

Abstract
Plant-based medicines are useful in the treatment of cancer. Many breast cancer patients use complementary and alternative medicine in parallel with conventional treatments. Neem is historically well known in Asia and Africa as a versatile medicinal plant with a wide spectrum of biological activities. The experiments reported herein determined whether the administration of an ethanolic fraction of Neem leaf (EFNL) inhibits progression of chemical carcinogen-induced mammary tumorigenesis in rat models. Seven-week-old female Sprague Dawley rats were given a single intraperitoneal injection of N-methyl-N-nitrosourea (MNU). Upon the appearance of palpable mammary tumors, the rats were divided into vehicle-treated control groups and EFNL-treated groups. Treatment with EFNL inhibited MNU-induced mammary tumor progression. EFNL treatment was also highly effective in reducing mammary tumor burden and in suppressing mammary tumor progression even after the cessation of treatment. Further, we found that EFNL treatment effectively upregulated proapoptotic genes and proteins such as p53, B cell lymphoma-2 protein (Bcl-2)-associated X protein (Bax), Bcl-2-associated death promoter protein (Bad) caspases, phosphatase and tensin homolog gene (PTEN), and c-Jun N-terminal kinase (JNK). In contrast, EFNL treatment caused downregulation of anti-apoptotic (Bcl-2), angiogenic proteins (angiopoietin and vascular endothelial growth factor A [VEGF-A]), cell cycle regulatory proteins (cyclin D1, cyclin-dependent kinase 2 [Cdk2], and Cdk4), and pro-survival signals such as NFκB, mitogen-activated protein kinase 1 (MAPK1). The data obtained in this study demonstrate that EFNL exert a potent anticancer effect against mammary tumorigenesis by altering key signaling pathways.
Keywords: Neem, breast cancer, apoptosis, cell cycle, angiogenesis
Introduction
Breast cancer is one of the most commonly diagnosed cancers in women. According to the Surveillance, Epidemiology, and End Results (SEER) statistics in 2012, approximately 226 870 women will be diagnosed with breast cancer in the US, with an associated mortality of 39 920.1 Earlier and current literature shows that over 60% of breast cancer patients use some form of complementary and alternative medicine.2,3 Many natural products possess antitumorigenic properties and induce tumor regression in xenograft models. Phytochemicals are major components of alternative medicine. Studies have shown that phytochemicals that display potent anticancer effects in both in vitro and in vivo rodent models can be potential candidates for chemoprevention in humans.4,5 These phytochemicals have different chemical properties and can block tumorigenesis by multiple mechanisms that include prevention of pro-carcinogen activation, inhibition of cell proliferation, invasion, angiogenesis, and stimulation of apoptosis.6
Azadirachta indica, commonly known as Neem, has a wider array of uses than many other plants. Various parts of the Neem tree like the leaves, bark, fruit, flowers, oil, and gum have been associated with medicinal properties.7 In vitro studies have demonstrated the antiproliferative effects of Neem.8 Further, it has also been shown that Neem leaf extracts have potent anti-tumorigenic activity against 7, 12-dimethylbenz(a)anthracene (DMBA)-induced hamster buccal pouch carcinogenesis and mammary tumorigenesis.9,10 Others have demonstrated that the antioxidant components of Neem leaf fractions may modulate cancer cell proliferation, angiogenesis, and apoptosis.9 The mechanisms by which Neem inhibits mammary cancer growth is not well studied. The chemical carcinogen-induced rat mammary model is one of the best known in vivo model systems for investigating the efficacy of chemopreventive agents. The aims of our study were to explore the antitumorigenic properties of A. indica against mammary tumor progression in the in rodent model of N-methyl-N-nitrosourea (MNU)-induced mammary tumorigenesis and to investigate the molecular mechanisms through which it inhibits mammary tumorigenesis.
Results
EFNL inhibits mammary tumor progression and multiplicity
To determine whether EFNL possesses anti-tumorigenic effects against MNU-induced mammary tumorigenesis, rats bearing mammary tumors were treated with EFNL for 4 weeks and observed for another 8 weeks after the end of EFNL treatment. EFNL treatment significantly inhibited the progression of MNU-induced mammary tumors (Fig. 1A). A dose-dependent anti-tumorigenic effect of EFNL was observed. The palpable mammary tumors in the control group continued to grow, whereas administration of EFNL blocked the growth and resulted in significantly reduced mammary tumor volume (P < 0.001) compared with the controls. Some of the mammary tumors in the EFNL treatment group completely disappeared. Interestingly, even after 8 weeks of EFNL withdrawal, there was no regrowth of the mammary tumors observed, whereas mammary tumors in untreated control animals continued to grow. Further, EFNL treatment remarkably inhibited mammary tumor multiplicity (Fig. 1B). Control animals developed 4 ± 0.8 mammary tumors during the experimental observation period, whereas EFNL-treated animals did not develop any further mammary tumors during the same observation period (P < 0.001). These findings clearly demonstrate that EFNL not only inhibits progression of established mammary tumors but also reduces mammary tumor multiplicity. For all molecular analysis mammary tissue obtained from 4 mg/kg BW of EFNL treatment was used alongside mammary tissue obtained from vehicle treated controls.
