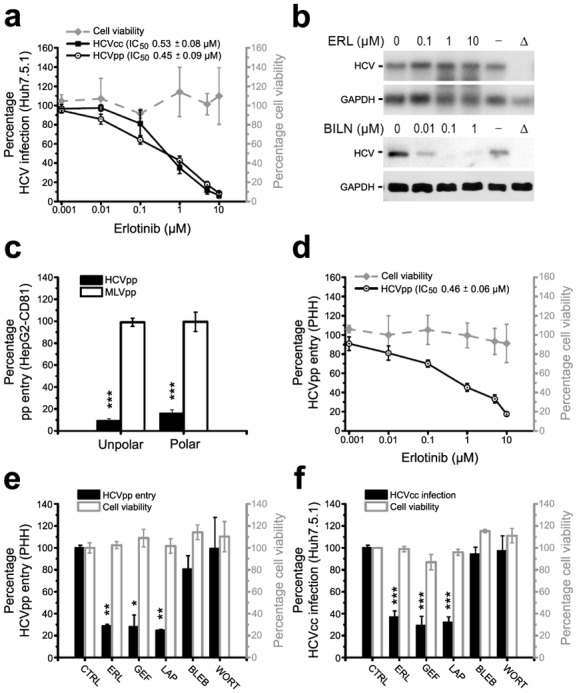
(a,b) HCV entry factor expression after RTK silencing or PKI treatment. (a) Cell surface expression of entry factors in EGFR or EphA2-silenced Huh7.5.1 cells assessed by flow cytometry. SR-BI silencing served as positive control. (b) Western blot analysis of HCV entry factor expression in PKI- or siRNA-treated Huh7.5.1 cells. (c–e) Effect of Erlotinib and EGFR-specific mAb on HCV binding and postbinding steps. (c) Flow cytometric analysis of HCV glycoprotein sE2-binding to Huh7.5.1 cells incubated with EGFR-specific mAb or transfected with siEGFR. SR-BI-specific reagents served as positive controls. (d) HCVcc infection of Huh7.5.1 cells and (e) HCVpp entry into PHH after inhibition of binding and postbinding steps by the indicated compounds (EGFR-specific mAbs: 10 and 50 μg mL−1) (f,g) Effect of Erlotinib and EGF on HCV entry kinetics. Time-course of HCVcc infection of Huh7.5.1 cells following incubation with (f) Erlotinib or indicated compounds or (g) EGF at different time-points during infection (Supplementary Methods). (h) Effect of Erlotinib and EGFR silencing on CD81-CLDN1 association(s). FRET of CD81-CLDN1 co-receptor associations in HepG2-CD81 cells incubated with Erlotinib or EGFR-specific siRNA (means ± SEM). (i) Role of EGFR in viral membrane fusion. Viral glycoprotein-dependent fusion of 293T with Huh7 cells incubated with EGF, Erlotinib or EGFR-specific siRNA was assessed as described25]. *, P<0.05; ***, P<0.0005. Unless otherwise indicated: EGFR-specific and control mAb: 10 μg mL−1, EGF: 1 μg mL−1, Erlotinib: 10 μM.
Next, we aimed to fine-map the entry step(s) affected by the RTKs. Viral attachment is the first step of viral entry. To ascertain whether PKI-inhibition of RTK function modulates HCV binding we used a surrogate model that measures association of the recombinant soluble form of HCV envelope glycoprotein E2 with cells [18]. RTK-specific antibodies or silencing RTK expression by siRNAs had no significant effect on E2 binding to target cells, whereas pre-incubation with SR-BI-specific antibodies or silencing SR-BI expression markedly reduced E2 binding (Fig. 4c and Supplementary Fig. 7a). Furthermore, in contrast to CD81 [19] and SR-BI [19], RTKs did not confer cellular E2 binding when expressed on the cell surface of CHO cells (data not shown). These data suggest that (i) RTKs are not required for HCV binding and that (ii) direct E2-RTK binding is not required for RTK-mediated HCV entry.
