

Abstract
Abnormal biometal homeostasis is a central feature of many neurodegenerative disorders including Alzheimer's disease (AD), Parkinson's disease (PD), and motor neuron disease. Recent studies have shown that metal complexing compounds behaving as ionophores such as clioquinol and PBT2 have robust therapeutic activity in animal models of neurodegenerative disease; however, the mechanism of neuroprotective action remains unclear. These neuroprotective or neurogenerative processes may be related to the delivery or redistribution of biometals, such as copper and zinc, by metal ionophores. To investigate this further, we examined the effect of the bis(thiosemicarbazonato)-copper complex, CuII(gtsm) on neuritogenesis and neurite elongation (neurogenerative outcomes) in PC12 neuronal-related cultures. We found that CuII(gtsm) induced robust neurite elongation in PC12 cells when delivered at concentrations of 25 or 50 nM overnight. Analogous effects were observed with an alternative copper bis(thiosemicarbazonato) complex, CuII(atsm), but at a higher concentration. Induction of neurite elongation by CuII(gtsm) was restricted to neurites within the length range of 75–99 µm with a 2.3-fold increase in numbers of neurites in this length range with 50 nM CuII(gtsm) treatment. The mechanism of neurogenerative action was investigated and revealed that CuII(gtsm) inhibited cellular phosphatase activity. Treatment of cultures with 5 nM FK506 (calcineurin phosphatase inhibitor) resulted in analogous elongation of neurites compared to 50 nM CuII(gtsm), suggesting a potential link between CuII(gtsm)-mediated phosphatase inhibition and neurogenerative outcomes.
Introduction
Altered biometal homeostasis is a central feature of many neurodegenerative disorders including Alzheimer's disease (AD), Parkinson's disease (PD) and motor neuron diseases. In AD, copper (Cu) and zinc (Zn) associate with extracellular amyloid-beta (Aβ) plaques, while intracellular Cu levels are decreased (Reviewed in [1] & [2]). As Cu is critical for many neuronal functions including synaptogenesis, normal synaptic activity and intracellular signaling [3], [4], targeting abnormal Cu homeostasis represents a potential therapeutic treatment for AD. Although a recent study found no cognitive improvements in AD patients receiving Cu supplements in their diet [5], early clinical trials of Cu/Zn ionophores including clioquinol (CQ) and PBT2 have demonstrated promising outcomes [6], [7].
Rather than acting as metal chelators to remove metal ions, the 8-hydroxyquinoline bidentate ligands, CQ and PBT2 can exert their effects in cell culture and animal models of AD by redistributing metal ions. CQ delivered Cu and Zn into cultured cells and stimulated Aβ degrading metallo-enzymes including matrix metalloprotease 2 (MMP2) [8]. PBT2 removed Zn from aggregated Aβ, redistributed the Zn into neuronal-like SH-SY5Y cells, inhibited glycogen synthase kinase 3 beta (GSK3β) and decreased calcineurin activity [9]. These effects may provide neuroprotective signaling changes in neurons.
Animal models of Aβ accumulation such as APP/PS1 and Tg2576 treated with CQ and PBT2 showed reductions in Aβ accumulation together with enhanced cognitive performance [10], [11]. However, it is not certain if these two observations are directly related. Our studies have provided evidence that metal delivery, or redistribution by ionophores such as CQ, PBT2 or metal complexes of bis(thiosemicarbazones) promote neuroprotective actions that are independent on changes to Aβ burden. Treatment of APP/PS1 mice with glyoxal-bis(N(4)-methylthiosemicarbazonato)-copper(II) (CuII(gtsm)) resulted in enhanced cognitive performance compared to sham-treated animals without changes to Aβ deposition (although there was a correlation with decreased Aβ trimer levels) [12]. Malm et al. demonstrated that the metal ionophore pyrrolidine dithiocarbamate (PDTC) elevated brain Cu levels and improved cognition in APP/PS1 mice without altered Aβ levels [13]. We found that another bis(thiosemicarbazone) diacetyl-bis(N4-methylthiosemicarbazonato)-copper(II) (CuII(atsm)), which is closely related to CuII(gtsm), had a potent neuroprotective action in multiple models of Parkinson's disease and motor neuron disease, two diseases not associated with the Aβ peptide [14], [15]. CuII(atsm) could also modify TAR-DNA binding protein 43 (TDP-43) metabolism, a protein central to neuropathology of amyotrophic lateral sclerosis and a sub-set of frontotemporal dementia cases [15], [16]. Combined, these studies provide strong evidence for a broad neuroprotective action of metal ionophores and Cu or Zn-complexes in models of neurodegeneration.