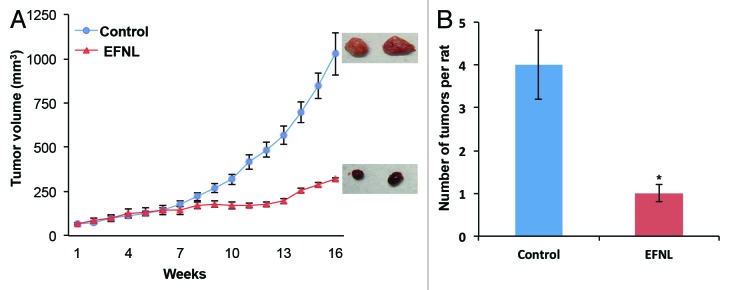
Figure 1. EFNL treatment inhibited the growth and multiplicity of mammary tumors. (A) EFNL treatment significantly reduced mammary tumor volume. Representative pictures of the mammary tumors from each group is also shown. Tumor volume was calculated using the equation 4/3π × r12 × r2 where r1 is the minor radius and r2 is the major radius. (B) EFNL treatment also reduced the number of mammary tumors compared with vehicle treated controls. *Represents statistical significance of P < 0.001 between control and EFNL treatment.
EFNL alters apoptosis, angiogenesis, and cell cycle signaling
We performed pathway-focused microarray analysis for genes using the Rat Cancer Pathway Finder PCR array, PI3K/AKT signaling PCR array, and apoptosis PCR array. EFNL treatment resulted in differential regulation of several genes in these pathways (Figs. 2 and 3). Expression levels of proapoptotic genes like Pten (86-fold), p53 (7.3-fold), Bax (6.8-fold), Bad (10-fold), and caspases death domain-containing protein (Cradd) (7.9-fold), Fas-associated protein with death domain (Fadd) (3.5-fold), and c-jun N-terminal kinase (JNK) (8.2-fold) (Fig. 2) were significantly upregulated by EFNL treatment compared with expression levels in the controls. On the contrary, we also observed that ENLF treatment remarkably downregulated the expression of antiapoptotic genes Bcl-2 (−20-fold), nucleolar protein 3 (Nol3) (−4.4-fold), Mcl-1 (−2.5-fold), and telomerase reverse transcriptase (Tert) (−2.5-fold) (Fig. 3). These findings demonstrate that EFNL inhibits mammary tumor progression by inducing apoptosis.

Figure 2. Significantly upregulated genes in the EFNL-treated mammary tumors. Apoptosis, angiogenesis, and PI3K/AKT pathway focused PCR array analysis was performed and the genes upregulated in EFNL-treated group are shown with their corresponding fold difference (mean ± SD). Three mammary tumor samples from each group were used to perform PCR array analysis and P < 0.05 was considered as statistically significant.

Figure 3. Significantly downregulated genes in the EFNL-treated mammary tumors. Three mammary tumor samples from vehicle-treated and EFNL-treated rats are used to perform apoptosis, angiogenesis, and PI3K/AKT pathway-focused PCR array analyses. The fold difference of downregulated genes in the EFNL-treated group are represented as mean ± SD. P < 0.05 was considered as statistically significant.
Angiogenesis is a fundamental requirement for the development of cancer. Inhibition of angiogenesis is considered to be one of the effective ways of blocking cancer growth. In our experiments we found that EFNL treatment effectively inhibited the expression of proangiogenic genes, vascular endothelial growth factor A (Vegfa [−10.5-fold]) and angiopoietin (−4.1-fold), indicating the antiangiogenic potential of EFNL (Fig. 3). Inhibition of angiogenesis by EFNL could be a reason for reduction in mammary tumor volume and for blocked development of new tumors as observed in our studies.
Cell cycle regulatory genes play a vital role in maintaining normal cell proliferation. Loss of cell cycle regulation could lead to the development of cancer. Genes that promote cell cycle activity (cyclin D1 [−5-fold], cdk2 [−5.3-fold], and cdk4 [−5.9-fold]) and proto-oncogenes (c-jun [−3.5-fold], and c-myc [−4.2-fold]) were significantly downregulated in EFNL treated mammary tumors (Fig. 3).
Mitogen-activated protein kinases (MAPKs) control fundamental processes like growth and proliferation. Inhibition of MAPK leads to decreased cell proliferation. EFNL treatment downregulated MAPK1 (−3.3-fold) gene expression compared with controls. Expression of NFκB (−4.6-fold) was also downregulated in EFNL-treated mammary tumors compared with mammary tumors from untreated control animals (Fig. 3).
