

Abstract
Conventional orthosteric drug development programs targeting G protein-coupled receptors (GPCRs) have focused on the concepts of agonism and antagonism, in which receptor structure determines the nature of the downstream signal and ligand efficacy determines its intensity. Over the past decade, the emerging paradigms of “pluridimensional efficacy” and “functional selectivity” have revealed that GPCR signaling is not monolithic, and that ligand structure can “bias” signal output by stabilizing active receptor states in different proportions than the native ligand. Biased ligands are novel pharmacologic entities that possess the unique ability to qualitatively change GPCR signaling, in effect creating “new receptors” with distinct efficacy profiles driven by ligand structure. The promise of biased agonism lies in this ability to engender “mixed” effects not attainable using conventional agonists or antagonists, promoting therapeutically beneficial signals while antagonizing deleterious ones. Indeed, arrestin pathway-selective agonists for the type 1 parathyroid hormone and angiotensin AT1 receptors, and G protein pathway-selective agonists for the GPR109A nicotinic acid and μ-opioid receptors, have demonstrated unique, and potentially therapeutic, efficacy in cell-based assays and preclinical animal models. Conversely, activating GPCRs in “unnatural” ways may lead to downstream biological consequences that cannot be predicted from prior knowledge of the actions of the native ligand, especially in the case of ligands that selectively activate as-yet poorly characterized G protein-independent signaling networks mediated via arrestins. Although much needs to be done to realize the clinical potential of functional selectivity, biased GPCR ligands nonetheless appear to be important new additions to the pharmacologic toolbox.
Despite the fact that heptahelical G protein-coupled receptors (GPCRs) are by far the most successfully exploited class of drug targets, accounting for nearly half of all pharmaceuticals in current use (1), the conceptual framework guiding GPCR drug discovery programs for decades has been remarkably simple. Dating back to the original application of allosteric models to membrane receptor function in the 1960s (2, 3), the basic concepts are that GPCRs exist in equilibrium between conformationally discrete “off” and “on” states that are distinguished by their ability to trigger downstream responses, and that ligands act by perturbing this equilibrium (4, 5). Within this framework, the actions of a ligand can be fully described by only 2 terms; the equilibrium dissociation constant of the ligand-receptor complex (Kd), and the maximal observed change in receptor activity (Vmax). Hence, GPCR ligands are classified as agonists if they can elicit a maximal response, partial agonists if they only generate a submaximal response at saturating ligand concentration, and antagonists if they lack intrinsic efficacy but competitively inhibit agonist responses. Later refinements of this “2-state” model, such as the “extended ternary complex” (6) and “cubic ternary complex” (7) models that were developed to explain the capacity of “inverse agonists” to reduce the basal activity of constitutively active mutated GPCRs, simply added terms accounting for the probability that the receptor might spontaneously transition to the active state in the absence of ligand. They did not consider the possibility of multiple active states.
According to the American psychologist Abraham Maslow, “if all you have is a hammer, everything looks like a nail” (8). The pharmacologic equivalent of Maslow's hammer is shown in Figure 1A. If GPCRs can only be off or on, then all ligands can do is change the conformational equilibrium, increasing the proportion of receptors in the on state in settings in which receptor activity is insufficient and decreasing it in the presence of excess endogenous agonist. Thus, conventional agonists and antagonists change the quantity of receptor activity, but only the receptor determines what signals are transmitted by the on state. Partial agonists, by virtue of their inability to completely shift the receptor equilibrium at saturating concentration, may exert “protean” effects (9) in systems with differing levels of constitutive basal receptor activity, but even they do not qualitatively change signaling.
Figure 1.
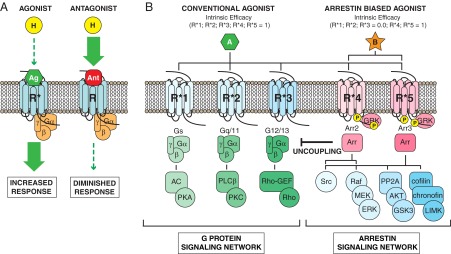
Evolving concepts of orthosteric GPCR ligand action. A, The conventional view of ligand efficacy assumes that all downstream GPCR signaling arises from a single “on” state. In this case, agonists (Ag) can increase receptor activity (R*) when levels of the endogenous ligand (H) are insufficient, and antagonists (Ant) can decrease receptor activity (R) in the face of endogenous ligand excess, but only the intensity of signaling is changed, not its character. B, Schematic depicting a hypothetical GPCR with 5 conformationally distinct “active” states (R*1–R*5), each of which couples the receptor to downstream G protein (Gs; Gq/11; G12/13) and non-G protein (arrestin2 [Arr2]; arrestin3 [Arr3]) effectors with different efficiency. Note that the 1:1 coupling between active state and effector depicted is an oversimplification. In such a system, a full agonist (A) will produce a full system response in all downstream effectors, just as in the conventional model. In contrast, “biased” agonists (B) engage different active receptor conformations with variable intrinsic efficacy, a property that permits them to activate some downstream pathways, eg, arrestin-dependent signals, while antagonizing others. The ability to engender “mixed” effects permits biased agonists to qualitatively change GPCR signaling. AC, adenylyl cyclase; GEF, guanine nucleotide exchange factor; LIMK, lim domain-containing kinase; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; MEK, MAPK kinase.
If all you have is a hammer, then the only way forward is to find new nails to drive. By the mid-1990s, innovative GPCR drug discovery was limited to finding new targets for conventional agonist and antagonist ligands, or focusing on subtype selectivity for known drug targets like the muscarinic cholinergic and serotonin receptors. With the knowledge that more than half of the nearly 1000 GPCRs present in the human genome were orphan receptors with unknown ligands and physiological functions, “deorphanizing” GPCRs became the major hope for finding new drugs (10, 11), and many promising drug targets, like ghrelin, orexins/hypocretins, and endocannabinoid receptors (12, 13) have emerged from these efforts. But what have been missing are new tools. More than just hammers, we have needed pharmacologic tools to expand the scope of efficacy beyond conventional agonism and change GPCR signaling qualitatively, not just quantitatively.
The Basis of Bias
Two concepts that together form the foundation for a novel class of GPCR-targeted therapeutics have gained attention over the last decade. The first of these, termed “pluridimensional efficacy” by Michel Bouvier in 2006 (14), recognizes that GPCRs signal by engaging multiple G protein and non-G protein effectors. Although it had long been recognized that GPCRs can couple to 2 or more unrelated G protein families at physiological levels of expression, enabling a single receptor to engage multiple signaling pathways simultaneously or activate them differentially in different tissues (15–17), it was the recognition that a robust G protein-independent signaling network lay beneath the surface of heterotrimeric G protein signaling that revealed the true breadth of GPCR efficacy (18, 19). The best-characterized non-G protein effectors are the arrestins, a family of 4 cytosolic proteins first described for their role in GPCR desensitization and the modulation of phototransduction (20). Arrestins bind tightly to agonist-occupied receptors that have been phosphorylated by GPCR kinases (GRKs) (21). Arrestin binding sterically precludes further G protein activation, producing homologous desensitization, and in the case of the nonvisual arrestins, arrestin2 and 3 (β-arrestin1 and 2), the arrestin C terminus binds elements of the clathrin endocytic machinery, leading to the clustering and sequestration of desensitized receptors (22, 23).
But the end is also the beginning, because arrestins also function as ligand-regulated scaffolds, recruiting a host of arrestin-bound proteins to agonist-occupied GPCRs and initiating a “second wave” of signaling that begins as G protein signaling wanes and persists as receptors coalesce and transit the intracellular compartment (19). Beginning in 1999 with the report that arrestin2 bound the nonreceptor tyrosine kinase, c-Src, and recruited it to activated β2-adrenergic receptors, studies linking arrestins to GPCR activation of novel enzymatic effectors began appearing with regularity (24). Among others, putative arrestin-regulated effectors now include Src family kinases (24–26), components of the ERK1/2 and c-Jun N-terminal kinase 3 MAPK cascades (27–29), the E3 ubiquitin ligase, Mdm2 (30), the cAMP phosphodiesterases, PDE4D3/5 (31), diacylglycerol kinase (32), the inhibitor of nuclear factor-κB, IκBα (33), the Ral-GDP dissociation stimulator, Ral-GDS (34), the actin filament-severing protein, cofilin (35), and the Ser/Thr protein phosphatase 2A (PP2A) (36, 37). Although the full extent of physiologically relevant arrestin signaling remains inadequately characterized, it is increasingly apparent that arrestin-dependent GPCR signaling performs numerous functions, among them enhancing second messenger degradation, regulating cytoskeletal dynamics controlling vesicle trafficking, exocytosis and cell migration, and promoting cell survival, growth, and hyperplasia (19, 38).