Following viral envelope binding, HCV enters its target cell in a multistep temporal process. To identify the time in which the PKIs exert their effect(s), we used a well characterized HCV binding/postbinding assay [19–21]. In contrast to heparin (an inhibitor of HCV binding) but similarly to CD81- and SR-BI-specific antibodies and Concanamycin A (ConA — an inhibitor of endocytosis) PKIs inhibited HCVcc infection when added after virus binding to target cells (Fig. 4d). Similar results were obtained for HCVpp entry into PHH after treatment with an EGFR-specific antibody (Fig. 4e). These data suggest that the RTKs act at postbinding steps of viral entry.
To further elucidate the entry steps targeted by the RTKs, we performed a kinetic entry assay [19,21] (Supplementary Fig. 7b). Interestingly, the half-maximal times (t1/2) for Erlotinib (t1/2=+20 min) and Dasatinib (t1/2=+26 min) to inhibit HCV entry were similar to the half-maximal time of a CD81-specific antibody (t1/2=+26 min) (Fig. 4f and Supplementary Fig. 7d). Moreover, similar to ConA, PKIs also had an inhibitory effect when added at late times post-infection. The role of EGFR as a postbinding factor was further confirmed by kinetic assays under serum-free conditions. In line with previous reports [22], HCV entry kinetics are delayed under serum-free conditions (Fig. 4g). EGF significantly (P<0.05) reduced the time for HCVcc to escape the inhibiting effects of an CD81-specific antibody in serum-starved cells from 44 ± 8 min to 27 ± 6 min (mean ± SD of three independent experiments), suggesting that EGF markedly and significantly (P<0.05) accelerates the rate of HCV entry (Fig. 4g). In summary, these data suggest that EGFR is required for efficient viral entry by modulating early and late steps of postbinding events.
Postbinding steps of HCV entry are mediated by HCV co-entry factors SR-BI, CD81, CLDN1 and OCLN. Since PKIs inhibited HCV entry at similar time-points as an CD81-specific antibody we investigated whether PKIs interfere with CD81-CLDN1 co-receptor interactions using a FRET-based assay [15,23,24]. PKIs significantly (P<0.0005) reduced CD81-CLDN1 FRET in polarized HepG2 cells (Fig. 4h and Supplementary Fig. 7e). Similar results were obtained using RTK-specific siRNAs (Fig. 4h and Supplementary Fig. 7e) confirming that the observed inhibition is RTK-specific and not mediated by off-target effects of the PKIs. These results suggest that EGFR and EphA2 regulate the formation of the CD81-CLDN1 co-receptor complexes that are essential for HCV entry [23] and that Erlotinib and Dasatinib inhibit HCV entry by interfering with the CD81-CLDN1 co-receptor association(s).
Since kinetic assays demonstrated that PKIs inhibited late steps of viral entry (Fig. 4f and Supplementary Fig. 7d), we investigated the impact of these kinases in a viral glycoprotein-dependent cell-cell fusion assay [25]. Both PKIs significantly (P<0.05) inhibited membrane fusion of cells expressing glycoproteins derived from genotypes 1a (H77), 1b (Con1) and 2a (J6) (Fig. 4i and Supplementary Fig. 7f), whereas the EGFR ligand EGF enhanced membrane fusion of cells expressing HCV envelope glycoproteins (Fig. 4i). In contrast, neither Erlotinib nor EGF had a marked effect on the membrane fusion of cells expressing measles virus envelope glycoproteins. Comparable results were obtained in EGFR and EphA2 silenced cells (Fig. 4i, data not shown) confirming a functional role of the RTKs during viral fusion.