Overall, the gene expression analysis revealed that EFNL differentially regulates genes that influence apoptosis, angiogenesis, cell cycling, and proliferation to cause inhibition of mammary tumor progression.
Altered expression of proteins involved in cell cycle progression, such as Cdks and cyclins, can drive abnormal proliferation in cancer. Using western blot and immunohistochemistry analyses, we examined the expression of some key proteins that regulate apoptosis, cell cycle, and signaling (Figs. 4, 5, 6, 7, and 8). Our data clearly demonstrates that EFNL induces significant reduction in proteins responsible for promotion of the cell cycle and inhibition of apoptosis in mammary tumors. Cdk2, cdk4, and cyclin D1 were significantly lowered in EFNL-treated mammary tumors compared with untreated control tumors (Figs. 4, 6, and 8). EFNL treatment also decreased the levels of the antiapoptotic protein Bcl-2. Total levels of Akt were not altered by EFNL treatment, whereas phosphorylation of Akt was significantly reduced, indicating that EFNL can inhibit cell survival and proliferation (Fig. 6).

Figure 4. Expression of cell cycle regulators and anti-apoptotic proteins are downregulated by EFNL. Expression of cell cycle regulatory proteins CDK2, cyclin D1, and anti-apoptotic protein BCL2 were analyzed in the vehicle and EFNL treated rat mammary cancer tissues using IHC. The number of positive cells and the intensity of staining for CDK2, cyclin D1, and BCL2 were lower in EFNL treated mammary cancer tissue compared with vehicle treated controls.

Figure 5. Immunohistolochemical analysis reveals that tumor suppressor proteins (PTEN and p53) and pro apoptotic protein (BAX) are upregulated by EFNL treatment. Rats that received EFNL treatment clearly showed increased expression of these proteins in their mammary cancers.

Figure 6. Activation and expression of signaling proteins were altered in EFNL treatment. Western blot analysis of cell proliferation, cell cycle, and apoptosis signaling proteins revealed reduced phosphorylation of AKT, decreased expression of CDK2, cyclinD1, and BCL2 in the EFNL-treated group. Representative blots were shown with their corresponding densitometric values normalized with internal control β actin.

Figure 7. EFNL treatment increased the tumor suppressor (p53 and PTEN) and pro-apoptotic (BAX) protein expressions. Western blot analysis showed that EFNL treatment increased the active form of tumor suppressor p53 along with increased expression of PTEN and Bax. Densitometric analyses were performed and the values were plotted after normalization with internal control β actin.

Figure 8. EFNL differentially regulated the levels of proteins involved in apoptosis, angiogenesis, and cell cycle. Induction of pro-apoptotic proteins BAD and JNK in the mammary cancer tissue of EFNL-treated rats are shown by western blot analysis. Further, a decrease in the levels of angiogenic factor VEGF and cell cycle protein CDK4 in the EFNL treatment are also shown.
Conversely, EFNL treatment enhanced the levels and activation of proteins regulating cell cycle arrest and apoptosis in mammary tumors. Total and phospho p53 levels were increased by EFNL treatment (Fig. 7). PTEN protein levels were higher in EFNL-treated mammary tumors than in untreated control mammary tumors (Figs. 5 and 7). C-Jun N-terminal kinase (JNK) a MAPK, which is involved in inducing apoptosis was markedly upregulated in the EFNL treatment (Fig. 8). Levels of the proapoptotic proteins Bax and Bad were also upregulated by EFNL (Figs. 7 and 8). Our results demonstrate that EFNL inhibits mammary tumor growth by differential regulation and activation of proteins involved in apoptosis, cell cycle, and signaling.
Discussion
Nature has been a main source of medical treatments from ancient times. Even today plant-based medicine continues to play a vital role in the primary health care of approximately 80% of the world’s population.11 Azadirachta indica, commonly known as Neem, is a versatile plant with multiple medicinal properties.12 Various parts of the Neem tree have been used as remedies for numerous ailments. In our current experiments we tested the antitumorigenic effect of EFNL. Our results demonstrate that EFNL has potent antitumorigenic effects against MNU-induced mammary tumorigenesis. Further, our data also indicate that EFNL exerts its antitumorigenic effects through alteration of transcription and translation of genes and proteins involved in key regulatory pathways that control cell proliferation and death.
