

Abstract
Increasing evidence suggests tumor-associated macrophages (TAMs) are polarized M2 subtype of macrophage that exerts pro-tumor effects and promote the malignancy of some cancers, but the concrete mechanism is not well defined. Our previous research exhibited that proto-oncogene AP-1 regulated IL-6 expression in macrophages and promoted the formation of M2 macrophages. In this study, we investigate whether extra-cellular stimulus M-CSF help this process or nuclear factor NFκB has a synergistic role in the activation state of polarized M2 subtype of macrophage. RAW 264.7 macrophage and 4T1 mouse breast cancer cells were co-cultured to reconstruct tumor microenvironment. Being co-cultured with 4T1 or its supernatant, the expression of c-Jun, the member of AP-1 family, has a dramatically increase both on the level of gene and on the protein in RAW 264.7 macrophages, but the expression of c-Fos does not increase neither on the level of gene nor on the protein. After co-cultured with 4T1, RAW 264.7 has a higher consumption of M-CSF than RAW 264.7 macrophages alone. With the stimulation of M-CSF, the mRNA of c-Jun increased significantly, but decreased remarkably after adding the anti-M-CSF. And at the same time, p50, the member of NFκB family, has a similar tendency to c-Jun. WB results suggest that with the stimulation of M-CSF, p-Jun in nuclear increases heavily but decreases after the neutralizing antibody added. Coimmunoprecipitation and immunoblotting techniques confirmed that c-Jun and p50 NFκB coprecipitated, and c-Jun protein expression is properly enhanced with rM-CSF effect. In conclusion, M-CSF induces macrophage transformation by upregulating c-Jun with a certain synergy of NFκB. Our study may present a novel therapeutic strategy against cancer.
Keywords: M-CSF, NFκB, c-Jun, co-culture, transformation
Introduction
In tumor cells, abnormal signal transduction is closely related to tumor metastases. The activated CSF-1/CSF-1R pathway is found critical in the growth and metastasis in breast, ovarian and liver cancer,1 and blocking the CSF-1/CSF-1R can inhibit cancer recurrence and metastasis.2 So the M-CSF/CSF-1/CSF-1R signaling pathway has attracted much attention in recent years. The transcription factor activator protein-1 (AP-1) is a downstream molecule in the M-CSF/MAPK signaling pathway, which is composed of Jun, Fos, ATF, and the JDP subfamily.3 It is proved involving in cell proliferation, differentiation and transformation, and essential for tumor formation, metastasis and invasion.4
Our previous study reported that we could mimic the tumor microenvironment in vitro using co-cultured murine 4T1 breast carcinoma cells and RAW 264.7 mouse macrophages. We confirmed that 4T1 cells were able to induce RAW 264.7 macrophages from M1 to M2 macrophage in vitro. And the transcription factor Fos family member Fra-1 induced this transformation.5 However, the specific molecular mechanisms are still unclear. Many recent researches have denoted that M-CSF has other important roles in the tumor microenvironment. It acts as an autocrine growth factor in promoting the deterioration of tumor cells.6,7 For example, it can recruit and activate M2 macrophages, which in turn contribute to promote metastasis. Inflammatory cell infiltration has been demonstrated highly correlating with poor prognosis in breast cancer.8 The vast majority of these infiltrated cells are macrophages, and M-CSF is the major cytokine that attracts these cells. This is a relatively new hypothesis and has been confirmed in molecular level recently. It is proved that M2 transformation is triggered by upstream M-CSF in vitro.9 As an initiator, M-CSF mediates this process through the downstream signaling pathways Fos/ERK and Jun/JNK.10 Similar to AP1, nuclear factor κB (NFκB) can mediate the relationship between inflammation and cancer through the upstream protein IKKβ.11 Active NFκB in the tumor tissue can activate the stromal cells that produce VEGF and degrade enzymes, promote tumor invasion and metastasis.12
In this study, we explore the mechanism by inquiring the upstream and midstream of the signaling pathways. We focus on two areas. First, we study whether M-CSF mediates the downstream signaling pathways Fos/ERK and Jun/JNK to recruit M2 macrophages. Second, we investigate whether AP-1 facilitates M-CSF inducing M2 macrophage transformation and whether NFκB has a synergistic role in this process.
Results
c-Jun participates in the transformation of M1 to M2 macrophages, but the function of c-Fos is not clear
It has been shown that after co-culture of RAW 264.7 and 4T1 cells, the number of M2 macrophages (F4/80+CD206+) in the co-culture increased obviously with time compared with the RAW 264.7 cells alone.5 This phenomenon was also observed when the supernatant of the 4T1 cells (TSN) and the RAW 264.7 macrophages were co-cultured. The expression of c-Fos and c-Jun in the cells (Fig. 1A and B) is detected under the same conditions. The mRNA levels of c-Jun were significantly increased (~2.9-fold assessed by real-time PCR) in the RAW 264.7 co-culture group but not in that of cultured alone. This result was further confirmed by immunocytochemistry (ICC) staining in the cultured RAW 264.7 macrophages and 4T1 cells (Fig. 1C). As expected, in the co-cultured group, more RAW 264.7 macrophages were immunoreactive with c-Jun-specific antibody, indicating that the level of c-Jun protein increased in this group.