Impact of RTKs in cell-cell transmission and viral spread
To investigate the relevance of RTK-mediated virus-host interactions for cell-cell transmission and viral spread, we used a cell-cell transmission assay [26](Fig. 5a–c). Erlotinib and Dasatinib significantly (P<0.0005) blocked HCV cell-cell transmission during short-term co-culture experiments (24 h) (Fig. 5d–f and Supplementary Fig. 8a–c). A marked inhibition of cell-cell transmission was also observed when EGFR and EphA2 were silenced using specific siRNAs: infection of GFP-positive target cells directly correlated with levels of RTK cell surface-expression (Fig. 5g,h and Supplementary Fig. 8d,e). Since PKIs markedly inhibited cell-cell transmission, we investigated whether Erlotinib and Dasatinib inhibit viral spread in the HCVcc system when added post-infection during long-term experiments. Both PKIs dose-dependently inhibited viral spread when added 48 h post-infection to HCV-infected cells for up to 14 days (Fig. 5f and Supplementary Fig. 8c). Cell viability was not affected by long term PKI treatment. A specific decrease in viral spread was also observed in cells with silenced RTK expression (Fig. 5i and Supplementary Fig. 8f). Taken together, these data demonstrate that PKIs reduce viral spread and suggest an important functional role of these RTKs in cell-cell transmission and dissemination.
Fig. 5. Functional role of EGFR in viral cell-cell transmission and spread.
(a) Experimental set-up. HCV producer cells (Pi = HCV RNA-electroporated Huh7.5.1) co-cultivated with non-infected target cells (T = GFP-expressing Huh7.5) [26] were incubated with siEGFR or PKIs. Cell-free HCV transmission was blocked by an E2-neutralizing antibody (25 μg mL−1) [26]. HCV-infected target cells (Ti = GFP+, HCV NS5A+) were quantified by flow cytometry [26]. (b) Immunofluorescence analysis of Pi, T and Ti cells stained with an NS5A-specific antibody. (c) Infectivity of Pi-T cell co-cultivation supernatants (cell-free HCV transmission). (d,e) Quantification of infected Ti cells during Erlotinib (ERL, 10 μM) treatment in the absence (total transmission) and presence (cell-cell transmission) of E2-specific antibody by flow cytometry. (f) Effect of PKIs on viral spread. Long-term HCVcc infection of Huh7.5.1 cells incubated with Erlotinib 48 h post-infection at the indicated concentrations. Medium with solvent (CTRL) or PKI was replenished every 2nd day. Cell viability was assessed using MTT test. (g) EGFR expression in target cells with silenced EGFR expression. Cell surface EGFR expression was analyzed by flow cytometry and target cells were divided in three groups displaying high, medium and low EGFR expression. (h) HCV infection in GFP-positive target cells expressing EGFR at high, medium and low levels (see panel g) assessed as described above. (i) Effect of EGFR silencing on viral spread. Long-term analysis of HCVcc infection in Huh7.5.1 cells transfected with EGFR-specific or control siRNA 24 h post-infection. Cell viability was assessed using MTT test.*, P<0.05; **, P<0.005; ***, P<0.0005.
Erlotinib inhibits HCV infection in vivo
To address the in vivo relevance of the identified virus-host interactions, we assessed the effect of Erlotinib on HCV infection in vivo in the chimeric uPA-SCID mouse model [27–29]. Erlotinib dosing and administration was performed as described previously for cancer xenograft models [30]. Erlotinib treatment caused a marked and significant (P<0.05) delay in the kinetics of HCV infection (Fig. 6). The median time to reach steady-state levels of infection increased from 15 days (placebo group) to 30 days (Erlotinib group) (median of pooled data from 6 placebo- and 8 Erlotinib-treated animals). Furthermore, Erlotinib treatment decreased steady-state HCV RNA levels 11.4 fold (mean of pooled data from 6 placebo- and 8 Erlotinib-treated animals; P<0.05). Following discontinuation of treatment, viral load reached similar levels as in control animals (Fig. 6). The treatment was well tolerated and did not induce any marked changes in safety parameters such as alanine transaminase (ALT), albumin or body weight (data not shown). Erlotinib plasma concentrations were similar as described previously in preclinical studies of cancer mouse models [30] (data not shown). Taken together, these data support a functional role of EGFR as a co-factor for HCV entry and dissemination in vivo and demonstrate that Erlotinib has antiviral activity in vivo.
Fig. 6. Erlotinib modulates HCV kinetics and inhibits infection in vivo.