Apoptosis is a key process that regulates cancer promotion and progression. Cancer cells avoid apoptosis and continue to proliferate without any control. Developing drugs that could promote apoptosis in cancer cells will be effective in reducing cancer burden. EFNL treatment significantly altered the expression of proapoptotic and antiapoptotic factors. Increased mRNA expression of death domain-containing protein (CRADD), Fas-associated protein with death domain (FADD),13 Bax, and caspases has been shown to induce apoptosis in several cancer cells.14 CRADD and FADD are death domain-containing adaptor proteins that activate caspases, which leads to apoptosis. In addition, Bax induces apoptosis through the release of cytochrome c and activation of caspases.15 Caspase-mediated proteolysis of cellular substrates is one of the hallmarks of apoptotic cell death. Increased expression of these genes in EFNL-treated animals could result in apoptosis leading to inhibition of mammary tumor progression.
Bcl-2 is an independent predictor of breast cancer outcome.16-18 Reduced Bcl-2 mRNA expression has been demonstrated to inhibit mammary tumor growth,19 whereas increased expression inhibits most kinds of programmed cell death and facilitates survival of mammary tumor cells.20 Therefore, downregulation of Bcl-2 expression by EFNL treatment would result in reduced survival and increased apoptosis in the mammary tumors.
Another antiapoptotic factor, nucleolar protein 3 (NOL3) inhibits apoptosis by interfering with death-inducing signaling complex (DISC) formation. NOL3 has also been demonstrated to confer both chemo- and radioresistance in breast cancer cells.21 NOL3 can also inhibit apoptosis by direct interaction with Bax and p53.22 These interactions disable Bax and p53 transcriptional function.
The first tumor suppressor gene to be identified was p53,23 which prevents neoplastic development by inhibiting the proliferation of abnormal cells and is one of the most commonly mutated genes in many human cancers. In many breast cancers, the steady-state level of p53 mRNA is lower than that observed in normal breast epithelium.24 Upregulation of c-jun N-terminal kinase (JNK) stabilizes and activates p53, which leads to apoptosis and G2/M cell cycle arrest.25,26 We observed that EFNL treatment not only reduced the mRNA expression of NOL3 but also increased expression of Bax and JNK. The increase in JNK in EFNL-treated groups could have led to p53 stabilization and activation, leading to increased apoptosis and to the resulting inhibition of mammary tumor growth. These findings reconfirm that EFNL is a potent inducer of apoptosis. In addition, EFNL inhibited Mcl-1 expression. It has been demonstrated that inhibition of Mcl-1 in breast cancer cells promotes cell death both in vitro and in vivo.27 In our current study, we observed that EFNL treatment significantly reduced mammary tumor volume and multiplicity. This could be attributed to the increased expression of proapoptotic factors like CRADD, FADD, caspases Bax, and p53, and to decreased expression of antiapoptotic factors like Bcl-2, Mcl-1, and NOL3.
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway mediates several cellular functions vital to tumor initiation and progression, including proliferation, motility, migration, invasion, and angiogenesis.28 Deregulation of this pathway has been implicated in breast cancer development and progression.29 The PI3K-dependent phosphorylation and activation of Akt is a key regulator of cell survival mechanisms. Remarkable reduction of Akt phosphorylation by EFNL treatment could play a critical role in the inhibition of mammary tumorigenesis. The PTEN gene was identified as a candidate tumor suppressor gene frequently deleted in brain, breast, and prostate cancer.30-32 PTEN opposes the effects of PI3K by dephosphorylating its lipid products. The products of PI3K activity are required for activation of Akt, which when constitutively activated has transforming potential.33 EFNL treatment significantly increased the expression of PTEN, which could inhibit mammary tumorigenesis through its inhibitory effect on Akt.
Cyclin D1 plays an important role in the regulation of progression of the cell cycle from the G1 to the S phase through the formation of active enzyme complexes with cyclin-dependent kinases, Cdk4 and Cdk6.34 Overexpression of cyclin D1, Cdk4, and Cdk6 could contribute to loss of normal cell cycle control, leading to tumorigenesis.34 Early studies reported cyclin D1 gene amplification in 10–15% of mammary carcinomas.35,36 When anti-cyclin D1 antibodies became available, it was discovered that cyclin D1 protein overexpression is found in the majority of human breast cancers.37 The overexpression of cyclin D1 protein correlates with poor prognosis and it also distinguishes malignant breast carcinomas from premalignant breast lesions.38,39 Consistent with the oncogenic role of cyclin D1 are the observations that transgenic mice engineered to overexpress this cyclin in their breast tissue are prone to mammary adenocarcinomas.40 Altered expression of cdk2 is a critical factor in the transition of cells from G1 to S phase, and its deregulation in cancer could be causative of oncogenesis. Our data demonstrate that EFNL drastically reduces the levels of cyclin D1, cdk2, and cdk4, which could play a critical role in inhibition of mammary tumorigenesis.