The second critical thread in the bias story is the formulation of a general allosteric model of GPCR signaling by Kenakin and colleagues beginning in the 1990s (39–41, 43). As more techniques to measure GPCR efficacy were developed, it became apparent that GPCRs do not behave as though they have a single “active” conformation that transmits all downstream responses with equal efficiency (44, 45). Reports of “reversal of potency,” in which 2 ligands exhibit opposite rank order of potency for 2 downstream responses measured in the same system (46–49); or “reversal of efficacy,” in which a single ligand exhibits opposing efficacy toward 2 different downstream responses (50–52), offered definitive proof that structurally distinct ligands can activate the same GPCR in different ways, something that can only occur if there is more than one “active” receptor state. Biochemical (53–55) and biophysical (56–58) approaches provided additional evidence that GPCRs adopt multiple active states that couple the receptor to different downstream effectors with different efficiency.
Kenakin's allosteric model (40, 44, 59) envisions GPCRs not as off-on switches, but as “ensembles” of tertiary conformations, some subset of which elicit measurable outcomes that operationally define them as active states. At any instant, the biological activity of the system thus reflects the distribution of the receptor population across each of the component microstates that make up the conformational ensemble. Any molecular interaction that changes the energy landscape of the system has the potential to affect the conformational ensemble in a way that affects signaling. Considered as allosteric proteins, GPCRs are thus susceptible to numerous inputs that modify their signaling properties. In addition to orthosteric effects exerted via the ligand-binding site, lateral allosteric effects arising from protein-protein interactions within the plasma membrane or cytosol, and allosteric modulation arising from the interaction of small molecules with sites outside the ligand-binding pocket, all affect downstream signaling. In effect, orthosteric ligands and allosteric modulators work the same way and can be described in the same mathematical terms (43).
The concepts of “agonist trafficking,” “functional selectivity,” and “biased agonism” merge the principles of pluridimensional efficacy with allosteric models of GPCR efficacy to produce a comprehensive description of orthosteric ligand behavior (39, 59–64). If receptors are conformationally flexible, there is no a priori reason that the conformational ensemble stabilized by one ligand should duplicate either the distribution of spontaneously formed active states or those generated by a structurally distinct ligand. Figure 1B illustrates the differences between conventional and biased agonism using a hypothetical GPCR with 5 conformationally distinct “active” states (R*1–R*5), each of which couples the receptor to downstream G protein and non-G protein effectors with different efficiency. In such a system, a conventional full agonist (A) can be viewed as possessing equivalent intrinsic efficacy for R*1–R*5, ie, it does not discriminate between them. Although there are exceptions (65), native ligands are mostly full agonists, because they coevolved with the receptor to stabilize active receptor conformations in the correct proportions to elicit the most physiologically adaptive set of downstream signals. Like full agonists, conventional partial agonists would not discriminate between active states; they simply possess lower intrinsic efficacy such that only a fraction of the receptors will be in an active state at full receptor occupancy. In contrast, the intrinsic efficacy of a biased agonist (B) differs between active states, such that at full receptor occupancy some active states are favored (R*4–R*5) over others (R*1–R*3).
It is important to recognize that under the right circumstances both partial and biased agonists can activate only a subset of the receptor's full signaling repertoire. In the case of a partial agonist, responses that are tightly coupled or highly amplified will be preserved because fractional receptor activation is sufficient to generate a maximal response, whereas responses that are weakly coupled or that have an activation threshold, in which full receptor activation is necessary for a maximal response, will be disproportionally decreased or absent. Similarly, unamplified responses, such as many of those transmitted via stoichiometric receptor-arrestin complexes, may be undetectable due to the reduced fraction of receptors in an active conformation. This form of variability, arising from differences in “signal strength,” does not require the existence of multiple active states, and the rank order of potency of a series of conventional full and partial agonists will not vary when multiple responses are compared, because the intrinsic efficacy of each ligand is the same across all active states (59).
On the other hand, biased agonists, because they only stabilize a subset of active states, possess the ability to activate some pathways while antagonizing others, leading to qualitative differences in signaling. Soon after the discovery of arrestin-dependent signaling, it became apparent that the G protein- and arrestin-coupled modes of GPCR signaling were pharmacologically dissociable, ie, ligands could discriminate between receptor conformations responsible for these 2 distinct forms of GPCR signaling (66). Ligand bias can range from relatively modest deviations in rank order of potency, to frank reversal of efficacy, wherein the characterization of a ligand as agonist, antagonist, or inverse agonist is completely assay dependent. Among the more dramatic examples of functional selectivity are ligands like ICI118551 and [D-Trp12,Tyr34]-PTH(7–34), that function as inverse agonists for heterotrimeric G protein signaling while acting as partial agonists for arrestin-dependent GPCR sequestration and arrestin signaling (67, 68). Such ligands are clearly activating the same receptor in different ways than the native agonist; thus it is the ligand-receptor complex, not the receptor alone, that is specifying the cellular response. In effect, the receptor bound to a biased ligand is a different functional entity, with different signaling characteristics, than the same receptor bound to a conventional agonist or partial agonist.
The Faces of Bias
The goal of any pharmaceutical agent is to modify a biological response in target cells. An orthosteric ligand, conventional or biased, can only effect such change via the ligand-binding pocket of its cognate receptor. Everything that follows, from activation of effectors and second- messenger generation to integrated biological responses like cell proliferation, differentiation, survival, and death, is encoded at that one point.
Biased efficacy in vitro
What range of effects can be obtained through functional selectivity? Allosteric models of GPCR function are based on the concept of conformational selection, which posits that structurally distinct ligands may stabilize active receptor conformations in different proportions than the native ligand, but do not force receptors into sterically unfavorable “novel” conformations or couple them to novel effectors. If the native ligand is truly pluripotent, ie, couples the receptor to all possible effectors, then the actions of a biased ligand must comprise a subset of the whole. Experimental data examining the actions of biased ligands in vitro using a wide range of readouts tend to bear this out.
The most immediate consequence of ligand binding is a change in receptor conformation, which can be monitored at different points within the receptor using intramolecular fluorescence probes (56, 57). In contrast to rhodopsin, in which covalently bound trans-retinal is sufficient to stabilize the active state of the receptor even in the absence of a G protein (69, 70), ligand-induced activation of the β2-adrenergic receptor involves generation of a number of intermediate conformations. Using 13CH3-ϵ-Met nuclear magnetic resonance, Nygaard et al (71) found that whereas agonist binding destabilizes the inactive state, it does not stabilize the fully active conformation observed in the β2-adrenergic receptor-Gs complex (72). This higher degree of conformational heterogeneity may permit structurally distinct ligands to direct receptor coupling to different downstream effectors. When measured using site-specific one-dimensional 19F-nuclear magnetic resonance spectroscopy, agonist, inverse agonist, and arrestin-selective β2-adrenergic receptor agonists were found to differentially affect the movement of transmembrane helices VI and VII (73), such that G protein activation correlated with movement of helix VI, whereas coupling to arrestins depended upon movement of helix VII. Importantly, helix VI and helix VII were shown to move independently, providing a physical basis for biased agonism and a conformational “signature” predictive of arrestin selectivity. Dynamic studies performed using atomic force microscopy-based single-molecule force spectroscopy similarly demonstrated that ligands differentially perturb the conformational equilibrium of the receptor (74), presumably thereby biasing the probability of coupling to different effectors.
The first step in signal transduction occurs when the activated GPCR touches its proximal effectors, events that can be monitored in live cells using bioluminescence resonance energy transfer-based technologies (75, 76). A recent study by Saulière et al (77) using a comprehensive panel of bioluminescence resonance energy transfer-based sensors encompassing all heterotrimeric G protein families and arrestins to compare the efficacy of the native angiotensin AT1A receptor agonist, angiotensin II (AngII), with that of the arrestin-biased agonist, [Sar1,Ile4 Ile8]-AngII, offers insights into physical basis of signal bias. In this study, [Sar1,Ile4,Ile8]-AngII was found to behave as a weak partial agonist for heterotrimeric G proteins, proportionally activating the same G protein species as AngII, albeit with much lower efficacy than the native ligand. Reflective of its arrestin signal bias, [Sar1,Ile4,Ile8]-AngII recruited arrestin3 to the AT1A receptor with nearly comparable efficacy. Consistent with the theory of conformational selection, the data suggested that biased ligands select receptor conformations that couple the receptor to downstream effectors with different efficiency than the native ligand, but do not drive the receptor to engage otherwise unrecognized effectors.