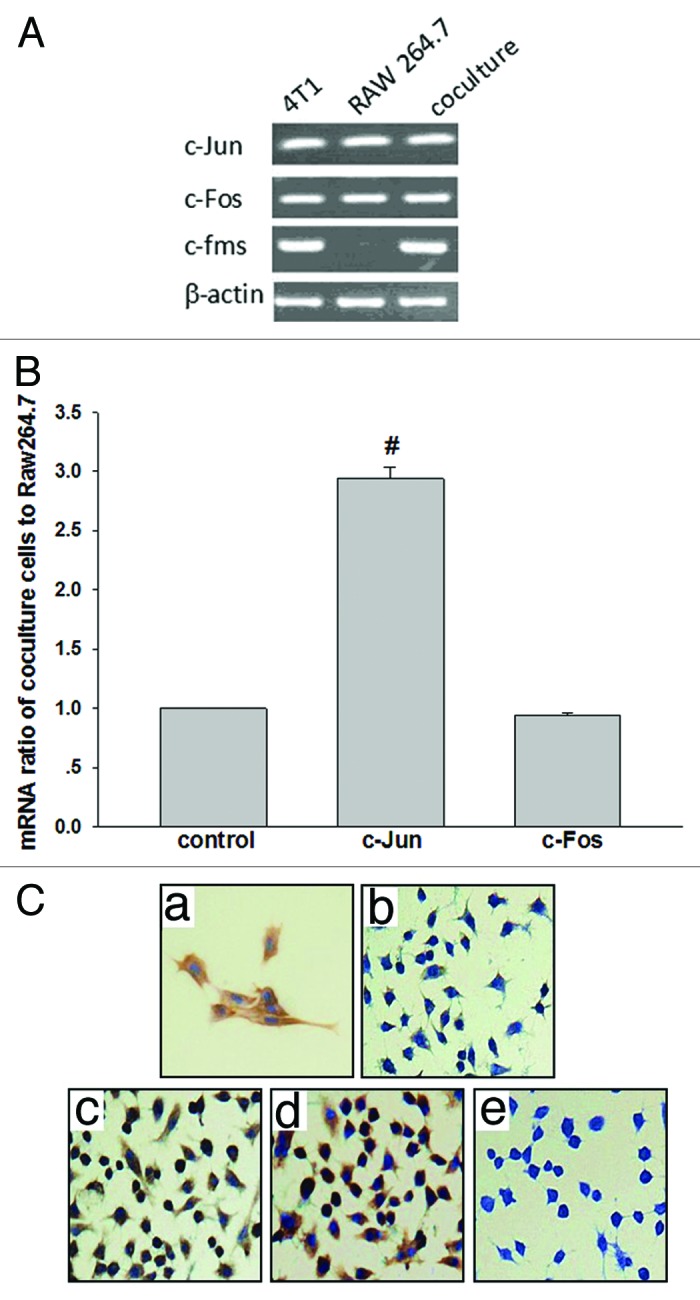
Figure 1. Increased c-Jun mRNA and protein levels in co-cultured cells. (A) Transcript levels of c-Jun, c-Fos, and c-fms in 4T1, RAW 264.7, and co-culture cells were shown by RT-PCR analysis. β-actin was used as an internal control. (B) Co-culture-indcued (RAW 264.7 macrophages: 4T1 cells = 1:4; 48 h) upregulation of c-Jun and c-Fos in mRNA in RAW 264.7 macrophages was verified by real-time PCR (quantitative RT-PCR). GAPDH was used to normalize the c-Jun and c-Fos levels. (C) c-Jun immunocytochemical staining in Raw 264.7 macrophages after treatment with different time of co-culture cells. (a) Positive control. (b) Weak cytoplasticimmunostaining was observed after 6 h. (c) Strong cytoplasticimmunostaining was observed after 24 h. (d) Strong cytoplasticimmunostaining was very pronounced after 48 h. (e) Nonspecific immunoglobulin was used as a negative control. 400× magnification.
M-CSF participates in the transformation of M1 to M2 macrophages by directly increasing the expression of c-Jun via the MAPK signaling pathway
An ELISA assay was used to detect the content of M-CSF in the co-cultured and cultured alone cells (Fig. 2). 4T1 cells secreted high amounts of M-CSF. According to the standard curve created (y = 0.0228x + 0.0312), the M-CSF levels were as high as 6.21 ng/ml in supernatant of 4T1 cells and the RAW 264.7 macrophages secreted a certain amount of M-CSF, which decreased with time. The M-CSF in the supernatant of the co-cultured cells was less than that of the RAW 264.7 macrophages alone. The 4T1 cells barely express M-CSF receptor (c-fms) mRNA (Fig. 1A), which suggests that exogenous M-CSF (from the 4T1 cells) did not balance the survival need of the RAW 264.7 macrophages in the co-culture.
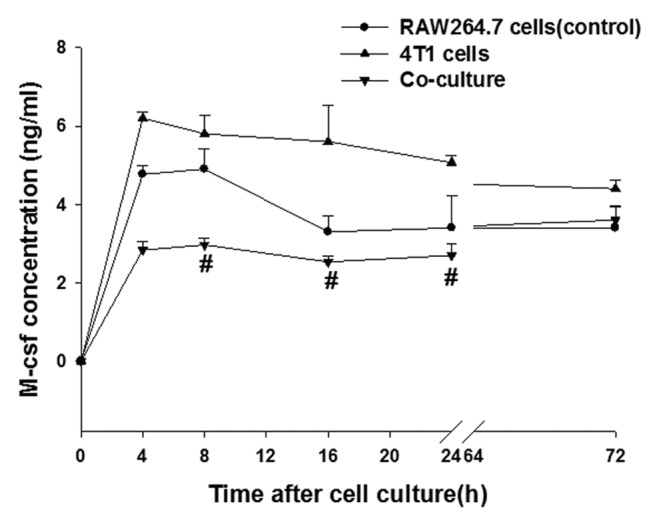
Figure 2. Production of the cytokine M-CSF in the RAW 264.7, 4T1, and co-culture cells was determined by enzyme-linked immunosorbent assay (ELISA). RAW 264.7, 4T1 were cultured or cocultured for the indicated time periods, after which the cytokine concentrations were measured by ELISA. The data on M-CSF production represent the mean (standard error [SE]) of 3 experiments. Statistical differences between groups were calculated by the Student t test. #P < 0.05 vs. RAW 264.7 cells.