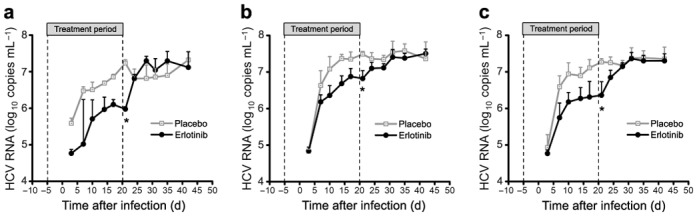
Chimeric uPA/SCID mice repopulated with PHH [27,28] were treated with Erlotinib or placebo during infection with patient-derived HCV as indicated by the bar and dashed lines. Erlotinib administration and dosage were performed as previously described for xenograft tumor mouse models [30]. Serum HCV load was analyzed at the time points indicated. Results are shown as median viral load of Erlotinib- (n=4) or placebo-treated control mice (n=3). (a,b) Two independent studies (with a total of 2 × 7 mice, respectively) are shown. (c) Pooled data of results shown in a,b (n=14); *, P<0.05.
DISCUSSION
Using RNAi screening we uncovered a network of kinases with functional impact for HCV entry and identified EGFR and EphA2 as novel co-factors for HCV entry. The implications of our results are two-fold: (i) the identification of kinases as novel HCV entry factors advances our knowledge on the molecular mechanisms and cellular requirements of HCV entry and (ii) the discovery of PKIs as antivirals defines a novel strategy for prevention and treatment of HCV infection.
EGFR is a RTK that regulates a number of key processes, including cell proliferation, survival, differentiation during development, tissue homeostasis, and tumorigenesis [31]. EphA2 mediates cell positioning, cell morphology, polarity and motility [32]. Since PKIs had no effect on HepG2 polarization (Supplementary Fig. 9), it is unlikely that changes in polarity explain their mode of action. Our results rather highlight a role of these RTKs in the formation of HCV entry factor complexes and membrane fusion. EGF accelerated HCV entry suggesting that EGFR plays a key role in the HCV entry process allowing HCV to efficiently enter its target cell (Fig. 4g). Applying FRET proximity analysis we found that inhibition of EGFR or EphA2 activity reduced CD81-CLDN1 association(s) (Fig. 4h and Supplementary Fig. 7e). Since EGFR activation has been reported to promote CLDN1 redistribution [33,34] and CD81 or CLDN1 cell surface expression levels were not modulated by EGFR silencing (Fig. 4a,b), we hypothesize that EGFR activation modulates CLDN1 and/or CD81 trafficking that are necessary to form receptor complexes.
The observations that Erlotinib inhibited late steps in the kinetic infection assay (Fig. 4f and Supplementary Fig. 7d) and HCV cell fusion assay [25] suggest a functional role for EGFR in pH-dependent fusion of viral and host cell membranes [25,35].
Functional experiments using specific ligands, antibodies and kinase inhibitors demonstrate that both ligand-binding and kinase domains are required for EGFR to promote HCV entry. EGFR ligands enhance HCV infection and an EGFR-specific antibody inhibits HCV infection (Fig. 3). This antibody binds between ligand binding domain III and the autoinhibition (tether) domain IV of the extracellular part of EGFR [36] and prevents EGF and TGF-α-induced receptor dimerization [37]. Thus, it is likely that the domain and/or receptor dimerization targeted by the antibody are required for HCV entry. Taken together, these findings support a model in which EGFR-ligand binding activates EGFR kinase function that is required for HCV entry.
Similar results were obtained for EphA2 where antibodies specific for the extracellular domain of EphA2 inhibited HCV entry into PHH and EphA2 surrogate ligands decreased viral entry (Supplementary Fig. 6). Since addition of surrogate ligands only reduced HCV entry to a small extent it is conceivable that the effect of EphA2 on HCV entry could be mediated as well in a ligand-independent and ligand-dependent manner. This is consistent with other well characterized EphA2 functions such as cell invasion and migration [38].