The next level of signal propagation occurs as G proteins, arrestins, and potentially other non-G protein effectors, activate second messenger pathways and protein phosphorylation cascades. This is the level at which most in vitro studies of ligand bias have been performed, and, to date, most have failed to find unique signaling events produced by biased agonists. Rather, biased ligands appear to activate a subset of the downstream second messenger and protein kinase cascades regulated by their conventional counterparts (19, 78). In their systematic comparison of AngII and [Sar1,Ile4,Ile8]-AngII, Saulière et al (77) were able to discern relatively subtle and cell-type specific differences in the production of second messengers, but these probably arose from “signal strength” effects that led to differential activation of graded and threshold responses. Nonetheless, their results at least admit the possibility that biased agonists can qualitatively change information flow by coupling the receptor to effectors with different efficiency.
Unbiased surveys of conventional and arrestin-selective agonism to date have likewise found little in the way of “novel” signaling events triggered by biased agonists. A side-by-side global phosphoproteomic comparison of AngII and [Sar1,Ile4,Ile8]-AngII performed using LTQ-Orbitrap high-resolution mass spectrometry was able to detect more than 1183 regulated protein phosphorylation sites of 10 000 surveyed, but of these 756 (64%) were unique to AngII, 369 (34%) were regulated by both AngII and [Sar1,Ile4,Ile8]-AngII, and only 58 (5%) were unique to [Sar1,Ile4,Ile8]-AngII (79). A less sensitive gel-based phosphoproteomic comparison of the same 2 ligands uncovered novel arrestin-dependent AT1A receptor pathways leading to regulation of PP2A-Akt-glycogen synthase kinase 3β (GSK3β) signaling and stimulation of prostaglandin E2 synthesis by prostaglandin E synthase type 3, but still found that both AngII and [Sar1,Ile4,Ile8]-AngII activated them (80).
The same story appears to hold when examining integrated biological responses such as cell proliferation or migration. For example, both AngII and [Sar1,Ile4,Ile8]-AngII have been reported to produce arrestin-dependent inotropic and lusitropic effects in isolated murine cardiomyocytes (81). Likewise, the ability of [Sar1,Ile4,Ile8]-AngII to stimulate proliferation of neonatal rat cardiomyocytes is a property shared with AngII; its lack of a hypertrophic response reflects its inability to activate Gq/11-dependent pathways, not its ability to engender novel responses (82). Similarly, the capacity to promote arrestin-dependent cell survival and stimulate migration in primary murine osteoblasts is a property shared by the arrestin-selective type 1 PTH receptor agonist, [D-Trp12,Tyr34]-PTH(7–34), and its conventional counterpart PTH(1–34) (83).
Spatial and temporal bias
Most discussions of ligand bias focus on the capacity of ligands to change the efficiency with which GPCRs activate different downstream effectors. But cellular responses are also affected by the kinetics and spatial compartmentalization of pathway activation (84, 85). Another facet of bias relates to the ability of structurally distinct ligands to change the duration and/or location of signaling, ie, not changing the effector per se, but rather the dynamics of its activation.
Variation in ligand structure can affect both the duration and subcellular location of heterotrimeric G protein signaling. For example, both PTH(1–34) and a corresponding fragment of PTHrp, PTHrp(1–36), robustly activate Gs-cAMP signaling via the PTH1 receptor, but the 2 ligands differ markedly in the kinetics of receptor association and dissociation, with PTHrp(1–36) exhibiting a slower on rate and faster off rate. Consequently, the cAMP response to PTH(1–34) is prolonged, and live cell imaging demonstrates that although cAMP generation in response to PTHrp(1–36) is limited to the plasma membrane, PTH(1–34) continues to stimulate cAMP production from within the endosomal compartment after the receptor has internalized (86–88). These differences appear to arise from conformational selection, in that ligands that exhibit high affinity for the Gs-uncoupled PTH1 receptor, including PTH(1–34) and a series of synthetic N-terminal PTH analogs, possess higher binding affinity, increased potency, and prolonged Gs activation kinetics, than ligands, like PTHrp(1–36), that bind with high affinity only to the precoupled receptor-G protein complex (89, 90).
Similarly, ligand structure can change the stability of the GPCR-arrestin complex, leading to alterations in desensitization, trafficking, and signaling kinetics, and even admitting the possibility that arrestin-dependent signaling can be dissociated from arrestin-dependent desensitization. Zimmerman et al (91) found that a series of angiotensin peptide analogs that all supported arrestin2 recruitment and caused AT1A receptor-arrestin complexes to traffic to endosomes differed widely in their ability to promote arrestin-dependent signaling. Using fluorescence recovery after photobleaching, they demonstrated ligand-specific differences in the stability of the receptor-arrestin complex, such that ligands that promoted tighter arrestin binding were effective activators of ERK1/2, whereas ligands with faster off rates were severely compromised in their ability to generate arrestin-dependent signals. Interestingly, they found ligand-specific differences in the degree to which arrestin recruitment, activation, and signaling were sensitive to knockdown of GRK2 and GRK6, suggesting that ligand-induced changes in receptor conformation affected arrestin conformation and function by eliciting different patterns of receptor phosphorylation, consistent with the “phosphorylation code” hypothesis proposed by Tobin and colleagues (92).
Bias In Vivo
If biased GPCR agonists are to find a prominent place in the pharmacopoeia of the future, it will be because they can produce biological effects in vivo that are not attainable using conventional agonist or antagonist ligands. Although some drugs approved for clinical use, eg, the β-adrenergic receptor antagonists propranolol and carvedilol, have been shown in retrospect to exhibit a degree of bias (67, 93), no current pharmaceuticals are known to possess unique clinical efficacy based on their ability to bias GPCR signaling. Nonetheless, data from several preclinical studies using arrestin- or G protein-biased ligands offer encouragement.
Arrestin pathway-selective bias
In bone, the conventional PTH1 receptor agonist, PTH(1–34), stimulates bone-forming osteoblasts, increasing both osteoblast number and activity, while at the same time accelerating bone turnover by causing osteoblasts to secrete soluble factors that increase the number and activity of bone-resorbing osteoclasts (94). When administered intermittently, PTH(1–34) produces a net increase in bone mass, a property that is exploited clinically for the treatment of severe osteoporosis. Traditionally, the actions of PTH in bone have been attributed to Gs-cAMP signaling (95), so the expectation would be that a ligand like (d-Trp12,Tyr34)-PTH(7–34), which antagonizes PTH1 receptor-Gs coupling, would at best have no effect on bone mass. Paradoxically, mice treated with (d-Trp12,Tyr34)-PTH(7–34) also exhibit increased bone formation, with greater trabecular bone volume, increased osteoblast number, and accelerated mineral apposition, but with no effect on osteoclast number or markers of bone turnover (96). The key to this unexpected phenotype apparently lies in the ability of (d-Trp12,Tyr34)-PTH(7–34) to expand the osteoblast pool through arrestin-dependent cell cycle regulation and antiapoptotic signaling, while “uncoupling” the PTH1 receptor from Gs-cAMP-dependent activation of osteoclasts (83, 96).
In the cardiovascular realm, the positive inotropic and lusitropic responses of isolated murine cardiomyocytes to [Sar1,Ile4,Ile8]-AngII (81) appear to translate into improved cardiac contractility in vivo. An analogous arrestin pathway-selective AT1A receptor agonist, TRV120027 [Sar-Arg-Val-Tyr-Ile-His-Pro-(D)-Ala-OH], that also stimulates arrestin-dependent activation of Src, ERK1/2, and endothelial nitric-oxide synthase in vitro, similarly improves cardiomyocyte contractility (97). Administration of TRV120027 to rats reduced mean arterial blood pressure, as did the nonpeptide AT1A receptor antagonists losartan and telmisartin, but unlike the neutral antagonists, which decrease cardiac performance, TRV120027 increased cardiac performance and preserved cardiac stroke volume. In a canine rapid-pacing model of heart failure, TRV120027 produced cardiac unloading while preserving renal function, suggesting that it may have utility in heart failure treatment (98, 99).