To further study the survival effect of M-CSF on the RAW 264.7 macrophages, the supernatant from the 4T1 cells (TSN, tumor culture supernatant) and recombinant rM-CSF were added to the co-culture system. The mRNA expression levels of c-Jun, c-Fos, p65, and p50 NFκB at 12 h, 24 h, and 48 h were detected. At 12–48 h, the expression levels of c-Jun and p50 increased to varying extents (Fig. 3A). Additionally, the stimulation was neutralized with anti-M-CSF at 24 h. In the rM-CSF-stimulated group, the expression of c-Jun and p50 mRNA increased up to 86.22 and 9.51 times higher than that in the rM-CSF non-stimulated group (Fig. 3B).

Figure 3. JNK and NFκB pathway were activated by M-CSF stimulation. (A) RT-PCR analysis showing that the level of c-Jun and p50 NFκB in RAW 267.4 macrophages treated by 40% TSN (tumor culture supernatant) and rM-CSF (30 ng/ml) begin to rise gradually as incubation time extended, control cells were treated with the same medium at 0 h. (B) Real-time PCR claiming a dramatically effect of neutralizing antibodies to rM-CSF (anti-rM-CSF) on the mRNA expression of c-Jun, c-Fos, p50, and p65 in RAW 264.7 macrophages after 24 h incubation, and the effect occur basically simultaneous. (C) Western blot analysis demonstrating that the expression of c-Jun phosphorylation protein (P-Jun) in RAW 264.7 macrophages nucleus (under) was markedly reduced after anti-rM-CSF treatment, and M-CSF having little effect on c-Jun phosphorylation protein in cytoplasm (upper). The blots were stripped and reprobed with anti-β-actin to confirm equal protein loading. #P < 0.05 vs. RAW 264.7, *P < 0.05 vs. RAW 264.7 + rM-CSF.
Based on these data, the level of the c-Jun protein in RAW 264.7 macrophages with or without rM-CSF stimuli (Fig. 3C) was evaluated. c-Jun is a natural substrate of JNK, and its phosphorylation indicates that JNK is in active state. From the nucleoprotein analysis of the RAW 264.7 macrophages with rM-CSF stimulation, the phosphorylation of c-Jun was increased compared with the group without rM-CSF stimulation. Therefore, M-CSF stimuli activate the JNK/MAPK signal transduction pathways in RAW 264.7 macrophages. After M-CSF stimulated, the expression of the c-Jun whether in gene or in protein increased obviously in the nuclear of RAW 264.7 macrophages, which may be performed through activation of the JNK/MAPK signal transduction pathways. However, the impact of M-CSF stimulation on cytoplasmic c-Jun expression is minimal.
NFκB may be synergistically involved in the regulation of c-Jun in RAW 264.7 macrophages transformation
Simultaneously, similar to c-Jun, the mRNA expression of p50 NFκB also increased after M-CSF stimulation. In the rM-CSF-stimulated group, the expression of p50 mRNA increased up to 9.51 times higher than that in the rM-CSF non-stimulated group and that dropped sharply in the anti-M-CSF group with values of 0.18 (Fig. 3A and B). This result suggested that activation of NFκB and JNK pathway were involved in M-CSF-mediated M2 polarization, though the expression of p65 NFκB showed little change. In order to prove the claim, to determine whether NFκB cooperating with c-Jun induce M2 polarization, we proceed in three phases. First, we performed immunoblotting experiments with anti-p50 NFκB under different treatment conditions to determine how M-CSF-mediated induction of macrophage alternative activation is linked to the NFκB pathway. The p50 is a subunit of NFκB, we chose this subunit randomly to stand for NFκB. We detected a strong protein band not only in TSN, but in M-CSF-treated group. Obviously, this strong band appeared after pretreatment with M-CSF and TSN (Fig. 4A). Second, we continued to perform immunoprecipitation with anti-c-Jun and anti-p50 (Fig. 4B) to further define the interaction between c-Jun and p50 in the absence and presence of M-CSF or a p50 inhibitor, andrographolide. After andrographolide inhibited p50 NFκB, c-Jun protein expression decreased as well as the interaction between c-Jun and p50 NFκB weakened, the results suggested that interaction between c-Jun and p50 NFκB was affected by M-CSF and was impaired after inhibition of p50 using andrographolide treatment. Third, to further confirm that p50 synergistically upregulates c-Jun expression, we used retroviral shRNA strategies to knock down c-Jun protein levels and to assess p50 expression in RAW 264.7 macrophages induced by M-CSF. As shown in (Fig. 4C), c-Jun protein levels were stably knocked down, c-Jun shRNA-mediated silencing was associated with an appropriate decrease in p50 expression. The result suggested that c-Jun and p50 could influence each other, and the underlying mechanism need to be elucidated in future. However, it strengthened this agreement with the certain synergy of p50 to upregulate c-Jun protein expression.