Since functional and mechanistic studies demonstrate that the expression and activity of EGFR and EphA2 appear to be involved in similar entry steps, it is likely that both RTKs are part of the same entry regulatory pathway. Since Erlotinib and EGF modulated entry of HCVpp but showed minimal effects on other viruses studied (Supplementary Fig. 10), it is likely that the uncovered molecular mechanisms on co-receptor interactions are most relevant for HCV entry.
Finally, our results have important clinical implications for the prevention and treatment of HCV infection as they demonstrate for the first time that clinically licensed PKIs have significant antiviral activity in vitro and in vivo. Furthermore, the identification of a RTK-specific antibody inhibiting HCV infection (Fig. 3) provides a new strategy to target RTKs to prevent and treat HCV infection. Taken together, these results demonstrate that small molecules or antibodies targeting RTKs as HCV entry factors hold promise as a novel class of antivirals and may offer a perspective for urgently needed antiviral strategies for prevention and treatment of resistant HCV infection.
METHODS
For more detailed Methods see Supplementary Methods. Unless otherwise stated, data are presented as means ± SD of at least three independent experiments performed in duplicate or triplicate.
Infection of cell lines and primary human hepatocytes with HCVpp, HCVcc and serum-derived HCV
Pseudotyped particles (pp) expressing envelope glycoproteins from different HCV strains (Supplementary Methods), VSV, MLV, influenza, measles and endogenous feline leukemia virus (RD114) and HCVcc were generated as described [14,15,21,41–45]. Infection of Huh7, Huh7.5.1 cells and PHH with HCVpp, HCVcc (TCID50 103–104 mL−1 for Huh7.5.1 experiments, TCID50 105–106 mL−1 for PHH experiments) and serum-derived HCV (genotype 1b) [46] was performed as described [14,19,21,47]. Polarization of HepG2-CD81, determination of TJ integrity and cell polarity index were performed, measured and calculated as previously described [15]. Gene silencing was performed 3 d prior to infection as described for the RNAi screen (Supplementary Methods). Inhibitors, antibodies or ligands were added 1 h prior to HCVpp or HCVcc infection and during infection unless otherwise stated. Experiments with RTK ligands were conducted with serum-starved cells. Unless otherwise stated, HCV entry and infection was assessed by luciferase reporter gene expression.
Analysis of HCV replication
Electroporation of RNA derived from plasmid pSGR-JFH1 or replication-deficient mutant pSGR-JFH1/GND (Δ) was performed as described [42]. Four hours after electroporation, cells were incubated with inhibitors. Total RNA was isolated and HCV RNA was analyzed by Northern blot as described [48].
Rescue of gene silencing
To assess whether silencing of endogenous RTKs could be rescued by expression of RNAi-resistant RTK expression, 4 × 106 Huh7.5.1 cells were co-electroporated with 10 μg siRNA targeting the 3′UTR of the endogenous cellular mRNA (siEGFR si3, siEphA2 si4, HS-CDC2_14) and a RTK encoding plasmid expressing siRNA-resistant mRNA containing a deletion of the 3′UTR (pEGFR, pEphA2, pCDC2) [40,49,50]. 2.5 × 104 cells cm−2 were seeded 72 h prior to infection with HCVcc (Luc-Jc1; genotype 2a/2a) or HCVpp (H77; genotype 1a). EGFR rescue in PHH was performed by co-transduction with lentiviruses expressing shEGFR [40] and/or EGFR [40] 72 h prior to infection with HCVpp (HCV-J; genotype 1b).
Analysis of EGFR phosphorylation in PHH and Huh7.5.1 cells
EGFR phosphorylation was assessed in cell lysates using the Human Phospho-RTK Array Kit (R&D Systems Inc.), where RTKs are captured by antibodies spotted on a nitrocellulose membrane. Levels of phospho-RTK were assessed using an HRP-conjugated pan phospho-tyrosine-specific antibody followed by chemiluminescence detection as described by the manufacturer. Phospho-tyrosine (P-Tyr) and phosphorylation of the unrelated c-mer proto-oncogene tyrosine kinase (MERTK) served as internal positive and negative controls. PHH were incubated in EGF-free William’s E medium. Huh7.5.1 cells were serum-starved overnight prior to addition of ligands, inhibitors and antibodies.