G protein pathway-selective bias
Bias goes both ways, and G protein pathway-selective ligands have also shown promise. The most obvious benefit of G protein selectivity is that such ligands would escape arrestin-dependent desensitization. Prolongation of opiate-induced analgesia was the first phenotype of arrestin3-knockout mice to be described, demonstrating the critical role of arrestin3 in desensitization of central nervous system opioid receptors (100, 101). Recently, TRV130, a biased μ-opioid receptor agonist that demonstrates G protein signaling potency and efficacy similar to morphine, but with reduced arrestin recruitment and receptor sequestration, was shown in mice to produce comparable analgesia with less gastrointestinal dysfunction and respiratory depression than equianalgesic doses of morphine (102). Such results suggest that circumventing opioid receptor desensitization may lead to longer lasting or safer analgesics.
The other application of G protein bias would be avoiding unwanted arrestin-dependent side effects. For example, the niacin receptor, GPR109A, has been shown to decrease serum-free fatty acids by activating Gi/o while at the same time producing cutaneous flushing through arrestin-dependent activation of phospholipase A2 (103). Predictably, a G protein pathway-selective GPR109A agonist, MK-0354, has been shown to decrease serum-free fatty acids in vivo without causing flushing, making it a potentially useful agent for treating hyperlipidemia (104). Another highly promising area relates to the mechanism of action of antipsychotics. β-Catenin and Akt signaling in the D2 dopamine receptor-rich striatum of mice is regulated by an arrestin3 scaffolded PP2A-Akt-GSK3β complex (36, 37). Within the complex, PP2A dephosphorylates Akt Thr308, keeping it tonically inactive. Because phosphorylation by Akt inhibits GSK3α/β, keeping arrestin-bound Akt quiet increases GSK3α/β activity. GSK3β, in turn, phosphorylates β-catenin, accelerating its proteosomal degradation and thereby reducing its transcriptional activity. Predictably, striatal extracts from arrestin3-null mice show higher levels of β-catenin expression. The mood stabilizer, lithium, disrupts the interaction between arrestin, Akt, and PP2A, relieving PP2A-mediated inhibition of Akt allowing it to phosphorylate and inactivate GSK3β (105). Interestingly, the clinical efficacy of all classes of antipsychotic drug correlates directly to their D2 receptor binding affinity and ability to antagonize the receptor, and it was found that although different classes of antipsychotics exhibit complex efficacy profiles with respect to D2 receptor-G protein coupling, they share the property of antagonizing the D2 receptor-arrestin3 interaction (106), suggesting that antagonism of arrestin signaling contributes to the activity of all antipsychotics.
Predicting Biased Outcomes
When dealing with conventional agonism or antagonism, a reasonable prediction of drug effects can be derived from study of the native ligand. Biased agonists, on the other hand, activate receptors in “unnatural” ways, and their biological effects may be less predictable. Whereas a concern with allosteric modulators, which bear no structural relationship to the orthosteric ligand, is unexpected, “off-target” effects (107), developers of biased therapeutics must be aware of the possibility of unpredicted “on-target” effects; not side effects arising from unexpected drug-protein interactions, but rather from the drug affecting its intended target in unintended ways. This is particularly true for arrestin pathway-selective agonists, because our appreciation of the scope physiologically relevant GPCR signals transmitted via arrestins remains incomplete (19, 38, 108).
By way of illustration, parametric gene set enrichment analysis of cDNA microarray data from bone of mice treated with PTH(1–34) or the arrestin-biased ligand, [d-Trp12,Tyr34]-PTH(7–34), found a striking degree of nonoverlap in the genome level response (83). As expected of a conventional PTH1 receptor agonist, PTH(1–34) affected gene clusters classically associated with embryologic skeletal patterning and PTH actions in bone, including Wnt/β-catenin signaling, bone morphogenic protein signaling, TGF-β signaling, phosphatidylinositol 3-kinase/Akt signaling, and ERK/MAPK signaling, that corresponded to regulation of processes linked to the control of skeletal morphogenesis, osteoblast differentiation, matrix biosynthesis and mineralization, and bone turnover (109–111). In contrast, [d-Trp12,Tyr34]-PTH(7–34) primarily affected pathways regulating apoptosis, eg, ataxia telangectasia-mutated signaling and p53 signaling; cell survival, eg, phosphatidylinositol 3-kinase/Akt signaling; and cell cycle checkpoint regulation. Thus, although both agents increased trabecular bone mass, they appeared to do so in largely different ways.
Unexpected actions of [Sar1)-Ile4-Ile8)]-AngII have been reported as well. In a murine model of heart failure, [Sar1-Ile4-Ile8]-AngII was found to promote aldosterone production via arrestin2- and ERK1/2-dependent up-regulation of the steroidogenic acute regulatory protein, StAR, a mitochondrial transport protein that is the rate-limiting step in the biosynthesis of steroid hormones. In this model, the resultant elevation in circulating aldosterone levels led to salt and volume retention, adverse cardiac remodeling, and heart failure progression (112). Arrestin-mediated signaling also appears to promote vascular smooth muscle cell hyperplasia and an exaggerated vascular injury response. In primary vascular smooth muscle, [Sar1-Ile4-Ile8]-AngII stimulates proliferation by promoting AT1A receptor-dependent epidermal growth factor receptor transactivation (113–115) and activating antiapoptotic signaling pathways (116).
Expanding the Pharmacologic Tool Kit
Rather than an occasional anomaly, it is now clear that ligand bias is a general property in GPCR signaling, even being found in nature, as for example between CCL19 and CCL21, the 2 endogenous ligands for the CC chemokine receptor 7 (65). Table 1 lists several GPCRs for which biased ligands have been described and the therapeutic arenas in which ligand bias may prove useful. Conditions caused by mutated constitutively active GPCRs, eg, familial male precocious puberty (LH receptor; Ref. 131), hyperthyroidism due to toxic follicular adenomas (TSH receptor; Ref. 132), and some cases of familial nephrogenic diabetes insipidus (V2 vasopressin receptor; Ref. 133), offer additional settings in which ligands that favor G protein- or arrestin coupling might offer therapeutic advantage. Even more promising, arrestin-mediated signaling pathways have been implicated in the pathogenesis of many common clinical conditions; eg, asthma (134, 135), inflammatory bowel disease (136, 137), certain malignancies (138), and autoimmune disorders (139), suggesting that ligands that bias GPCR signaling to favor or oppose arrestin coupling may have widespread application.
Table 1.
Potential Targets for Biased GPCR Therapeutics
| Receptor | Ligand(s) | Observed Bias | Therapeutic Arena | References |
|---|---|---|---|---|
| β1/2-Adrenergic | Carvedilol; ICI118551 | Arrestin-selective agonism | Congestive heart failure | 14, 67, 93, 117 |
| AT1 angiotensin | Sar1,Ile4,Ile8-AngII; TRV120027 | Arrestin-selective agonism | Congestive heart failure | 81, 97–99 |
| α2-Adrenergic | Desipramine | Arrestin-selective agonism | Hypertension | 118 |
| PTH1 parathyroid hormone | [d-Trp12,Tyr34]-bPTH (7–34) | Arrestin-selective agonism | Osteoporosis | 83, 96 |
| D1 dopamine | A-77636 | Arrestin-selective agonism | Parkinson's disease | 119 |
| CCR5 chemokine | AOP-RANTES | Arrestin-selective agonism | HIV infection | 120 |
| CB1 cannabinoid | ORG27969 | Arrestin-biased allosteric modulator | Anxiety, depression, pain, obesity | 121 |
| GPR109A nicotinic acid | MK-0354 | Gi/o-selective agonism | Hyperlipidemia | 103, 104 |
| μ-Opioid | TRV130 | Gi/o-selective agonism | Analgesia | 102 |
| κ-Opioid | 6′-Guanidinonaltrindole | Gi/o-selective agonism | Analgesia, dysphoria | 122–124 |
| D2 dopamine | Aripiprazole | Gi/o-selective agonism | Schizophrenia, psychosis | 106, 125 |
| 5-HT2A serotonin | 2,5-Dimethoxy-4 iodoamphetamine | Gi/o-selective agonism | Schizophrenia, hallucinations | 126, 127 |
| GLP-1 glucagon-like peptide | Oxyntomodulin | Gs-selective agonism | Diabetes, β-cell mass/function | 128–130 |
Beyond the challenges that pluridimensional efficacy poses in terms of understanding the molecular pathogenesis of disease and defining the ligand efficacy profile most likely to prove therapeutic, the phenomenon of ligand bias has implications for the design of assays to characterize GPCR signaling in the research or drug discovery settings (44, 140–142). The traditional approach of using unidimensional high-throughput assays for ligand classification is inadequate in an environment in which ligand classification is assay dependent (44, 45). Instead, drug discovery approaches need to be flexible. If the goal is to define efficacy in a limited number of known dimensions, eg, G protein and arrestin coupling, then multiplexing assays may be adequate to identify functional selectivity. Beyond assays based on ligand binding, G protein activation, or second-messenger generation, high-content assays based on imaging techniques that use fluorescent signals can provide both temporal and spatial information about signaling (143, 144). Considering just one such event, the interaction of GPCRs with arrestins, responses can be monitored through direct visualization of GPCR/arrestin-green fluorescent protein complexes (145, 146), with bioluminescence resonance energy transfer (147), with enzyme fragment complementation (148), or with protease-activated transcriptional reporter genes (149). Greater efficiency can be obtained by multiplexing green fluorescent protein- and immunofluorescence-based assays to provide simultaneous readouts of multiple signaling pathways within the same cell (150). Once empiric data on the potency and efficacy of a series of ligands across 2 or more dimensions of efficacy are generated, the extent of ligand bias, eg, ΔΔLog(τ/KA) or ΔΔLog (Relative Activity) (43, 151), can be quantified relative to a reference agonist based on adaptations of the Back-Leff operational model (4, 5).