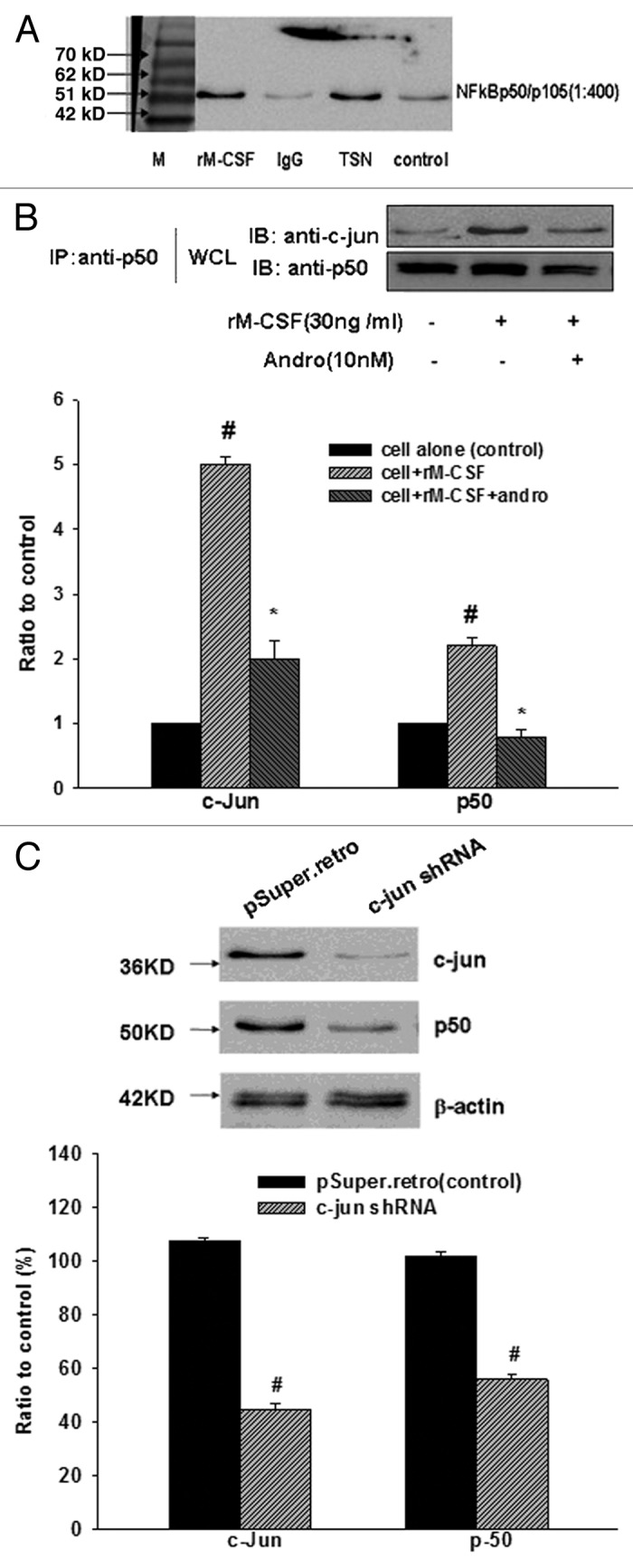
Figure 4. Andro and c-Jun shRNA damage the interaction of c-Jun and p50 NFκB in M-CSF-stimulated cells. (A) Whole cell lysates (WCL) were prepared for immunoprecipitation with anti-p50 NFκB. RAW 267.4 macrophages were pretreated with 40% TSN and stimulated by M-CSF (30 ng/ml) for 24 h. After SDS-PAGE, Coomassie brilliant blue G-250/sliver staining was performed. Strong brands were found with TSN and rM-CSF treatment. (B) Coimmunoprecipitation and immunoblotting of c-Jun and p50 NFκB, RAW 267.4 macrophages were pretreated with rM-CSF (30 ng/ml) for 24 h and stimulated by andrographolide (Andro, 10 nM) for another 12 h. #P < 0.05 vs. cell alone (control), *P < 0.05 vs. cell + rM-CSF. (C) Infection of RAW 264.7 macrophages with c-Jun shRNA leads to decreased p50 expression. Immunoblot analysis of RAW 264.7 macrophages infected with pSuper.retro (vector control) or pSuper-c-Jun.shRNA (c-Jun shRNA). β-actin was used as an internal loading control.
Next, we demonstrate that the blockade of NFκB pathway by adding andrographolide affected M-CSF-mediated M2 polarization state including polarized cytokine production, characteristic surface marker expression, arginase 1 (Arg1) and iNOS expression using real time-PCR. M-CSF-exposed Raw 264.7 macrophages exhibited the classical M2 activation pattern, such as the upregulation of CCL22, and Arg1, and an augmented production of IL-10. We further examined how those cells responded to a NFκB pathway inhibitor by adding andrographolide. In contrast, M-CSF-exposed Raw 264.7 macrophages after andrographolide stimuli induced completely absent in the patterns that had ever been exposed to M-CSF (Fig. 5). Additionally, Figure 5 showed the defective expression of M1 phenotype genes (IL-12p35, TNF-α, and iNOS) in M-CSF-treated Raw 264.7 macrophages, and the defect could be compensated by adding andrographolide to some extent. These results clearly indicate that NFκB may affect the regulation of M-CSF in RAW 264.7 macrophages transformation with a markedly altered phenotype, particularly for this phenotype transformation process contributes to tumor progression, blocking it could be further developed for the clinical treatment of tumors.