Analysis of HCV binding, postbinding and entry kinetics
HCV glycoprotein E2 binding to cells was performed as described [18] using polyclonal SR-BI [21]- or monoclonal EGFR-specific antibodies (100 μg mL−1) or SR-BI- or EphA2-specific serum (1:100) and corresponding controls. HCV postbinding and entry kinetics were performed as described [19,21] (Supplementary Methods and Supplementary Fig. 7).
Receptor association using fluorescence resonance energy transfer (FRET)
Homotypic and heterotypic interactions of CD81 and CLDN1 were analyzed as described [15,23,24]. The data from 10 cells were normalized and the localized expression calculated.
Membrane fusion
HCV membrane fusion during viral entry was investigated using a cell-cell fusion assay as described [25].
Cell-cell transmission of HCV
Cell-cell transmission of HCV was assessed as described [26]. Briefly, producer Huh7.5.1 cells were electroporated with HCV Jc1 RNA and co-cultured with gene-silenced or naïve target Huh7.5-GFP cells in the presence or absence of PKIs (10 μM). An HCV E2-neutralizing antibody (25 μg mL−1) was added to block cell-free transmission [26]. After 24 h of co-culture cells were fixed with PFA, stained with a NS5A-specific antibody (0.1 μg mL−1), and analyzed by flow cytometry [26]. Total and cell-cell transmission were defined as percentage HCV infection of Huh7.5-GFP+ target cells (Ti) in the absence (total transmission) or presence (cell-cell transmission) of an HCV E2-specific antibody.
HCV infection and treatment of chimeric uPA/SCID mice
Chimeric mice repopulated with PHH [27,28] were infected with serum-derived HCV (genotype 2a, 1 × 104 HCV IU per mouse) via the orbital vein during isofluoran anesthetization (PhoenixBio Inc., Japan). Erlotinib administration and dosage (50 mg kg−1 day−1) were performed as previously described in xenograft tumor mouse models [30]. Four mice received 50 mg kg−1 day−1 Erlotinib and three mice placebo (p. o.) from day −10 until day 20 of infection in two independent experiments (total 2 × 7 animals). Plasma HCV RNA, ALT, albumin and Erlotinib were monitored as described [28,51]. All experimental procedures used to treat live animals in this study had been approved by the Animal Ethics Committee of PhoenixBio in accordance with the Japanese legislation.
Toxicity assays
Cytotoxic effects on cells were assessed in triplicates by analyzing the ability to metabolize 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) [52]. Formazan crystals were solubilized 5 h after adding MTT (0.6 mg mL−1) as described [52].
Supplementary Material
Acknowledgments
This work was supported by ERC-2008-AdG-233130-HEPCENT, INTERREG-IV-Rhin Supérieur-FEDER-Hepato-Regio-Net 2009, ANR-05-CEXC-008, ANRS 2008/354, Région Alsace, INCa, NIH, MRC and the Wellcome Trust. The authors acknowledge A-L. Morand, L. Froidevaux, A. Weiss and L. Poidevin (IGBMC, France), S. Durand and E. Soulier (Inserm U748, France), for excellent technical work. The authors thank R. Bartenschlager (University of Heidelberg, Germany), J. Ball (University of Nottingham, UK), T. Wakita (National Institute of Infectious Diseases, Japan), C. M. Rice (The Rockefeller University, USA), F. V. Chisari (The Scripps Research Institute, USA), E. Harlow (Harvard University, USA), M. Tanaka (Hamamatsu University, Japan) for providing reagents.