If, on the other hand, the objective is to detect whether compounds exert any form of activity against a receptor, then technology designed to detect integrated whole-cell responses may be necessary. Resonant wave guide grating technology has led to the development of optical biosensors that can measure dynamic mass redistribution signals in living cells (152, 153). This technology can detect interactions of GPCRs with cytosolic signaling molecules at a depth of 150–200 nM and detect receptor internalization. The resulting dynamic mass redistribution signal is a noninvasive cell-based technology that can measure virtually any receptor activation in any cell type in real time. The approach has been applied to the detection and quantification of functional selectivity in intact cells (154). Another technology that can be used for the same purpose involves measuring changes in the electrical impedance of cell monolayers caused by receptor-mediated changes in cell mass redistribution (155, 156). The principal limitation of these “label-free” systems is that because whole-cell responses represent the integration of numerous pathways, it may be difficult or impossible to deconvolute the kinetic pattern of the response in a manner that allows identification of the specific signaling pathway(s) being affected. On the other hand, label-free systems can be used with primary cells, circumventing the potential for signaling artifacts in engineered cell systems and allowing ligands to be screened in therapeutically relevant cell types.
Finally, the recent emergence of high-resolution crystal structures of GPCRs in their agonist-bound, antagonist-bound, and effector-coupled states has begun to enable model-based or virtual in silico approaches to drug discovery (157, 158). Virtual screens have identified novel orthosteric and allosteric ligands for the D1, D2 (159), and D3 (160) dopamine, CB1 (161) and CB2 (162) cannabinoid, and CXCR7 (163) receptors, among others. Importantly, a large library virtual screen against the activated β2-adrenergic receptor crystal structure identified “hits” that exhibited arrestin-selective signaling bias (164), suggesting that virtual structure-based approaches might be used to fashion ligands with specific efficacy.
Conclusions
In the native context, GPCRs and their natural ligands coevolved to control signaling in the most physiologically adaptive manner. Signaling, reflected in the distribution of GPCR-active states and the efficiency with which these states engage downstream effectors, is “balanced” to meet the needs of the organism. Biased agonists, on the other hand, alter the distribution of active states. The resulting signals are not qualitatively different, but are “unbalanced” compared with the native ligand. As these unbalanced signals propagate, differences in the tissue response may arise from relatively subtle differences in signal strength or the kinetics of pathway activation and deactivation. Certainly, the presence of disease can cause adaptive responses to become maladaptive, as in the case of endogenous catecholamine signaling in the setting of congestive heart failure (165), and this is one arena in which biased agonists hold promise as therapeutics. The ability to “recalibrate” GPCR signaling to adapt to the needs of a perturbed system is something that cannot be achieved using conventional agonist or antagonists (166). But we must remain cognizant of the converse. The same properties that make them valuable as potential therapeutics may also make them more prone to produce unanticipated effects.
It is clear that the concepts of pluridimensional efficacy and GPCR allosterism hold the keys to new drugs. Small molecule allosteric modulators have already found their way into the clinic for the treatment of secondary hyperparathyroidism (167) and advanced HIV disease (168). Biased agonists, tools that can qualitatively change GPCR signaling for therapeutic benefit, seem to be poised on the horizon. The future will tell whether they prove to be the utilitarian wrenches, saws, and screwdrivers of the pharmacologist's tool kit or a collection of left-handed monkey wrenches with limited utility or specialized application.
Acknowledgments
This work was supported by National Institutes of Health Grants DK055524 and R01 GM095497 (to L.M.L.), and the Research Service of the Charleston, South Carolina Veterans Affairs Medical Center.
The contents of this article do not represent the views of the Department of Veterans Affairs or the United States Government.
Disclosure Summary: The author has nothing to disclose.
Footnotes
- AngII
- angiotensin II
- GPCR
- G protein-coupled receptor
- GRK
- GPCR kinase
- GSK3β
- glycogen synthase kinase 3β
- PP2A
- protein phosphatase 2A.
References
- 1. Ellis C. The state of GPCR research in 2004. Nat Rev Drug Discov. 2004;3:577–626 [DOI] [PubMed] [Google Scholar]
- 2. Karlin A. On the application of “a plausible model” of allosteric proteins to the receptor for acetylcholine. J Theor Biol. 1967;16:306–320 [DOI] [PubMed] [Google Scholar]
- 3. Thron CD. On the analysis of pharmacological experiments in terms of an allosteric receptor model. Mol Pharmacol. 1973;9:1–9 [PubMed] [Google Scholar]
- 4. Black JW, Leff P. Operational models of pharmacological agonist. Proc R Soc Lond [Biol]. 1983;220:141–162 [DOI] [PubMed] [Google Scholar]
- 5. Black JW, Leff P, Shankley NP. An operational model of pharmacological agonism: The effect of E/[A] curve shape on agonist dissociation constant estimation. Br J Pharmacol. 1985;84:561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the β 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636 [PubMed] [Google Scholar]
- 7. Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model. III. Resurrecting efficacy. J Theor Biol. 1996;181:381–397 [DOI] [PubMed] [Google Scholar]
- 8. Maslow AH. The Psychology of Science. New York, NY: Harper Row; 1996:15 [Google Scholar]
- 9. Kenakin T. Pharmacological proteus? Trends Pharmacol Sci. 1995;16:256–258 [DOI] [PubMed] [Google Scholar]
- 10. Wilson S, Bergsma DJ, Chambers JK, et al. Orphan G-protein-coupled receptors: the next generation of drug targets? Br J Pharmacol. 1998;125:1387–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Howard AD, McAllister G, Feighner SD, et al. Orphan G-protein-coupled receptors and natural ligand discovery. Trends Pharmacol Sci. 2001;22:132–140 [DOI] [PubMed] [Google Scholar]
- 12. Kojima M, Hosoda H, Matsuo H, Kangawa K. Ghrelin: discovery of the natural endogenous ligand for the growth hormone secretagogue receptor. Trends Endocrinol Metab. 2001;12:118–122 [DOI] [PubMed] [Google Scholar]
- 13. Xu YL, Jackson VR, Civelli O. Orphan G protein-coupled receptors and obesity. Eur J Pharmacol. 2004;500:243–253 [DOI] [PubMed] [Google Scholar]
- 14. Galandrin S, Bouvier M. Distinct signaling profiles of β1 and β2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol. 2006;70:1575–1584 [DOI] [PubMed] [Google Scholar]
- 15. Offermanns S, Wieland T, Homann D, et al. Transfected muscarinic acetylcholine receptors selectively couple to Gi-type G proteins and Gq/11. Mol Pharmacol. 1994;45:890–898 [PubMed] [Google Scholar]
- 16. Laugwitz KL, Allgeier A, Offermanns S, et al. The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families. Proc Natl Acad Sci USA. 1996;93:116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin LQ, Wang HY, Friedman E. Stimulated D(1) dopamine receptors couple to multiple Gα proteins in different brain regions. J Neurochem. 2001;78:981–990 [DOI] [PubMed] [Google Scholar]
- 18. Luttrell LM, Lefkowitz RJ. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465 [DOI] [PubMed] [Google Scholar]
- 19. Luttrell LM, Gesty-Palmer D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. [published correction appears in Pharmacol Rev. 2010;62:564]. Pharmacol Rev. 2010;62305–62330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharm Rev. 2001;53:1–24 [PubMed] [Google Scholar]
- 21. Lefkowitz RJ. G protein-coupled receptor kinases. Cell. 1993;74:409–412 [DOI] [PubMed] [Google Scholar]
- 22. Goodman OB, Jr., Krupnick JG, Santini F, et al. β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383:447–450 [DOI] [PubMed] [Google Scholar]
- 23. Laporte SA, Oakley RH, Zhang J, et al. The β2-adrenergic receptor/β-arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luttrell LM, Ferguson SS, Daaka Y, et al. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661 [DOI] [PubMed] [Google Scholar]