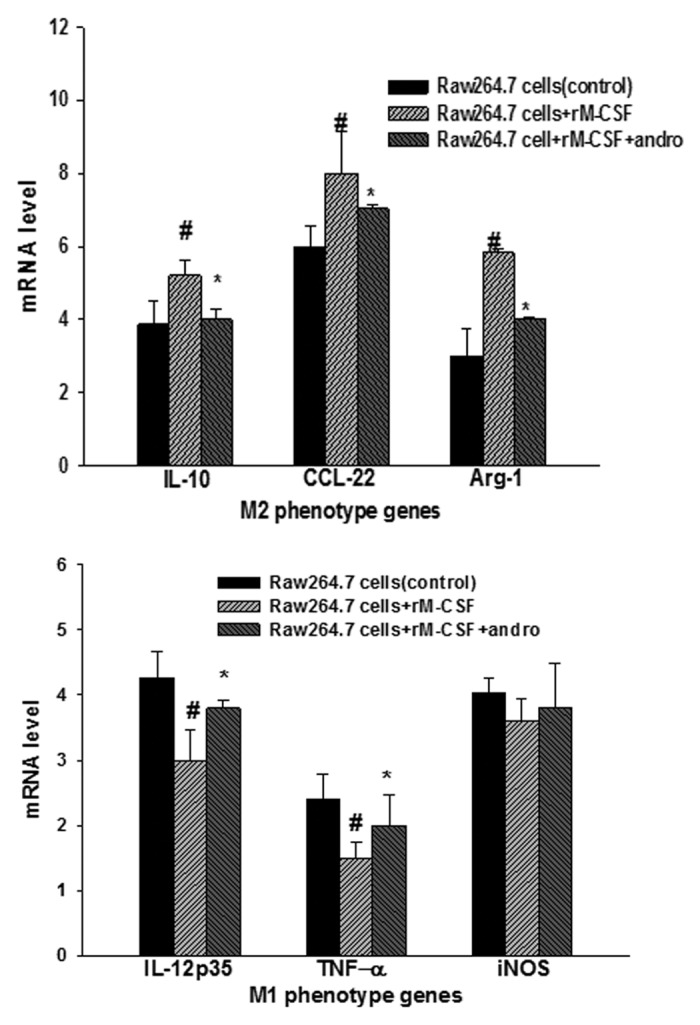
Figure 5. Phenotype identification of surface markers (M1 phenotype genes [IL-12p35, TNF-α, and iNOS] and M2 phenotype genes [IL-10, CCL22, and Arg1]) was determined by real-time PCR. Blocking NFκB by adding Andro affected M-CSF-mediated M2 polarization state including polarized cytokine production. RAW 267.4 macrophages untreated, stimulated with M-CSF for 24 h, or stimulated with andro for another 12 h after 24 h of M-CSF treatment and were subsequently analyzed. Results are representative of 10 separate experiments. Statistical differences between groups was calculated by one way ANOVA. #P < 0.05 vs. RAW 264.7, *P < 0.05 vs. RAW 264.7+rM-CSF.
Taken together, these data suggested that a cascade of molecular events initiated by M-CSF may be the mechanism of TAMs formation. That is, c-Jun may involve in the NFκB activation after M-CSF treatment in macrophages. Then c-Jun and p50 NFκB may cooperatively induce a series of events, such as upregulating expression of the IL-6/JAK1/STAT6, thereby attenuating M2 polarization and promoting tumor growth.
Discussion
Our previous study demonstrated that co-cultured RAW 264.7 macrophages could confer a M2 macrophage phenotype. This phenotype is characterized by high levels of IL-10, CCL2, CCL22, and Arg-1 secretion with a decrease in secretion of IL-12p35, TNF-α, and iNOS. AP-1 is a key molecule that facilitates this transformation.5 Both c-Fos and c-Jun are immediate-early genes (IEG), and their high expression and transactivation play an important role in cell proliferation and phenotype differentiation. In cells, c-Fos and c-Jun dimerize to form AP-1. A co-cultured cell environment is similar to the tumor microenvironment. Therefore, detecting c-Jun and c-Fos expression in RAW 264.7 macrophages under co-cultured conditions is the best way to study macrophage transformation in vitro. Using real-time PCR, c-Jun mRNA levels in the co-cultured cells were found 2.9 times higher than those in RAW 264.7 macrophages alone. In contrast, the levels of c-Fos mRNA were almost the same in both conditions (a ratio less than 1). Otherwise the levels of c-Fos protein were hardly detected by ICC. Some research showed that when c-Fos combined with c-Jun, the half-life of c-Fos often shortened to 45–120 min, and c-Fos degraded more quickly because it contained AU rich elements in its 3′ untranslated regions and some fragments in the protein coding region were very unstable.13,14 This is perhaps the reason we couldn’t capture the signal change of c-Fos expression.
But the question is: How does c-Jun work in the transformation of M1 to M2 macrophages? With this question, we detected the M-CSF content in the co-cultured cells and the supernatant using ELISA assay. In the 4T1 cell supernatant, M-CSF secretion was as high as 6.21 ng/ml. and the RAW 264.7 macrophages could secrete a certain amount of M-CSF. The concentration of M-CSF was reduced with the incubation time increased. In the RAW 264.7 macrophages that were co-cultured, the levels of M-CSF were decreased compared with that of the RAW 264.7 macrophages cultured alone, and the 4T1 cells did not express M-CSF receptor mRNA in Figure 1A. This finding suggested an exogenous M-CSF supply (4T1 cells secrete) is much less than the consumption for the RAW 264.7 macrophages when the cells were co-cultured. The results of others found that leukemia inhibitory factor (LIF) and IL-6 promoted the conversion of mononuclear cells to macrophages, and IL-6 promoted M2 macrophage generation, which was dependent on autocrine M-CSF consumption.15 M-CSF is known able to stimulate many signaling pathways (including the MAPK pathway) to promote the proliferation and differentiation of macrophages. As a central hub, AP-1 happened to be in this molecular pathway. So, M-CSF stimulation and neutralization were used in the study to detect the sensitivity of c-Jun and c-Fos to M-CSF in the 4T1 and RAW 264.7 cell co-culture system.
At the mRNA level, real-time PCR revealed that c-Fos, c-Jun, c-fms, p50, and p65 reached their highest expression when stimulated in 24 h, on very reliable premises that c-Fos, c-Jun, c-fms, p50, p65, c-rel, IKKα, IKKβ, and IL-6 (data not shown) were expressed to varying degrees at different time points (12–48 h) in RAW 264.7 macrophages. At the protein level, phosphorylated c-Jun in both the cytoplasmic and nuclear fractions was investigated. In the resting state, phosphorylated c-Jun was expressed at low levels in the nucleus. After the rM-CSF stimulus was added, the expression increased significantly, and the growth could be suppressed by a specific antibody. However, the phosphorylated protein in the cytoplasm did not decrease. There are two possible reasons for this result. First, M-CSF may not play a switching function, so the neutralizing antibody may affect the efficiency of phosphorylation and the protein’s ability to translocate to the nucleus. Therefore, the protein in the cytoplasm did not change greatly, but significantly increased in the nucleus. Second, there are other factors that influence the production, aggregation and transfer into the nucleus of phosphorylated c-Jun.