To further understand the neuroprotective action of metal ionophores in vivo, we studied dendritic spine density in a transgenic mouse model of Alzheimer's disease, Tg2576 mice, treated with PBT2 [17]. Dendritic spine density is a measure of synaptic strength, as dendritic spines are the site of interneuron synaptic connections in the brain. We observed a significant improvement in apical spine density in older Tg2576 mice treated with PBT2 [17]. A role for a general neurotherapeutic action by PBT2 in these mice was supported by elevation of several important neurotrophic proteins in the treated mice including pro-BDNF and TrkB. Cell culture studies demonstrated that PBT2 promoted neurite elongation[17]. This could be enhanced by the addition of Cu or Zn and prevented by co-incubation with the cell-impermeable Cu/Zn chelator, diAmsar [17]. These studies suggested that the neuroprotective action of PBT2 involved the redistribution of biometals such as Cu and/or Zn.
As CuII(gtsm) and CuII(atsm) have also demonstrated neuroprotective activity in vivo in murine models of neurodegeneration, we have now examined whether these metal-complexes induce similar changes in cultured cells. The metal complexes of bis(thiosemicarbazones) provide some advantages over 8-hydroxyquinoline compounds by being stable, pre-formed complexes that release Cu under specific intracellular environments [18]. This enabled us to determine if the delivery of Cu as a stable CuII complex results in beneficial action upon the intracellular release of the metal as CuI. We investigated changes to total neurite numbers and neurite elongation (i.e., neurogenerative outcomes) in nerve growth factor-treated PC12 cell cultures. This neurosecretory cell line was chosen because it is the most well characterized model for neurite formation and growth, providing a useful model to examine how and where Cu complexes act. CuII(gtsm) potently induced neurite elongation at concentrations as low as 25 nM. Treatment with CuII(atsm), which is structurally comparable but does not readily release Cu inside cells under basal cellular redox conditions [19], required a 10-fold higher concentration to elicit a similar response. The potential mechanisms underlying the increased metal-associated neurite elongation were investigated. There was no specific association with kinase activation, but there was a significant association between inhibition of phosphatases and neurite elongation. Promotion of neurogenerative outcomes could potentially be a valuable in vitro biomarker for the neuroprotective action of these metal-complexes.
Materials and Methods
Materials
(CuII(gtsm), CuII(atsm), (gtsm)H2, diAmsar (1,8-diminosarcophagine) were synthesized as previously reported [20]–[23]. All other chemicals were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia) unless specified otherwise.
Pheochromocytoma (PC12) cell culture
PC12 cells (a kind gift from Dr Ann Turnley, Centre for Neuroscience, The University of Melbourne, Victoria, 3010, Australia) [17] were maintained in DMEM (Invitrogen, Mulgrave, Victoria, Australia) supplemented with 10% (v/v) foetal bovine serum, 5% (v/v) horse serum and 5 mL Penicillin/Streptomycin sulfate (solution contains 5000 units of penicillin and 5000 µg of streptomycin per mL, Invitrogen). Culture plates (Nunc, ThermoFischer Scientific, Scoresby, Victoria, Australia) were coated with 5 µg/cm2 collagen (Type I collagen, Rat tail derived, dissolved in 0.02 M acetic acid (Invitrogen)) for 1 hr at 37°C and then rinsed with PBS pH 7.4 before plating of cells, or stored at 4°C until used. For neurite elongation assays, cells were plated at low density (17×103 cells/cm2) onto collagen-coated plates and allowed to settle for 24 hr. Medium was then replaced with serum-free medium containing nerve growth factor (NGF) (50 ng/mL (Invitrogen)) and cultured for a further 48 hr prior to treatments with metal-complexes (added into existing medium). Cells were examined for neurite elongation after a further 18 hr treatment. This protocol was employed to ensure that cells were at an early stage of neurite development to determine if this could be improved by treatment with Cu-complexes. Cells were not examined after longer periods because extensive neurite networks precluded quantitation of changes.