Angiogenesis plays an important role in breast cancer growth and metastasis.41 Angiopoietin-1 (Angpt-1) mRNA and protein expression positively correlates with the degree of angiogenesis in vivo. Earlier studies have suggested that increased expression of Ang-1 promotes42 as well as inhibits tumor growth.43 Ang-1 has been shown to act in synergy with VEGF to enhance angiogenesis and increase vessel density.44 Overexpression of VEGF gives rise to increased vascular branching and leaky vessels; but the vessels induced in the presence of Ang-1 are not leaky.45 Reduced mRNA expression of Ang-1 and VEGF in the EFNL-treated animals could have inhibited mammary tumorigenesis by inhibiting angiogenesis. Furthermore, downregulation of NFκB, which upregulates the expression of VEGF, could have also contributed toward the inhibition of mammary tumorigenesis by EFNL.
In summary, these studies demonstrate that EFNL treatment induces a long lasting therapeutic effect on mammary tumorigenesis in the MNU-induced mammary tumorigenesis model. At least some effects of EFNL treatment are mediated through deregulation of multiple pathways (apoptosis, angiogenesis, cell cycle, and signaling) that are critical for mammary tumor promotion and progression. Further investigations will provide a rationale for considering the use of EFNL treatment as a chemotherapeutic modality against breast cancer.
Materials and Methods
Animals
Virgin Sprague Dawley rats were purchased from Harlan Sprague Dawley. The rats were housed in a temperature-controlled room with a 12-h light and 12-h dark schedule. They were given food and water ad libitum. All experiments were conducted in accordance with the guidelines established by the Texas Tech University Animal Care and Use Committee guidelines.
Ethanolic fraction of Neem leaves preparation
Ten grams of Neem leaf powder was dissolved in 100 ml of absolute ethanol. The extract was continuously stirred for 48 h at 4 °C. After 48 h, the solution was filtered and lyophilized. The lyophilized powder, designated below as ethanolic fraction of Neem leaves (EFNL), was dissolved in modified Eagle’s media for use in the treatment groups, as described below.
Mammary cancer induction
All rats were treated with a single dose of MNU (Sigma) at 50 mg/kg body weight, intraperitoneally, at 7 weeks of age. MNU was dissolved in physiological saline adjusted to pH 5.0.46,47
Mammary tumor volume
After carcinogen treatment, all animals were weighed and palpated once a week. The palpable mammary tumors were measured using a caliper. Tumor volume was calculated using the equation 4/3π × r12 × r2 where r1 is the minor radius and r2 is the major radius.
Ethanolic fraction of Neem leaves (EFNL) treatment, in vivo
Animals were randomized into experimental and control groups on the appearance of the first palpable mammary tumor, with 30 animals per group. Treatments with EFNL were initiated when a mammary tumor was first palpable. The treatment was given as a daily intraperitoneal injection for 4 weeks as follows: control, vehicle treatment; 4 mg/kg BW EFNL.
Molecular analysis
Animals were euthanized 12 weeks after the end of EFNL treatment. Mammary tumor tissue samples, as well as uterus, intestine, and normal mammary glands, were immediately dissected into smaller sections, some of which were fixed in formalin for histological analysis. The remaining samples were snap frozen in liquid nitrogen and processed for molecular analysis. All the molecular analyses were performed on 3 different mammary tumors from each group.
Pathway-focused PCR array
Total RNA was extracted from each tumor sample using Trizol reagent (Invitrogen) and was then treated with DNase using the Turbo DNA free kit (Ambion) as per the recommended protocol. Product was then reverse transcribed into cDNA using a first strand cDNA synthesis kit (SABiosciences). Apoptosis, cancer pathway finder, and PI3K/AKT pathway-focused PCR microarray analyses were performed as per the manufacturer’s protocols (SABiosciences).
Western blotting
Approximately 20 mg tissue samples were lysed in RIPA buffer, and the protein extraction protocol was performed. The homogenized lysates were centrifuged at 10 000 × g for 20 min at 4 °C, and the supernatant fraction was collected. Protein concentrations were determined using the BCA protein assay kit (Pierce). The proteins were resolved on SDS-PAGE gels (Bio-Rad) and transferred to PVDF membranes (Millipore). The membranes were incubated in 1× phosphate-buffered saline (PBS) containing 5% nonfat dry milk/5% bovine serum albumin (BSA), for 2 h, to block nonspecific binding sites. The blots were incubated with primary antibodies at 1:1000 dilutions with anti-AKT, -phospho-AKT, -B cell lymphoma-2 protein (Bcl-2)-associated X protein (BAX), -BCL2, -CCND1, -p53, -phosphatase and tensin homolog gene (PTEN) (all from Cell Signaling), anti-cyclin-dependent kinase 4 (CDK4; Abcam), anti-phospho-p53 (GeneTex), and anti-β-actin (Sigma) overnight at 4 °C. The blots were washed 3 times, 10 min each, in Tris-buffered saline (TBS). The blots were then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After washing three times in TBS for 10 min each, proteins were visualized using SuperSignal West Pico chemiluminescent substrate detection solution (Pierce). Densitometry was performed on a scanner and quantitated using Fujifilm, LAS 4000.