While choosing a phosphorylated antibody, we noticed that ser 73 affected the formation of AP-1 and could improve the ability to induce transcription.16 Using this antibody, our research made a connection between the MAPK/ERK pathway and the JNK/SAPK pathway. It is known that c-Fos and c-Jun mRNA levels are controlled by the ERK pathway. This pathway controls IEG expression and the JNK pathway. The JNK pathway activates c-Jun and enhances its transcription activity by phosphorylating the c-Jun protein at the N-terminal ser 63 and ser 73 phosphorylation sites could promote AP-1 dimer formation. Therefore, JNK may regulate the activation of c-Jun at the protein level. Thus, we can conclude that M-CSF controls AP-1 family transcription and activation of these transcription factors at the protein level through the MAPK pathway. This activation facilitates the conversion of RAW 264.7 to M2 macrophages.
Recently, more research shows that malignant tumors originated due to infection.17,18 Clinically, anti-inflammatory therapy can be used in the early stage of tumorigenesis to avoid malignant lesions, which proves the feasibility of TAMs as a target for cancer treatment.19 NFκB is closely related to inflammation and can often promote tumor metastasis and microvascular generation.20,21 Other research evidence has shown that TAMs accumulate a large amount p50 NFκB homologous dimer in the nucleus when responding to the M1 macrophage type cell signals. p50 NFκB overexpression does not only trigger macrophages that are LPS tolerant in a mice fibrosarcoma and human ovarian cancer,22 but also inhibit IL-12 promoter activity in RAW 264.7 macrophages.23-25 Our study found that expression of the NFκB family p50 increased after M-CSF stimulation, and IKKα, the kinase subunit that catalyzes IκB degradation showed diverse degrees of expression.26,27 Increased p50 NFκB expression suggests that the NFκB pathway is activated after M-CSF stimulation. other findings offers that NFκB and AP-1 can inhibit the production of MMP9 and proinflammatory cytokine.28,29 Our finding indirectly showed that NFκB might act a synergistic role in M-CSF-MAPK-AP-1-related RAW 264.7 to M2 macrophages transformation.
The co-immunoprecipitation experiments observed that c-Jun interacted with p50 NFκB in the presence of rM-CSF. After pretreating with andrographolide, this interaction was impaired. However, after immunoprecipitation with c-Jun, p50 protein expression had no significant difference among groups stimulated by rM-CSF. This may due to that p50 was only one part of NFκB, c-Jun could interact with other parts of NFκB. These results suggested that NFκB synergistically effected with c-Jun in RAW 264.7 macrophages transformation. In addition, our work found that andrographolide exerted its inhibitory on M2 macrophage alternative activation, that can inhibit the malignant, or cancerous changes of tumor microenvironment, but have no effect on normal cells, thus it might has a better selectivity in the treatment of cancer.
In summary, we demonstrated that M-CSF upregulates the expression of the transcriptional factor c-Jun, which in turn promote M2 macrophages transformation. And the nuclear factor NFκB synergistically works with c-Jun in the transformation of polarized M2 subtype of macrophage. What’s more, this transformation contributes to tumor progression, so blocking M-CSF,further NFκB and JNK pathway could be a promising target for the clinical tumor therapy at a totally new and advanced level.
Materials and Methods
Cell culture
The RAW 264.7 macrophages were purchased from the School of Basic Medicine, Peking Union Medical College. The 4T1 cells were kindly provided by Dr Ostrand-Rosenberg (University of Maryland). Andrographolide (Andro, a p50 inhibitor) was purchased from Sigma-Aldrich. The RAW 264.7 macrophages were co-cultured with 4T1 cells at 1:4 ratio for 24, 48, 72, or 96 h.5 In the cell stimulation test, RAW 264.7 macrophages were incubated with the supernatant from the 4T1 cells (TSN, tumor culture supernatant, diluted 1:2.5) and rM-CSF (30 ng/ml) for 24, 48, and 72 h. Anti-M-CSF (1 ug/ml) was used to quench the action of rM-CSF.