Treatment of cells with metal-complexes
Metal-complexes were dissolved in dimethyl sulphoxide (DMSO) to achieve a 10 mM stock concentration. Dilutions were then performed where necessary so that the final volume of DMSO added to cells did not exceed 0.001% (v/v) of the final volume of culture medium in each well. Complexes were added to cells at indicated concentrations for 18 hr. Where indicated, inhibitors of extracellular signal regulated kinase (ERK) (PD98059, 10 µM), c-Jun N-terminal kinase (JNK) (SP600125, 10 µM), glycogen synthase kinase 3 (GSK3) (GSK3 inhibitor VII, 10 µM) and calcineurin (FK506, 5 and 10 nM) were added to cells for 18 hr. No evidence of significant metal-complex interaction with collagen matrix was found.
Quantification of neurite formation and elongation
For assessment of neurite growth, cells were treated for 18 hr (after 48 hr NGF-differentiation) and fixed in 4% (v/v) paraformaldehyde (PFA) (ProSciTech, Thuringowa, QLD, Australia) in PBS (pH 7.4) for 20 min, then preserved in PBS containing 0.001% (w/v) sodium azide until bright field photographs were taken with a Leica inverted microscope with a Zeiss Axiocam digital camera at 500x magnification. ImageJ software (NIH) (with the neuronJ plugin) was used to quantify neuritogenesis and neurite elongation. Mean neurite length was calculated using NeuronJ by tracing the individual neurites of all the cells in 6 separate images taken randomly for each treatment (from three separate experiments). The length of each trace was measured, lengths were converted into µm and an average taken for each treatment. The proportion of cells with neurites 2x or longer than the width of the cell body was expressed as a percentage of the total. Neurite lengths were grouped into categories of 0–24, 25–49, 50–74, 75–99, 100–124 and greater than 125 µm and the mean number of neurites in each category was compared to assess if the distribution of neurite lengths was affected by treatment (n = 6 images from each of 3 separate cultures for each treatment). For assessment of neurite numbers and length, the investigator was blinded to the treatments.
SDS-PAGE and Western Blot Analysis
After treatment, the media was discarded and the cells were scraped into Phosphosafe buffer (Phosphosafe Extraction Reagent, Merck, Kilsyth, VIC, Australia) supplemented with 100x phenylmethylsulfonyl fluoride (PMSF) and 50x deoxyribonuclease I (DNAse I, Roche Diagnostics, Castle Hill, NSW, Australia), using 50 µL for each well of a 6-well plate or 100 µL for a 100 mm dish. The cell suspension was then centrifuged at 16,000×g for 5 min. The cell pellet was discarded and an aliquot of the supernatant/lysate was taken for protein determination using the Pierce BCA protein determination assay as per instructions (Thermo Scientific, Rockford, IL, USA). Cell lysates were standardized using PBS (pH 7.4) and 4x sample buffer (250 mM Tris, 20% (v/v) glycerol, 8% (w/v) sodium dodecyl sulphate (SDS), 2% (w/v) β-mercaptoethanol, 0.01% (w/v) bromophenol blue) and incubated at 100°C for 5 min. Samples (10–20 µg) were then loaded onto 12% tris-glycine gels. Electrophoresis was performed at 125 V for 1 hr 45 min and proteins transferred onto PVDF membrane (Roche Diagnostics) at 25 V for 1 hr 45 min using transfer buffer (prepared from 25x stock (Invitrogen)) with 20% (v/v) methanol. PVDF membranes were blocked for 1 hr at room temperature with PBS (pH 7.4) supplemented with 0.05% (v/v) Tween-20 detergent (PBST) and 5% (w/v) skim milk powder. Membranes were incubated with primary antibodies all purchased from Cell Signaling Technology (Arunel, Queensland, Australia) and diluted at 1∶5000) overnight at 4°C in blocking solution, washed 3 times for 5 min with PBST, incubated for 2–3 hr at room temperature with secondary anti-rabbit antibody (diluted at 1∶500) in the blocking solution and then washed 4 times for 5 min with PBST before chemiluminescence imaging of protein bands using ECL Advance (GE Healthcare Biosciences, Castle Hill, NSW, Australia) and a Fujifilm LAS-3000 imager. Densitometric protein quantification was performed using ImageJ software and protein levels standardized relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression.