Footnotes
AUTHOR CONTRIBUTIONS
J. L., M. B. Z. and T. F. B. wrote the manuscript. J. L., M. B. Z., F. X., D. L., F.-L. C, J. A. McK, and T. F. B. designed experiments and analyzed data. J. L., M. B. Z, F. X., C. T., I. F., L. Z., C. D., C. J. M., M. T., S. G., C. R., M. N. Z., D. L. and J. F. performed experiments. S. M. R., T. P., A. H. P., P. P. and M. D. contributed essential reagents, W. R. and O. P. performed bioinformatic analyses, J.L., B. F. and L. B. implemented the siRNA screen, T. F. B. designed and supervised the project.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests. Inserm and the University of Strasbourg have filed a patent application on–Host cell kinases as targets for antiviral therapy against HCV infection.
References
- 1.Tai AW, Chung RT. Treatment failure in hepatitis C: mechanisms of non-response. J Hepatol. 2009;50:412–420. doi: 10.1016/j.jhep.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hezode C, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 2009;360:1839–1850. doi: 10.1056/NEJMoa0807650. [DOI] [PubMed] [Google Scholar]
- 3.Timpe JM, et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology. 2008;47:17–24. doi: 10.1002/hep.21959. [DOI] [PubMed] [Google Scholar]
- 4.Zeisel MB, Cosset FL, Baumert TF. Host neutralizing responses and pathogenesis of hepatitis C virus infection. Hepatology. 2008;48:299–307. doi: 10.1002/hep.22307. [DOI] [PubMed] [Google Scholar]
- 5.von Hahn T, Rice CM. Hepatitis C virus entry. J Biol Chem. 2008;283:3689–3693. doi: 10.1074/jbc.R700024200. [DOI] [PubMed] [Google Scholar]
- 6.Barth H, et al. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J Virol. 2006;80:10579–10590. doi: 10.1128/JVI.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pileri P, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 8.Scarselli E, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans MJ, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 10.Ploss A, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hidalgo M, Bloedow D. Pharmacokinetics and pharmacodynamics: maximizing the clinical potential of Erlotinib (Tarceva) Semin Oncol. 2003;30:25–33. [PubMed] [Google Scholar]
- 12.Shepherd FA, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 13.Li J, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6:291–299. doi: 10.1038/nchembio.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fafi-Kremer S, et al. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J Exp Med. 2010;207:2019–2031. doi: 10.1084/jem.20090766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mee CJ, et al. Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J Virol. 2009;83:6211–6221. doi: 10.1128/JVI.00246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 17.Thoresen GH, et al. Response to transforming growth factor alpha (TGFalpha) and epidermal growth factor (EGF) in hepatocytes: lower EGF receptor affinity of TGFalpha is associated with more sustained activation of p42/p44 mitogen-activated protein kinase and greater efficacy in stimulation of DNA synthesis. J Cell Physiol. 1998;175:10–18. doi: 10.1002/(SICI)1097-4652(199804)175:1<10::AID-JCP2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 18.Dreux M, et al. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009;5:e1000310. doi: 10.1371/journal.ppat.1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krieger SE, et al. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology. 2010;51:1144–1157. doi: 10.1002/hep.23445. [DOI] [PubMed] [Google Scholar]
- 20.Koutsoudakis G, et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol. 2006;80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeisel MB, et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology. 2007;46:1722–1731. doi: 10.1002/hep.21994. [DOI] [PubMed] [Google Scholar]
- 22.Dreux M, et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J Biol Chem. 2006;281:18285–18295. doi: 10.1074/jbc.M602706200. [DOI] [PubMed] [Google Scholar]