Immunohistochemistry
Tissue samples were processed by our histology core facility for detection and localization of antigens of interest by use of immunohistochemistry. Standard immunohistochemistry staining protocols were performed. In brief, specific antibodies were diluted in blocking buffer to a dilution of 1:100–1:400. Mammary cancer sections, on slides, were deparaffinized and placed in three changes of xylene, then hydrated in graded alcohol series. Slides were washed extensively in tap water then rinsed in three changes of distilled water (DI). For aiding antigen retrieval, slides were placed in a decloaking chamber for 15 min. The slides were then washed gently with 5 changes of DI, rinsed once with PBS, and then incubated for 15 min with 1% fetal calf serum to block nonspecific binding. After 3 additional washes in DI, slides were incubated for 10 min with peroxidase-free blocking reagent, again rinsed in DI 3 times, and placed in PBS for 5 min. Tissue sections were then incubated with the primary antibody in a humidity chamber for 1 h, rinsed once with DI, and placed in PBS for 5 min. Tissue sections were then covered with Ultramarque Polyscan HRP label, placed in a humidity chamber for 30 min, rinsed in one change of DI, and placed in PBS for 5 min. Chromogen (3,3′-diaminobenzidine tetrahydrochloride [DAB]; Dako) solution was prepared (1:100 dilutions), and the intensity of staining was monitored under a microscope to ensure proper tissue staining. Slides were incubated for 5 min and then rinsed with 5 changes of DI. Counterstaining was achieved with modified Harris Hematoxylin solution for 45 s, and then slides were rinsed in tap water and placed in bluing agent for 1 min. Finally, tissue sections were rinsed in tap water, placed in xylene and coverslip was carefully placed on the tissue sections. Mammary epithelial cells with brown staining were counted as positive regardless of the intensity of the staining, while those with bluish purple nuclear staining from hematoxylin were counted as negative cells. The level of protein expression was scored using the Q score method.48-50 In brief, the Q score is calculated by multiplying the percentage of positive cells (P = 0 for <10%, 1 for 10–25%; 2 for 25–50%; 3 for 50–75%; 4 for >75%) by the intensity (I = 1 for weak staining; 2 for moderate staining; 3 for strong staining), Q = P × I.
Statistical analysis
The data are expressed as mean ± SEM. The Mann–Whitney test or the Student t test was used to analyze differences between the control and EFNL treated group using the GraphPad Prism 6 software package. P < 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
The financial and material help from the TTUHSC PLFSOM is greatly appreciated
Glossary
Abbreviations:
- EFNL
ethanolic fraction of Neem leaf
- MNU
N-methyl-N-nitrosourea
- MAPK
mitogen-activated protein kinase
- PI3K
phosphatidylinositol 3-kinase
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26604
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Henderson JW, Donatelle RJ. Complementary and alternative medicine use by women after completion of allopathic treatment for breast cancer. Altern Ther Health Med. 2004;10:52–7. [PubMed] [Google Scholar]
- 3.McLay JS, Stewart D, George J, Rore C, Heys SD. Complementary and alternative medicines use by Scottish women with breast cancer. What, why and the potential for drug interactions? Eur J Clin Pharmacol. 2012;68:811–9. doi: 10.1007/s00228-011-1181-6. [DOI] [PubMed] [Google Scholar]
- 4.Tan AC, Konczak I, Sze DM, Ramzan I. Molecular pathways for cancer chemoprevention by dietary phytochemicals. Nutr Cancer. 2011;63:495–505. doi: 10.1080/01635581.2011.538953. [DOI] [PubMed] [Google Scholar]
- 5.Singh M, Singh P, Shukla Y. New strategies in cancer chemoprevention by phytochemicals. Front Biosci (Elite Ed) 2012;4:426–52. doi: 10.2741/389. [Elite Ed] [DOI] [PubMed] [Google Scholar]