- 25. DeFea KA, Vaughn ZD, O'Bryan EM, Nishijima D, Déry O, Bunnett NW. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a β-arrestin-dependent scaffolding complex. Proc Natl Acad Sci USA. 2000;97:11086–11091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barlic J, Andrews JD, Kelvin AA, et al. Regulation of tyrosine kinase activation and granule release through β-arrestin by CXCRI. Nat Immunol. 2000;1:227–233 [DOI] [PubMed] [Google Scholar]
- 27. McDonald PH, Chow CW, Miller WE, et al. β-Arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577 [DOI] [PubMed] [Google Scholar]
- 28. DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luttrell LM, Roudabush FL, Choy EW, et al. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98:2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science. 2001;294:1307–1313 [DOI] [PubMed] [Google Scholar]
- 31. Perry SJ, Baillie GS, Kohout TA, et al. Targeting of cyclic AMP degradation to β 2-adrenergic receptors by β-arrestins. Science. 2002;298:834–836 [DOI] [PubMed] [Google Scholar]
- 32. Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK, Lefkowitz RJ. Targeting of diacylglycerol degradation to M1 muscarinic receptors by β-arrestins. Science. 2007;315:663–666 [DOI] [PubMed] [Google Scholar]
- 33. Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. β-Arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc Natl Acad Sci USA. 2004;101:8603–8607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhattacharya M, Anborgh PH, Babwah AV, et al. β-Arrestins regulate a Ral-GDS Ral effector pathway that mediates cytoskeletal reorganization. Nat Cell Biol. 2002;4:547–555 [DOI] [PubMed] [Google Scholar]
- 35. Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. β-Arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem. 2007;282:20634–20646 [DOI] [PubMed] [Google Scholar]
- 36. Beaulieu JM, Sotnikova TD, Yao WD, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273 [DOI] [PubMed] [Google Scholar]
- 38. Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17:126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238 [DOI] [PubMed] [Google Scholar]
- 40. Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374 [DOI] [PubMed] [Google Scholar]
- 41. Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discov. 2005;4:919–927 [DOI] [PubMed] [Google Scholar]
- 42. Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389 [DOI] [PubMed] [Google Scholar]
- 43. Kenakin TP. ′7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol Sci. 2009;30:460–469 [DOI] [PubMed] [Google Scholar]
- 44. Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luttrell LM, Kenakin TP. Refining efficacy: allosterism and bias in G protein-coupled receptor signaling. Methods Mol Biol. 2011;756:3–35 [DOI] [PubMed] [Google Scholar]
- 46. Spengler D, Waeber C, Pantaloni C, et al. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175 [DOI] [PubMed] [Google Scholar]
- 47. Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104 [PubMed] [Google Scholar]
- 48. Sagan S, Chassaing G, Pradier L, Lavielle S. Tachykinin peptides affect differently the second messenger pathways after binding to CHO-expressed human NK-1 receptors. J Pharmacol Exp Ther. 1996;276:1039–1048 [PubMed] [Google Scholar]
- 49. Takasu H, Gardella TJ, Luck MD, Potts JT, Jr., Bringhurst FR. Amino-terminal modifications of human parathyroid hormone (PTH) selectively alter phospholipase C signaling via the type 1 PTH receptor: Implications for design of signal-specific PTH ligands. Biochemistry. 1999;38:13453–13460 [DOI] [PubMed] [Google Scholar]
- 50. Gray JA, Roth BL. Paradoxical trafficking and regulation of 5-HT(2A) receptors by agonists and antagonists. Brain Res Bull. 2001;56:441–451 [DOI] [PubMed] [Google Scholar]
- 51. Holloway AC, Qian H, Pipolo L, et al. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777 [DOI] [PubMed] [Google Scholar]
- 52. Sneddon WB, Magyar CE, Willick GE, et al. Ligand-selective dissociation of activation and internalization of the parathyroid hormone (PTH) receptor: conditional efficacy of PTH peptide fragments. Endocrinology. 2004;145:2815–2823 [DOI] [PubMed] [Google Scholar]
- 53. Seifert R, Gether U, Wenzel-Seifert K, Kobilka BK. Effects of guanine, inosine, and xanthine nucleotides on β(2)-adrenergic receptor/G(s) interactions: evidence for multiple receptor conformations. Mol Pharmacol. 1999;56:348–358 [DOI] [PubMed] [Google Scholar]
- 54. Perez DM, Hwa J, Gaivin R, Mathur M, Brown F, Graham RM. Constitutive activation of a single effector pathway: evidence for multiple activation states of a G protein-coupled receptor. Mol Pharmacol. 1996;49:112–122 [PubMed] [Google Scholar]
- 55. Barroso S, Richard F, Nicolas-Ethève D, Kitabgi P, Labbé-Jullié C. Constitutive activation of the neurotensin receptor 1 by mutation of Phe(358) in helix seven. Br J Pharmacol. 2002;135:997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential binding of agonists to the β2 adrenoceptor. Kinetic evidence for intermediate conformational states. J Biol Chem. 2004;279:686–691 [DOI] [PubMed] [Google Scholar]
- 57. Swaminath G, Deupi X, Lee TW, et al. Probing the β2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J Biol Chem. 2005;280:22165–22171 [DOI] [PubMed] [Google Scholar]
- 58. Zürn A, Zabel U, Vilardaga JP, Schindelin H, Lohse MJ, Hoffmann C. Fluorescence resonance energy transfer analysis of α2a-adrenergic receptor activation reveals distinct agonist-specific conformational changes. Mol Pharmacol. 2009;75:534–541 [DOI] [PubMed] [Google Scholar]
- 59. Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401 [DOI] [PubMed] [Google Scholar]
- 60. Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissocation of μ-opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746 [DOI] [PubMed] [Google Scholar]
- 61. Jarpe MB, Knall C, Mitchell FM, Buhl AM, Duzic E, Johnson GL. [D-Arg1,D-Phe5,D-Trp7,9,Leu11]Substance P acts as a biased agonist toward neuropeptide and chemokine receptors. J Biol Chem. 1998;273:3097–3104 [DOI] [PubMed] [Google Scholar]
- 62. Kudlacek O, Waldoer M, Kassack MU, et al. Biased inhibition by a suramin analogue of A1-adenosine receptor/G protein coupling in fused receptor/G protein tandems: the A1 adenosine receptor is predominantly coupled to Goα in human brain. Naunyn Schmiedeberg's Arch Pharmacol. 2002;365:8–16 [DOI] [PubMed] [Google Scholar]
- 63. Manning DR. Measures of efficacy using G proteins as endpoints: differential engagement of G proteins through single receptors. Mol Pharmacol. 2002;62:451–452 [DOI] [PubMed] [Google Scholar]
- 64. Gurwitz D, Haring R, Heldman E, Fraser CM, Manor D, Fisher A. Discrete activation of transduction pathways associated with acetylcholine M1 receptor by several muscarinic ligands. Eur J Pharmacol. 1994;267:21–31 [DOI] [PubMed] [Google Scholar]
- 65. Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222 [DOI] [PubMed] [Google Scholar]
- 66. Wei H, Ahn S, Shenoy SK, et al. Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Azzi M, Charest PG, Angers S, et al. β-Arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003;100:11406–11411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gesty-Palmer D, Chen M, Reiter E, et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–10864 [DOI] [PubMed] [Google Scholar]
- 69. Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA. 2008;105:7439–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Choe HW, Kim YJ, Park JH, et al. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655 [DOI] [PubMed] [Google Scholar]
- 71. Nygaard R, Zou Y, Dror RO, et al. The dynamic process of β(2)-adrenergic receptor activation. Cell. 2013;152:532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rasmussen SG, DeVree BT, Zou Y, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zocher M, Fung JJ, Kobilka BK, Müller DJ. Ligand-specific interactions modulate kinetic, energetic, and mechanical properties of the human β2 adrenergic receptor. Structure. 2012;20:1391–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Coulon V, Audet M, Homburger V, et al. Subcellular imaging of dynamic protein interactions by bioluminescence resonance energy transfer. Biophys J. 2008;94:1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lohse MJ, Nuber S, Hoffmann C. Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol Rev. 2012;64:299–336 [DOI] [PubMed] [Google Scholar]
- 77. Saulière A, Bellot M, Paris H, et al. Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol. 2012;8:622–630 [DOI] [PubMed] [Google Scholar]
- 78. Gesty-Palmer D, Luttrell LM. Heptahelical terpsichory. Who calls the tune? J Recept Signal Transduct Res. 2008;28:39–58 [DOI] [PubMed] [Google Scholar]