- 23.Harris HJ, et al. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem. 2010;285:21092–21102. doi: 10.1074/jbc.M110.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris HJ, et al. CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol. 2008;82:5007–5020. doi: 10.1128/JVI.02286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavillette D, et al. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol. 2007;81:8752–8765. doi: 10.1128/JVI.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witteveldt J, et al. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J Gen Virol. 2009;90:48–58. doi: 10.1099/vir.0.006700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiraga N, et al. Absence of viral interference and different susceptibility to interferon between hepatitis B virus and hepatitis C virus in human hepatocyte chimeric mice. J Hepatol. 2009;51:1046–1054. doi: 10.1016/j.jhep.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Kamiya N, et al. Practical evaluation of a mouse with chimeric human liver model for hepatitis C virus infection using an NS3-4A protease inhibitor. J Gen Virol. 2010;91:1668–1677. doi: 10.1099/vir.0.019315-0. [DOI] [PubMed] [Google Scholar]
- 29.Meuleman P, et al. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology. 2005;41:847–856. doi: 10.1002/hep.20657. [DOI] [PubMed] [Google Scholar]
- 30.Higgins B, et al. Antitumor activity of erlotinib (OSI-774, Tarceva) alone or in combination in human non-small cell lung cancer tumor xenograft models. Anticancer Drugs. 2004;15:503–512. doi: 10.1097/01.cad.0000127664.66472.60. [DOI] [PubMed] [Google Scholar]
- 31.Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. 2009;218:460–466. doi: 10.1002/jcp.21635. [DOI] [PubMed] [Google Scholar]
- 32.Lackmann M, Boyd AW. Eph, a protein family coming of age: more confusion, insight, or complexity? Sci Signal. 2008;1:re2. doi: 10.1126/stke.115re2. [DOI] [PubMed] [Google Scholar]
- 33.Singh AB, Harris RC. Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J Biol Chem. 2004;279:3543–3552. doi: 10.1074/jbc.M308682200. [DOI] [PubMed] [Google Scholar]
- 34.Flores-Benitez D, et al. Control of tight junctional sealing: roles of epidermal growth factor and prostaglandin E2. Am J Physiol Cell Physiol. 2009;297:C611–620. doi: 10.1152/ajpcell.00622.2008. [DOI] [PubMed] [Google Scholar]
- 35.Blanchard E, et al. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiter JL, Maihle NJ. A 1.8 kb alternative transcript from the human epidermal growth factor receptor gene encodes a truncated form of the receptor. Nucleic Acids Res. 1996;24:4050–4056. doi: 10.1093/nar/24.20.4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCole DF, Keely SJ, Coffey RJ, Barrett KE. Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor-alpha. J Biol Chem. 2002;277:42603–42612. doi: 10.1074/jbc.M206487200. [DOI] [PubMed] [Google Scholar]
- 38.Miao H, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pestka JM, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A. 2007;104:6025–6030. doi: 10.1073/pnas.0607026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothenberg SM, et al. Modeling oncogene addiction using RNA interference. Proc Natl Acad Sci U S A. 2008;105:12480–12484. doi: 10.1073/pnas.0803217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pietschmann T, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lavillette D, et al. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology. 2005;41:265–274. doi: 10.1002/hep.20542. [DOI] [PubMed] [Google Scholar]
- 44.Frecha C, et al. Efficient and stable transduction of resting B lymphocytes and primary chronic lymphocyte leukemia cells using measles virus gp displaying lentiviral vectors. Blood. 2009;114:3173–3180. doi: 10.1182/blood-2009-05-220798. [DOI] [PubMed] [Google Scholar]
- 45.Sandrin V, et al. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. 2002;100:823–832. doi: 10.1182/blood-2001-11-0042. [DOI] [PubMed] [Google Scholar]
- 46.Fofana I, et al. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology. 2010;139:953–964. 964 e951–954. doi: 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- 47.Meunier JC, et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J Virol. 2008;82:966–973. doi: 10.1128/JVI.01872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohmann V, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 49.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, et al. Negative regulation of EphA2 receptor by Cbl. Biochem Biophys Res Commun. 2002;296:214–220. doi: 10.1016/s0006-291x(02)00806-9. [DOI] [PubMed] [Google Scholar]
- 51.Chahbouni A, den Burger JC, Vos RM, Sinjewel A, Wilhelm AJ. Simultaneous quantification of erlotinib, gefitinib, and imatinib in human plasma by liquid chromatography tandem mass spectrometry. Ther Drug Monit. 2009;31:683–687. doi: 10.1097/FTD.0b013e3181c05a14. [DOI] [PubMed] [Google Scholar]
- 52.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.