- 6.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Dasgupta T, Banerjee S, Yadava PK, Rao AR. Chemopreventive potential of Azadirachta indica (Neem) leaf extract in murine carcinogenesis model systems. J Ethnopharmacol. 2004;92:23–36. doi: 10.1016/j.jep.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Kumar S, Suresh PK, Vijayababu MR, Arunkumar A, Arunakaran J. Anticancer effects of ethanolic neem leaf extract on prostate cancer cell line (PC-3) J Ethnopharmacol. 2006;105:246–50. doi: 10.1016/j.jep.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Manikandan P, Letchoumy PV, Gopalakrishnan M, Nagini S. Evaluation of Azadirachta indica leaf fractions for in vitro antioxidant potential and in vivo modulation of biomarkers of chemoprevention in the hamster buccal pouch carcinogenesis model. Food Chem Toxicol. 2008;46:2332–43. doi: 10.1016/j.fct.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Vinothini G, Manikandan P, Anandan R, Nagini S. Chemoprevention of rat mammary carcinogenesis by Azadirachta indica leaf fractions: modulation of hormone status, xenobiotic-metabolizing enzymes, oxidative stress, cell proliferation and apoptosis. Food Chem Toxicol. 2009;47:1852–63. doi: 10.1016/j.fct.2009.04.045. [DOI] [PubMed] [Google Scholar]
- 11.Schuster BG. A new integrated program for natural product development and the value of an ethnomedical approach. J Altern Complement Med. 2001;7(Suppl 1):S61–72. doi: 10.1089/107555301753393823. [DOI] [PubMed] [Google Scholar]
- 12.Paul R, Prasad M, Sah NK. Anticancer biology of Azadirachta indica L (neem): a mini review. Cancer Biol Ther. 2011;12:467–76. doi: 10.4161/cbt.12.6.16850. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T, Alnemri ES. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res. 1997;57:615–9. [PubMed] [Google Scholar]
- 14.Fernández-Luna JL. Apoptosis regulators as targets for cancer therapy. Clin Transl Oncol. 2007;9:555–62. doi: 10.1007/s12094-007-0103-7. [DOI] [PubMed] [Google Scholar]
- 15.Lin JW, Chen JT, Hong CY, Lin YL, Wang KT, Yao CJ, Lai GM, Chen RM. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro Oncol. 2012;14:302–14. doi: 10.1093/neuonc/nor217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Treré D, Montanaro L, Ceccarelli C, Barbieri S, Cavrini G, Santini D, Taffurelli M, Derenzini M. Prognostic relevance of a novel semiquantitative classification of Bcl2 immunohistochemical expression in human infiltrating ductal carcinomas of the breast. Ann Oncol. 2007;18:1004–14. doi: 10.1093/annonc/mdm074. [DOI] [PubMed] [Google Scholar]
- 17.Callagy GM, Webber MJ, Pharoah PD, Caldas C. Meta-analysis confirms BCL2 is an independent prognostic marker in breast cancer. BMC Cancer. 2008;8:153. doi: 10.1186/1471-2407-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali HR, Dawson SJ, Blows FM, Provenzano E, Leung S, Nielsen T, Pharoah PD, Caldas CA. A Ki67/BCL2 index based on immunohistochemistry is highly prognostic in ER-positive breast cancer. J Pathol. 2012;226:97–107. doi: 10.1002/path.2976. [DOI] [PubMed] [Google Scholar]
- 19.Zhang N, Kong X, Yan S, Yuan C, Yang Q. Huaier aqueous extract inhibits proliferation of breast cancer cells by inducing apoptosis. Cancer Sci. 2010;101:2375–83. doi: 10.1111/j.1349-7006.2010.01680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomadaki H, Scorilas A. Molecular profile of the BCL2 family of the apoptosis related genes in breast cancer cells after treatment with cytotoxic/cytostatic drugs. Connect Tissue Res. 2008;49:261–4. doi: 10.1080/03008200802147829. [DOI] [PubMed] [Google Scholar]
- 21.Mercier I, Vuolo M, Madan R, Xue X, Levalley AJ, Ashton AW, Jasmin JF, Czaja MT, Lin EY, Armstrong RC, et al. ARC, an apoptosis suppressor limited to terminally differentiated cells, is induced in human breast cancer and confers chemo- and radiation-resistance. Cell Death Differ. 2005;12:682–6. doi: 10.1038/sj.cdd.4401631. [DOI] [PubMed] [Google Scholar]
- 22.Foo RS, Nam YJ, Ostreicher MJ, Metzl MD, Whelan RS, Peng CF, Ashton AW, Fu W, Mani K, Chin SF, et al. Regulation of p53 tetramerization and nuclear export by ARC. Proc Natl Acad Sci U S A. 2007;104:20826–31. doi: 10.1073/pnas.0710017104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gasco M, Shami S, Crook T. The p53 pathway in breast cancer. Breast Cancer Res. 2002;4:70–6. doi: 10.1186/bcr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000;405:974–8. doi: 10.1038/35016125. [DOI] [PubMed] [Google Scholar]
- 25.Fuchs SY, Adler V, Pincus MR, Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci U S A. 1998;95:10541–6. doi: 10.1073/pnas.95.18.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu J, Sun J, Xue Y. Involvement of JNK and P53 activation in G2/M cell cycle arrest and apoptosis induced by titanium dioxide nanoparticles in neuron cells. Toxicol Lett. 2010;199:269–76. doi: 10.1016/j.toxlet.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell C, Yacoub A, Hossein H, Martin AP, Bareford MD, Eulitt P, Yang C, Nephew KP, Dent P. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther. 2010;10:903–17. doi: 10.4161/cbt.10.9.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:121–30. doi: 10.1517/14728222.2011.644788. [DOI] [PubMed] [Google Scholar]