- 79. Christensen GL, Kelstrup CD, Lyngsø C, et al. Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol Cell Proteomics. 2010;9:1540–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kendall RT, Strungs EG, Rachidi SM, et al. The β-arrestin pathway-selective type 1A angiotensin receptor (AT1A) receptor agonist [Sar1Ile4Ile8]angiotensin II regulates a robust G protein-independent signaling network. J Biol Chem. 2011;286:19880–19891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rajagopal K, Whalen EJ, Violin JD, et al. β-Arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006;103:16284–16289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Aplin M, Christensen GL, Schneider M, et al. Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes. Basic Clin Pharmacol Toxicol. 2007;100:296–301 [DOI] [PubMed] [Google Scholar]
- 83. Gesty-Palmer D, Yuan L, Martin B, et al. β-Arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol Endocrinol. 2013;27:296–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Luttrell LM. 'Location, location, location': activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol. 2003;30:117–126 [DOI] [PubMed] [Google Scholar]
- 85. Stork PJ. ERK signaling: duration, duration, duration. Cell Cycle. 2002;1:315–317 [DOI] [PubMed] [Google Scholar]
- 86. Dean T, Vilardaga JP, Potts JT, Jr., Gardella TJ. Altered selectivity of parathyroid hormone (PTH) and PTH-related protein (PTHrP) for distinct conformations of the PTH/PTHrP receptor. Mol Endocrinol. 2008;22:156–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ferrandon S, Feinstein TN, Castro M, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vilardaga JP, Romero G, Friedman PA, Gardella TJ. Molecular basis of parathyroid hormone receptor signaling and trafficking: a family B GPCR paradigm. Cell Mol Life Sci. 2011;68:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Shimizu N, Dean T, Tsang JC, Khatri A, Potts JT, Jr., Gardella TJ. Novel parathyroid hormone (PTH) antagonists that bind to the juxtamembrane portion of the PTH/PTH-related protein receptor. J Biol Chem. 2005;280:1797–1807 [DOI] [PubMed] [Google Scholar]
- 90. Okazaki M, Ferrandon S, Vilardaga JP, Bouxsein ML, Potts JT, Jr., Gardella TJ. Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation. Proc Natl Acad Sci USA. 2008;105:16525–16530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zimmerman B, Beautrait A, Aguila B, et al. Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci Signal. 2012;5:ra33. [DOI] [PubMed] [Google Scholar]
- 92. Tobin AB, Butcher AJ, Kong KC. Location, location, location … site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;29:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wisler JW, DeWire SM, Whalen EJ, et al. A unique mechanism of β-blocker action: carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Qin L, Raggatt LJ, Partridge NC. Parathyroid hormone: A double-edged sword for bone metabolism. Trends Endocrinol Metab. 2004;15:60–65 [DOI] [PubMed] [Google Scholar]
- 95. Mohan S, Kutilek S, Zhang C, et al. Comparison of bone formation responses to parathyroid hormone(1–34), (1–31), and (2–34) in mice. Bone. 2000;27:471–478 [DOI] [PubMed] [Google Scholar]
- 96. Gesty-Palmer D, Flannery P, Yuan L, Spurney R, Lefkowitz RJ, Luttrell LM. A β-arrestin biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1:1ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Violin JD, DeWire SM, Yamashita D, et al. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335:572–579 [DOI] [PubMed] [Google Scholar]
- 98. Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC., Jr Cardiorenal actions of TRV120027, a novel ß-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail. 2011;4:770–778 [DOI] [PubMed] [Google Scholar]
- 99. Boerrigter G, Soergel DG, Violin JD, Lark MW, Burnett JC., Jr TRV120027, a novel β-arrestin biased ligand at the angiotensin II type I receptor, unloads the heart and maintains renal function when added to furosemide in experimental heart failure. Circ Heart Fail. 2012;5:627–634 [DOI] [PubMed] [Google Scholar]
- 100. Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science. 1999;286:2495–2498 [DOI] [PubMed] [Google Scholar]
- 101. Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in β-arrestin-2 knock-out mice. J Neurosci. 2002;22:10494–10500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. DeWire SM, Yamashita DS, Rominger DH, et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717 [DOI] [PubMed] [Google Scholar]
- 103. Walters RW, Shukla AK, Kovacs JJ, et al. β-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest. 2009;119:1312–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Semple G, Skinner PJ, Gharbaoui T, et al. 3-(1H-tetrazol-5-yl)-1,4,5,6-tetrahydro-cyclopentapyrazole (MK-0354): a partial agonist of the nicotinic acid receptor, G-protein coupled receptor 109a, with antilipolytic but no vasodilatory activity in mice. J Med Chem. 2008;51:5101–5108 [DOI] [PubMed] [Google Scholar]
- 105. Beaulieu JM, Marion S, Rodriguiz RM, et al. A β-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136 [DOI] [PubMed] [Google Scholar]
- 106. Masri B, Salahpour A, Didriksen M, et al. Antagonism of dopamine D2 receptor/β-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA. 2008;105:13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wang L, Martin B, Brenneman R, Luttrell LM, Maudsley S. Allosteric modulators of G protein-coupled receptors: future therapeutics for complex physiological disorders. J Pharmacol Exp Ther. 2009;331:340–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Luttrell LM. Arrestin pathways as drug targets. Prog Mol Biol Transl Sci. 2013;118:469–497 [DOI] [PubMed] [Google Scholar]
- 109. Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136:3632–3638 [DOI] [PubMed] [Google Scholar]
- 110. Deng ZL, Sharff KA, Tang N, et al. Regulation of osteogenic differentiation during skeletal development. Front Biosci. 2008;13;2001–2021 [DOI] [PubMed] [Google Scholar]
- 111. Lian JB, Stein GS, Javed A, et al. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:1–16 [DOI] [PubMed] [Google Scholar]
- 112. Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Soltys S, Koch WJ. An adrenal β-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc Natl Acad Sci USA. 2009;106:5825–5830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kim J, Zhang L, Peppel K, et al. β-Arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res. 2008;103:70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Miura S, Zhang J, Matsuo Y, Saku K, Karnik SS. Activation of extracellular signal-activated kinase by angiotensin II-induced Gq-independent epidermal growth factor receptor transactivation. Hypertens Res. 2004;27:765–770 [DOI] [PubMed] [Google Scholar]
- 115. Kim J, Ahn S, Rajagopal K, Lefkowitz RJ. Independent β-arrestin2 and Gq/protein kinase Cζ pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J Biol Chem. 2009;284:11953–11962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. β-Arrestin-2 mediates anti-apoptotic signaling through regulation of BAD phosphorylation. J Biol Chem. 2009;284:8855–8865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kim IM, Tilley DG, Chen J, et al. β-Blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA. 2008;105:14555–14560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cottingham C, Chen Y, Jiao K, Wang Q. The antidepressant desipramine is an arrestin-biased ligand at the α(2A)-adrenergic receptor driving receptor down-regulation in vitro and in vivo. J Biol Chem. 2011;286:36063–36075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ryman-Rasmussen JP, Griffith A, Oloff S, et al. Functional selectivity of dopamine D1 receptor agonists in regulating the fate of internalized receptors. Neuropharmacology. 2007;52:562–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hartley O, Gaertner H, Wilken J, et al. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc Natl Acad Sci USA. 2004;101:16460–16465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J Biol Chem. 2013;288:9790–9800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhou L, Lovell KM, Frankowski KJ, et al. Development of functionally selective, small molecule agonists at κ opioid receptors. J Biol Chem. 2013;288:36703–36716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM. Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at κ-opioid receptors in striatal neurons. J Biol Chem. 2013;288:22387–22398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Raehal KM, Bohn LM. β-Arrestins: regulatory role and therapeutic potential in opioid and cannabinoid receptor-mediated analgesia. Handb Exp Pharmacol. 2014;219:427–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Urban JD, Vargas GA, von Zastrow M, Mailman RB. Aripiprazole has functionally selective actions at dopamine D2 receptor-mediated signaling pathways. Neuropsychopharmacology. 2007;32:67–77 [DOI] [PubMed] [Google Scholar]