RT-PCR and real-time PCR
RT-PCR was performed to detect c-Jun, c-Fos, c-fms, p50, and p65 mRNA levels in the cells cultured with the 4T1 cell supernatant (diluted 1:2.5) and rM-CSF (30 ng/ml) for 0–48 h. The total cellular RNA (1 ug) was isolated from the cultured cells and reverse transcribed using oligo (dT) and M-MLV reverse transcriptase (Promega). Real-time PCR were performed to quantify c-Jun, c-Fos, p50, and p65 mRNA which was extracted from the cells cultured among 48 h. The cDNAs were amplified using the Real-Time PCR Detection System (Bio-Rad Laboratories). For each sample, the ΔΔCt values were calculated according to the derived equation 2−ΔΔCt (the Ct value of the target gene minus the Ct value of the housekeeping gene GAPDH). All real-time PCR results were expressed as fold changes in the mRNA expression compared with the control cells. The data represented are from three independent experiments performed in triplicate. Primers were exhibited as below:
c-Jun (forward: 5′-GTGCCAACTC ATGCTAACG-3′; reverse: 5′-GCAACCAGTC AAGTTCTCAA G-3′),
c-Fos (forward: 5′-CTCTAGTGCC AACTTTATCC C-3′; reverse: 5′-ATAGCTGCTC TACTTTGCCC-3′),
c-fms (forward: 5′-GCGAGGGTTC ATTATCCG-3′; reverse: 5′-ACTGTTTAGG GGGATTTCCA-3′),
β-actin (forward: 5′-ATATCGCTGC GCTGGTCGTC-3′; reverse: 5′-AGGATGGCGC TGGTCGTC-3′),
p50 NFκB (forward: 5′-GCATTCTGAC CTTGCCTATC-3′; reverse: 5′-TTGGATGCAT TCGGGG-3′),
p65 NFκB (forward: 5′-ATGTGCATCG GCAAGTGG-3′; reverse: 5′-TGCTGGGAAG GTGTAGGG-3′),
IL-10 (forward: 5′-GGGCCAGTAC AGCCGGGAAG-3′; reverse: 5′-CTGGCTGAAG GCAGTCCGCA-3′),
CCL22 (forward: 5′-GTGCCGATCC CAGGCAGGTC-3′; reverse: 5′-GGCGTCGTTG GCAAGGCTCT-3′),
Arg1 (forward: 5′-AGAGACCACG GGGACCTGGC-3′; reverse: 5′-TGGACCTCTG CCACCACACC-3′),
IL-12p35 (forward: 5′-GCACCCGCGT CGTGACCATC-3′; reverse: 5′-GCCCACCAGG CCAAGACCA C-3′),
TNF-α (forward: 5′-AAGGCCGGGG TGTCCTGGAG-3′; reverse: 5′-AGGCCAGGTG GGGACAGCTC-3′),
iNOS (forward: 5′-CCGCTGCCTT CCTGCTGTCG-3′; reverse: 5′-CCTCCGAGGG GGTGTGGTCC-3′),
GAPDH (forward: 5′-AGGCCGGTGC TGAGTATGTC-3′; reverse: 5′-TGCCTGCTTC ACCACCTTCT-3′).
Measurement of M-CSF concentration using ELISA
The supernatants were collected from the cells which cultured for 0 h, 4 h, 8 h, 16 h, 24 h, and 72 h, then they were used for measuring the murine M-CSF concentration through a mouse M-CSF-specific ELISA kit (R&D System) according to the manufacturer’s instructions. The results are from three independent experiments performed in triplicate.
Immunocytochemistry
The co-cultured cells (RAW 264.7 macrophages: 4T1 cells = 1:4) were applied on slides and fixed in a 4% paraformaldehyde solution. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide. To reduce the non-specific binding, the slides were treated with a solution of goat serum (ZSGB-BIO). Then, the sections were incubated overnight at 4 °C with a rabbit anti-c-Jun primary antibody (1:200, sc-44). After reacting with horseradish-peroxidase (HRP)-labeled goat anti-rabbit IgG (ZSGB-BIO), the immunoactivity was detected with diaminobenzidine (DAB; Boster). Lipopolysaccharide (LPS, 100 ng/mL)-treated RAW 264.7 macrophages were used as the positive control, and rabbit IgG substitute was used as the negative control. A half-quantitative evaluation of the immunostaining was applied by counting the immunopositive cells (identified by the presence of brown staining) in 10 randomly selected high power fields for each case.
Western blot analysis
The nuclear and cytoplasmic protein extraction kit was purchased from KeyGEN Biotech. The cells (5 × 106) were handled according to the manufacturer’s instructions. The total cell lysates were subjected to SDS-PAGE (10% acrylamide), and the proteins were transferred to PVDF membranes (Bio-Rad Laboratories). The membranes were blocked for 2 h at room temperature and incubated overnight with rabbit anti-c-Jun (phosphor-Ser 73) antibody (1:500, 11003-3; SAB). After washing, the membranes were incubated for 1 h at room temperature with HRP-conjugated goat anti-rabbit IgG (1:15 000, sc2004). Finally, the labeled proteins were visualized using the ECL plus reagent (Millipore).
Coimmunoprecipitation and immunoblotting analyses
The cell proteins were extracted as described above. For the coimmunoprecipitation studies, RAW 264.7 macrophages were cultured with rM-CSF (30 ng/ml) for 24 h. c-Jun was immunoprecipitated and detected by western blot analysis using either rabbit anti-p50 1:400 (Santa Cruz Biotechnology) or mouse anti-c-Jun 1:200 (Boster). The immuno-complex was captured by gently adding Protein A/G-Agarose Plus beads (Gendepot) and subsequently subjected to western blotting (10% SDS-PAGE). To investigate the mechanism of NFκB-AP-1 compound formation, we added andrographolide (10 nM, a non-phosphorylation site exposure blocker) to the medium for up to 12 h.
c-Jun knockdown using RNAi
RNAi-mediated c-Jun knockdown was accomplished by shRNA produced by the DNA-based shRNA-expressing retroviral vector (pSuper-Retro). The vectors were from GenePharma. The knockdown experiment was conducted with Lipofectamine (Invitrogen) according to the manufacturer's instructions. RAW 264.7 macrophages were cultured in full medium for 48 h before being treated under M-CSF (30 ng/ml). The c-Jun shRNA target sequence is GAUGGAAACG ACCUUCUAUT T.