Immunofluorescence
After treatment with metallo-complexes, cells were rinsed once in PBS, fixed in 4% PFA in PBS for 20 min at room temperature and rinsed three times with PBS. Cells were permeabilized for 5 min with 0.01% (v/v) Triton-X dissolved in 10% (v/v) goat serum in PBS, followed by 3 washes with PBS and blocked in 10% (v/v) goat serum in PBS overnight at 4°C. Cells were washed 3 times with PBS and incubated with rabbit anti-phospho-JNK primary antibody (at 1∶500 dilution, Cat. # 4668, Cell Signaling Technology) in 10% (v/v) goat serum in PBS for a minimum of 2 hr at room temperature or overnight at 4°C. Cells were washed again 3 times with PBS and incubated with goat anti-rabbit secondary antibody (Alexafluor 488) at 1∶500 dilution in 10% (v/v) goat serum in PBS overnight at 4°C. Following three washes in PBS, cells were incubated for 5 min with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) dissolved in PBS at 0.5 µg/mL. Cells were washed 4 times in PBS, mounted on coverslips with DAKO mounting medium (Dako, Campbellfield, VIC, Australia) and dried at room temperature.
Inductively Coupled Mass Spectrometry
ICP-MS analysis of metal levels was performed as reported previously [24]. Cell pellets collected for metal analysis by ICP-MS were re-suspended in 50 µL of concentrated nitric acid (Aristar, BDH, Kilsyth, VIC, Australia) and left to digest overnight. Samples were heated for 20 min at 90°C to complete the digestion. The volume of each sample was reduced to approximately 40–50 µL after digestion, then 1 mL of 1% (v/v) nitric acid diluent was added to each cell sample. Measurements were made using an Agilent ICPMS 7700x series ICP-MS instrument under operating conditions suitable for routine multi-element analysis. The instrument was calibrated using 0, 5, 10, 50, 100 and 500 parts per billion (ppb) of certified multi-element ICP-MS standard calibration solutions (ICP-MS-CA12-1, ICP-MS-CAL-3 and ICP-MS-CAL-4, Accustandard) prepared in 1% (v/v) nitric acid for Cu, Fe, Zn and Mn. A certified internal standard solution containing 200 ppb of Yttrium (Y89) via T-piece was used as an internal control (ICP-MS- IS-MIX1-1, Accustandard). Results were expressed as micromole per liter concentrations of metal (µmol/L). The concentrations of Cu and other metals were calculated as µg of metal per mg of protein based on the protein concentration of the aliquot taken from each sample.
Phosphatase assay
The SensoLyte FDP Protein Phosphatase Assay Kit Fluorimetric (Anaspec, Fremont, CA, USA) was used to quantify phosphatase activity. Following exposure to metal complexes, cells were rinsed twice in lysis buffer (supplied in kit) before scraping each sample into lysis buffer containing 0.2% (v/v) Triton-X as well as 10 µg/mL Aprotinin, 10 µg/mL Leupeptin, 100 µM PMSF and 10 µg/mL Pepstatin A (as instructed in assay kit protocol). The cell suspension was incubated at 4°C for 10 min with agitation and then centrifuged at 2,50×g for 10 min at 4°C. Supernatants were combined with the FDP-containing reaction solution (provided in the kit) in a black 96-well plate and fluorescence intensity was measured at Ex/Em = 490±20/520±20 nm using a WALLAC Victor2 plate reader (Perkin Elmer, Waltham, MA, USA). Data represented the mean ± standard error of the mean, n = 3 samples per treatment.
Calcineurin assay
A colourimetric calcineurin assay was used to detect calcineurin activity (Merck KGaA, Darmstadt, Germany). Cells grown and treated in T75 flasks (Nunc) were scraped into Hank's balanced salt solution (HBSS, Invitrogen), centrifuged at 720×g for 3 min before resuspension in 1 mL of HBSS and standardized according to cell numbers (5×105 cells/sample). Cells were pelleted by centrifugation at 720×g for 3 min before discarding the supernatant and lysing the cells with 100 µL (per sample) of calcineurin extraction buffer. The cell extracts were obtained by centrifuging the lysed cells at 16,000×g for 5 min. Resulting samples were desalted using spin columns (Corning, Tewksbury, MA, USA) containing Profinity™ IMAC uncharged resin (Bio-Rad Laboratories, Hercules, CA, USA). Assay reactions contained 25 µL of 2x assay buffer, 10 µL substrate and 15 µL of sample (or extraction buffer for negative control). The assay plate was incubated at 37°C for 30 min before termination of reactions by addition of 100 µL Malachite green reagent. A620 was measured after 20–30 minutes of colour development in a WALLAC Victor2 plate reader.