- 126. Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on β-arrestin-2 interactions in vivo. Proc Natl Acad Sci USA. 2008;105:1079–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Schmid CL, Bohn LM. Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ß-arrestin2/Src/Akt signaling complex in vivo. J Neurosci. 2010;30:13513–13524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jorgensen R, Kubale V, Vrecl M, Schwartz TW, Elling CE. Oxyntomodulin differentially affects glucagon-like peptide-1 receptor β-arrestin recruitment and signaling through Gα(s). J Pharmacol Exp Ther. 2007;322:148–154 [DOI] [PubMed] [Google Scholar]
- 129. Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. β-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic β cells. Proc Natl Acad Sci USA. 2008;105:6614–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dalle S, Ravier MA, Bertrand G. Emerging roles for β-arrestin-1 in the control of the pancreatic β-cell function and mass: new therapeutic strategies and consequences for drug screening. Cell Signal. 2011;23:522–528 [DOI] [PubMed] [Google Scholar]
- 131. de Roux N, Milgrom E. Inherited disorders of GnRH and gonadotropin receptors. Mol Cell Endocrinol. 2001;179:83–87 [DOI] [PubMed] [Google Scholar]
- 132. Duprez L, Parma J, Van Sande J, et al. TSH receptor mutations and thyroid disease. Trends Endocrinol Metab. 1998;9:133–140 [DOI] [PubMed] [Google Scholar]
- 133. Barak LS, Oakley RH, Laporte SA, Caron MG. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc Natl Acad Sci USA. 2001;98:93–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nichols HL, Saffeddine M, Theriot BS, et al. β-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci USA. 2012;109:16660–16665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Penn RB, Bond RA, Walker JK. GPCRs and Arrestins in Airways: Implications for Asthma. Handb Exp Pharmacol. 2014;219:387–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ramachandran R, Mihara K, Chung H, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem. 2011;286:24638–24648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lau C, Lytle C, Straus DS, DeFea KA. Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am J Physiol Cell Physiol. 2011;300:C113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sobolesky PM, Moussa O. The role of β-arrestins in cancer. Prog Mol Biol Transl Sci. 2013;118:395–411 [DOI] [PubMed] [Google Scholar]
- 139. Jiang D, Xie T, Liang J, Noble PW. β-Arrestins in the immune system. Prog Mol Biol Transl Sci. 2013;118:359–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kenakin TP. Ligand detection in the allosteric world. J Biomol Screen. 2010;15:119–130 [DOI] [PubMed] [Google Scholar]
- 141. Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216 [DOI] [PubMed] [Google Scholar]
- 143. Milligan G. High-content assays for ligand regulation of G-protein coupled receptors. Drug Disc Today. 2003;8:579–585 [DOI] [PubMed] [Google Scholar]
- 144. Ross DA, Lee S, Reiser V, et al. Multiplexed assays by high-content imaging for assessment of GPCR activity. J Biomol Screen. 2008;13:449–455 [DOI] [PubMed] [Google Scholar]
- 145. Ghosh RN, DeBiasio R, Hudson CC, Ramer ER, Cowan CL, Oakley RH. Quantitative cell-based high-content screening for vasopressin receptor agonists using transfluor technology. J Biomol Screen. 2005;10:476–484 [DOI] [PubMed] [Google Scholar]
- 146. Hanson BJ, Wetter J, Bercher MR, et al. A homogeneous fluorescent live-cell assay for measuring 7-transmembrane receptor activity and agonist functional selectivity through β-arrestin recruitment. J Biomol Screen. 2009;14:798–810 [DOI] [PubMed] [Google Scholar]
- 147. Hamdan FF, Audet M, Garneau P, Pelletier J, Bouvier M. High throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based β-arrestin2 recruitment assay. J Biomol Screen. 2005;10:463–475 [DOI] [PubMed] [Google Scholar]
- 148. Zhao X, Jones A, Olson KR, et al. A homogeneous enzyme fragment complementation-based β-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J Biomol Screen. 2008;13:737–747 [DOI] [PubMed] [Google Scholar]
- 149. Barnea G, Strapps W, Herrada G, et al. The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci USA. 2008;105:64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Henriksen U, Fog J, Loechel F, Praestegaard M. Profiling of multiple signal pathway activities by multiplexing antibody and GFP-based translocation assays. Comb Chem High Throughput Screen. 2008;11:537–544 [DOI] [PubMed] [Google Scholar]
- 151. Kenakin T. Quantifying biased β-arrestin signaling. Handb Exp Pharmacol. 2014;219:57–83 [DOI] [PubMed] [Google Scholar]
- 152. Fang Y, Ferrie AM, Fontaine NH, Yuen PK. Characteristics of dynamic mass redistribution of epidermal growth factor receptor signaling in living cells measured with label-free optical biosensors. Anal Chem. 2005;77:5720–5725 [DOI] [PubMed] [Google Scholar]
- 153. Cunningham BT, Li P, Schultz S, et al. Label free assays on the BIND system. J Biomol Screen. 2004;9:481–490 [DOI] [PubMed] [Google Scholar]
- 154. Fang Y, Ferrie AM. Label-free optical biosensor for ligand-directed functional selectivity acting on β2-adrenoceptor in living cells. FEBS Lett. 2008;582:558–564 [DOI] [PubMed] [Google Scholar]
- 155. McGuinness R. Impedance-based cellular assay technologies: recent advances, future promise. Curr Opin Pharmacol. 2007;7:535–540 [DOI] [PubMed] [Google Scholar]
- 156. Peters MF, Scott CW. Evaluating cellular impedance assays for detection of GPCR pleiotropic signaling and functional selectivity. J Biomol Screen. 2009;14:246–255 [DOI] [PubMed] [Google Scholar]
- 157. Congreve M, Langmead C, Marshall FH. The use of GPCR structures in drug design. Adv Pharmacol. 2011;62:1–36 [DOI] [PubMed] [Google Scholar]
- 158. Kooistra AJ, Roumen L, Leurs R, de Esch IJ, de Graaf C. From heptahelical bundle to hits from the haystack: structure-based virtual screening for GPCR ligands. Methods Enzymol. 2013;522:279–336 [DOI] [PubMed] [Google Scholar]
- 159. Kołaczkowski M, Bucki A, Feder M, Pawłowski M. Ligand-optimized homology models of D1 and D2 dopamine receptors: application for virtual screening. J Chem Inf Model. 2013;53:638–648 [DOI] [PubMed] [Google Scholar]
- 160. Lane JR, Chubukov P, Liu W, et al. Structure-based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol Pharmacol. 2013;84:794–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Kuang G, Hu G, Sun X, Li W, Liu G, Tang Y. In silico investigation of interactions between human cannabinoid receptor-1 and its antagonists. J Mol Model. 2012;18:3831–3845 [DOI] [PubMed] [Google Scholar]
- 162. Renault N, Laurent X, Farce A, et al. Virtual screening of CB(2) receptor agonists from bayesian network and high-throughput docking: structural insights into agonist-modulated GPCR features. Chem Biol Drug Des. 2013;81:442–454 [DOI] [PubMed] [Google Scholar]
- 163. Yoshikawa Y, Oishi S, Kubo T, Tanahara N, Fujii N, Furuya T. Optimized method of G-protein-coupled receptor homology modeling: its application to the discovery of novel CXCR7 ligands. J Med Chem. 2013;56:4236–4251 [DOI] [PubMed] [Google Scholar]
- 164. Weiss DR, Ahn S, Sassano MF, et al. Conformation guides molecular efficacy in docking screens of activated β-2 adrenergic G protein coupled receptor. ACS Chem Biol. 2013;8:1018–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Grassi G, Seravalle G, Quarti-Trevano F, Dell'oro R. Sympathetic activation in congestive heart failure: evidence, consequences and therapeutic implications. Curr Vasc Pharmacol. 2009;7:137–145 [DOI] [PubMed] [Google Scholar]
- 166. Maudsley S, Patel SA, Park SS, Luttrell LM, Martin B. Functional signaling biases in G protein-coupled receptors: game theory and receptor dynamics. Mini-Rev Med Chem. 2012;12:831–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Nemeth EF, Heaton WH, Miller M, et al. Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther. 2004;308:627–635 [DOI] [PubMed] [Google Scholar]
- 168. Muniz-Medina VM, Jones S, Maglich JM, et al. The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property? Mol Pharmacol. 2009;75:490–501 [DOI] [PubMed] [Google Scholar]