- 29.Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30:777–82. doi: 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416–30. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Ren Y, Wang L, Li B, Chen Y, Zhao W, Xu W, Li T, Dai F. PTEN mutation spectrum in breast cancers and breast hyperplasia. J Cancer Res Clin Oncol. 2010;136:1303–11. doi: 10.1007/s00432-010-0781-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conley-LaComb MK, Huang W, Wang S, Shi D, Jung YS, Najy A, Fridman R, Bonfil RD, Cher ML, Chen YQ, et al. PTEN regulates PDGF ligand switch for β-PDGFR signaling in prostate cancer. Am J Pathol. 2012;180:1017–27. doi: 10.1016/j.ajpath.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 35.Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A. 1993;90:1112–6. doi: 10.1073/pnas.90.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA, Sutherland RL. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–33. [PubMed] [Google Scholar]
- 37.Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, Barnes D, Peters G. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 1994;54:1812–7. [PubMed] [Google Scholar]
- 38.Wei M, Zhu L, Li Y, Chen W, Han B, Wang Z, He J, Yao H, Yang Z, Zhang Q, et al. Knocking down cyclin D1b inhibits breast cancer cell growth and suppresses tumor development in a breast cancer model. Cancer Sci. 2011;102:1537–44. doi: 10.1111/j.1349-7006.2011.01969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abramson VG, Troxel AB, Feldman M, Mies C, Wang Y, Sherman L, McNally S, Diehl A, Demichele A. Cyclin D1b in human breast carcinoma and coexpression with cyclin D1a is associated with poor outcome. Anticancer Res. 2010;30:1279–85. [PMC free article] [PubMed] [Google Scholar]
- 40.Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–71. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 41.Harfouche R, Echavarria R, Rabbani SA, Arakelian A, Hussein MA, Hussain SN. Estradiol-dependent regulation of angiopoietin expression in breast cancer cells. J Steroid Biochem Mol Biol. 2011;123:17–24. doi: 10.1016/j.jsbmb.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Shim WS, Teh M, Bapna A, Kim I, Koh GY, Mack PO, Ge R. Angiopoietin 1 promotes tumor angiogenesis and tumor vessel plasticity of human cervical cancer in mice. Exp Cell Res. 2002;279:299–309. doi: 10.1006/excr.2002.5597. [DOI] [PubMed] [Google Scholar]
- 43.Hayes AJ, Huang WQ, Yu J, Maisonpierre PC, Liu A, Kern FG, Lippman ME, McLeskey SW, Li LY. Expression and function of angiopoietin-1 in breast cancer. Br J Cancer. 2000;83:1154–60. doi: 10.1054/bjoc.2000.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thurston G. Complementary actions of VEGF and angiopoietin-1 on blood vessel growth and leakage. J Anat. 2002;200:575–80. doi: 10.1046/j.1469-7580.2002.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–4. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 46.Rajkumar L, Canada A, Esparza D, Collins K, Moreno E, Duong H. Decreasing hormonal promotion is key to breast cancer prevention. Endocrine. 2009;35:220–6. doi: 10.1007/s12020-009-9155-5. [DOI] [PubMed] [Google Scholar]
- 47.Rajkumar L, Arumugam A, Elsayed A, Schecter S, Kotkowski E, Castillo R, de la Torre A, Hernandez C. Long-term hormonal promotion overcomes genetic resistance to mammary cancer. Steroids. 2011;76:31–7. doi: 10.1016/j.steroids.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 48.McDonald JW, Pilgram TK. Nuclear expression of p53, p21 and cyclin D1 is increased in bronchioloalveolar carcinoma. Histopathology. 1999;34:439–46. doi: 10.1046/j.1365-2559.1999.00632.x. [DOI] [PubMed] [Google Scholar]
- 49.Shiao YH, Palli D, Caporaso NE, Alvord WG, Amorosi A, Nesi G, Saieva C, Masala G, Fraumeni JF, Jr., Rice JM. Genetic and immunohistochemical analyses of p53 independently predict regional metastasis of gastric cancers. Cancer Epidemiol Biomarkers Prev. 2000;9:631–3. [PubMed] [Google Scholar]
- 50.Charafe-Jauffret E, Tarpin C, Bardou VJ, Bertucci F, Ginestier C, Braud AC, Puig B, Geneix J, Hassoun J, Birnbaum D, et al. Immunophenotypic analysis of inflammatory breast cancers: identification of an ‘inflammatory signature’. J Pathol. 2004;202:265–73. doi: 10.1002/path.1515. [DOI] [PubMed] [Google Scholar]