MTT reduction and LDH assay
Cell health was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Amresco Inc., Ohio, USA) and the lactate dehydrogenase (LDH) assay (Roche Diagnostics). Cells were grown and treated exactly as in the neurite elongation assay. After treatment of cells as described, an aliquot of cell media was removed, centrifuged to remove any cell debris, and stored for LDH assay. MTT assay was performed by addition of a 10% volume of 5 mg/mL MTT dissolved in PBS pH 7.4 to the existing media, resulting in a final concentration of 0.5 mg/mL. Cells were incubated at 37°C until blue crystals were visible by eye (approximately 30 min). Media was aspirated and replaced with 100 µL of DMSO per well to dissolve crystals. 100 µL aliquots were distributed into wells of a 96 well plate and A570 was measured in a WALLAC Victor2 plate reader. LDH assays were performed by addition of equal volumes (50 µL) of cell media aliquots and LDH reagent to each well of a 96 well plate. Plates were incubated at room temperature until development of a strong red colour. LDH release was measured by A490–520 using a WALLAC Victor2 plate reader. MTT reduction was calculated by subtracting the blank from all the readings and then calculating percentages of the untreated controls (controls had 100% MTT reduction). LDH release was similarly calculated by setting Triton X-treated controls at 100% release and converting the test values to a percentage of the Triton X control.
Statistics
Data presented in graphs represents the mean ± standard error of the mean from analysis of phase contrast photomicroscopic images (n = 6 images from each of 3 separate cultures for each treatment). The investigator was blinded to the treatments. Densitometric analysis of western blots was performed using NIH ImageJ 1.43 software and adjusted according to loading control (GAPDH). Statistical analysis of results was performed using the two tailed student's t-test or ANOVA (one way or two way) with post hoc Bonferroni tests where appropriate. MTT and LDH data represented the mean ± standard error of the mean from the data calculated as a percentage of their respective controls (n = 6 wells for each treatment per experiment).
Results
CuII(gtsm) increased cellular copper levels resulting in enhanced neurite elongation in vitro
NGF-treated PC12 cells were used to examine the effects of CuII(gtsm) on total neurite numbers and neurite elongation (neurogenerative outcomes). Briefly, PC12 cells were treated with NGF for 48 hr and exposed to 50 nM CuII(gtsm), CuII(atsm), (gtsm)H2 (Cu-free ligand) or CuCl2, for 18 hr. Cells were then examined for: mean neurite length; mean number of neurites per cell; percentage of neurites that were at least twice (2x) the cell body width; and mean length of neurites when grouped according to length.
Metal accumulation and cell metabolism in response to treatments was first examined to determine the effect of CuII(gtsm) on metal delivery and cell health. MTT assay results indicated that there was a small but significant loss of MTT reduction with 25 and 50 nM CuII(gtsm) treatment (−17% and −18.5% respectively, with p<0.01 for both) (Figure 1A); however, an LDH assay showed that there was actually a small but significant decrease in LDH release with 100 nM CuII(gtsm) (−6%, p<0.05) (Figure 1B). The reason for the slight differences between MTT and LDH assays is not certain. Changes to LDH assessments were not due to extracellular interactions between Cu and LDH (data not shown). It's possible that CuII(gtsm) is able to induce some inhibition of cellular reductases without inducing cell damage. Whether there are some minor effects of Cu on lactate dehydrogenase export from cells is not known. However, the results demonstrate that no substantial changes to cells were found using these two common measures of cell health. No loss of MTT reduction was observed with the other compounds (CuII(atsm), CuCl2 or (gtsm)H2) at the concentrations tested (Figure 1A). ICP-MS analysis of cells treated with CuII(gtsm) (50 nM) indicated that cell-associated Cu increased significantly by 2.5-fold after 1 hr exposure and 3.1-fold after 18 hr exposure (p<0.001 for both) (Figure 1C).
Figure 1. The effect of CuII(gtsm) on MTT reduction, LDH release and Cu levels.