Statistical analysis
Results are mean ± SE and were analyzed with SigmaStat 10.0 software (SPSS). The differences between two groups were assessed using the Student t test. The differences among three or more groups were evaluated using a one-way ANOVA followed by the Dunnett test. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Qing-Shan Wang for the preliminary studies and Yongzhe Che (Nankai University) for technical assistance. This work was supported in whole by the National Science Foundation of China (NSFC) 81171975, Tianjin Institutes for Basic Sciences 09JCYBJC10800.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26718
References
- 1.Kavitha CV, Deep G, Gangar SC, Jain AK, Agarwal C, Agarwal R. Silibinin inhibits prostate cancer cells- and RANKL-induced osteoclastogenesis by targeting NFATc1, NF-κB, and AP-1 Activation in RAW264.7 cells. Mol Carcinog. 2012 doi: 10.1002/mc.21959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley ER, Guilbert LJ, Tushinski RJ, Bartelmez SH. CSF-1--a mononuclear phagocyte lineage-specific hemopoietic growth factor. J Cell Biochem. 1983;21:151–9. doi: 10.1002/jcb.240210206. [DOI] [PubMed] [Google Scholar]
- 3.Vesely PW, Staber PB, Hoefler G, Kenner L. Translational regulation mechanisms of AP-1 proteins. Mutat Res. 2009;682:7–12. doi: 10.1016/j.mrrev.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Edwards DR. Cell signalling and the control of gene transcription. Trends Pharmacol Sci. 1994;15:239–44. doi: 10.1016/0165-6147(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 5.Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20:701–12. doi: 10.1038/cr.2010.52. [DOI] [PubMed] [Google Scholar]
- 6.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–40. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambers SK. Role of CSF-1 in progression of epithelial ovarian cancer. Future Oncol. 2009;5:1429–40. doi: 10.2217/fon.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kacinski BM. CSF-1 and its receptor in ovarian, endometrial and breast cancer. Ann Med. 1995;27:79–85. doi: 10.3109/07853899509031941. [DOI] [PubMed] [Google Scholar]
- 9.Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de Waal-Malefyt R, Ottenhoff TH. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A. 2004;101:4560–5. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morandi A, Barbetti V, Riverso M, Dello Sbarba P, Rovida E. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PLoS One. 2011;6:e27450. doi: 10.1371/journal.pone.0027450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makarov SS. NF-kappaB as a therapeutic target in chronic inflammation: recent advances. Mol Med Today. 2000;6:441–8. doi: 10.1016/S1357-4310(00)01814-1. [DOI] [PubMed] [Google Scholar]
- 12.Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, Bottazzi B, Colombo MP, Mantovani A, Sica A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66:11432–40. doi: 10.1158/0008-5472.CAN-06-1867. [DOI] [PubMed] [Google Scholar]
- 13.Kovács KJ, Sawchenko PE. Sequence of stress-induced alterations in indices of synaptic and transcriptional activation in parvocellular neurosecretory neurons. J Neurosci. 1996;16:262–73. doi: 10.1523/JNEUROSCI.16-01-00262.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larsen PJ, Mikkelsen JD. Functional identification of central afferent projections conveying information of acute “stress” to the hypothalamic paraventricular nucleus. J Neurosci. 1995;15:2609–27. doi: 10.1523/JNEUROSCI.15-04-02609.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, Preisser L, Anegon I, Catala L, Ifrah N, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–30. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- 16.Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–57. doi: 10.1016/S0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 17.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coussens LM, Werb Z. Inflammatory cells and cancer: think different! J Exp Med. 2001;193:F23–6. doi: 10.1084/jem.193.6.F23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang LJ, Zhou X, Wang W, Tang F, Qi CL, Yang X, Wu S, Lin YQ, Wang JT, Geng JG. Andrographolide inhibits oral squamous cell carcinogenesis through NF-κB inactivation. J Dent Res. 2011;90:1246–52. doi: 10.1177/0022034511418341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YD, Ye BQ, Zheng SX, Wang JT, Wang JG, Chen M, Liu JG, Pei XH, Wang LJ, Lin ZX, et al. NF-kappaB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J Biol Chem. 2009;284:4473–83. doi: 10.1074/jbc.M806010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziegler-Heitbrock HW, Petersmann I, Frankenberger M. p50 (NF-kappa B1) is upregulated in LPS tolerant P388D1 murine macrophages. Immunobiology. 1997;198:73–80. doi: 10.1016/S0171-2985(97)80028-9. [DOI] [PubMed] [Google Scholar]
- 23.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 24.Bohuslav J, Kravchenko VV, Parry GC, Erlich JH, Gerondakis S, Mackman N, Ulevitch RJ. Regulation of an essential innate immune response by the p50 subunit of NF-kappaB. J Clin Invest. 1998;102:1645–52. doi: 10.1172/JCI3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Beller D. Aberrant production of IL-12 by macrophages from several autoimmune-prone mouse strains is characterized by intrinsic and unique patterns of NF-kappa B expression and binding to the IL-12 p40 promoter. J Immunol. 2002;169:581–6. doi: 10.4049/jimmunol.169.1.581. [DOI] [PubMed] [Google Scholar]
- 26.Kanarek N, Ben-Neriah Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol Rev. 2012;246:77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka H, Fujita N, Tsuruo T. 3-Phosphoinositide-dependent protein kinase-1-mediated IkappaB kinase beta (IkkB) phosphorylation activates NF-kappaB signaling. J Biol Chem. 2005;280:40965–73. doi: 10.1074/jbc.M506235200. [DOI] [PubMed] [Google Scholar]
- 28.Suh SJ, Kwak CH, Chung TW, Park SJ, Cheeeei M, Park SS, Seo CS, Son JK, Chang YC, Park YG, et al. Pimaric acid from Aralia cordata has an inhibitory effect on TNF-α-induced MMP-9 production and HASMC migration via down-regulated NF-κB and AP-1. Chem Biol Interact. 2012;199:112–9. doi: 10.1016/j.cbi.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y, Suh SJ, Li X, Liang JL, Chi M, Hwangbo K, Kwon O, Chung TW, Kwak CH, Kwon KM, et al. Citreorosein inhibits production of proinflammatory cytokines by blocking mitogen activated protein kinases, nuclear factor-κB and activator protein-1 activation in mouse bone marrow-derived mast cells. Biol Pharm Bull. 2012;35:938–45. doi: 10.1248/bpb.35.938. [DOI] [PubMed] [Google Scholar]