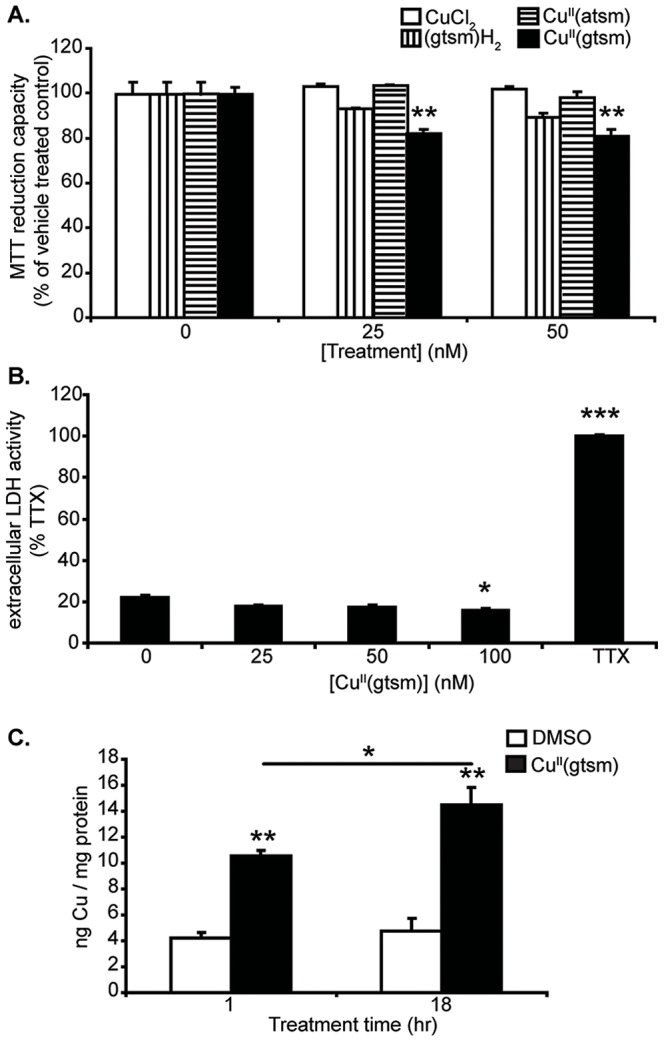
The effects of CuCl2, (gtsm)H2, CuII(atsm) and CuII(gtsm) (25 and 50 nM) on NGF-treated PC12 cells was assessed. (A) The treatments used did not affect MTT reduction with the exception of 25 and 50 nM CuII(gtsm) that inhibited MTT reduction slightly (n = 4/treatment). (B) LDH analysis of CuII(gtsm)-treated cell cultures indicates that concentrations of up to 100 nM can be used with no significant increase in LDH release (n = 4/treatment). (C) ICP-MS results showed that 1 hr exposure to 50 nM CuII(gtsm) increased cellular Cu content significantly and at 18 hr produced a significantly higher increase in Cu content than at 1 hr (n = 6/treatment). Values are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001.
Assessment of the mean number of neurites per cell indicated that 25, 50 and 100 nM CuII(gtsm) reduced neuritogenesis in a dose-dependent manner (−19%, −24% and −43% respectively, with p<0.001 for the three concentrations used) (Figure 2A). In contrast, CuII(gtsm) exposure significantly increased mean neurite length at these concentrations (+21%, +37% and +34% respectively, p<0.0001 for the three concentrations used) (Figure 2B), as well as the percentage of neurites that were 2x or longer than the cell body width (+112% p<0.01, +75% p<0.01 and +63% p<0.05 respectively) (Figure 3A). The ability of CuII(gstm) to induce increased neurite elongation was dependent on the delivery of Cu into cells, as treatment with the ligand alone (gtsm)H2, CuCl2 alone, or CuII(atsm) (which does not readily release Cu under basal cellular conditions [19]), had no effect on neurite elongation at the concentrations tested (Figure 3A). Calculating the combined length of all neurites per cell revealed that CuII(gtsm) had no significant effect on this parameter (data not shown). When neurites were grouped according to length, the number of neurites increased 2.3-fold (p, 0.01) in the size range of 75–99 µm while no change was observed in numbers of neurites in other size ranges (Figure 3B). This indicated that CuII(gtsm) promoted elongation within a particular subset of neurites (based on length). Treatment of cultures with CuII(gtsm) in the absence of NGF did not induce any neurite formation, consistent with the need for NGF to promote neurite outgrowth.
Figure 2. CuII(gtsm) effects on neurogeneration in NGF-treated PC12 cells.
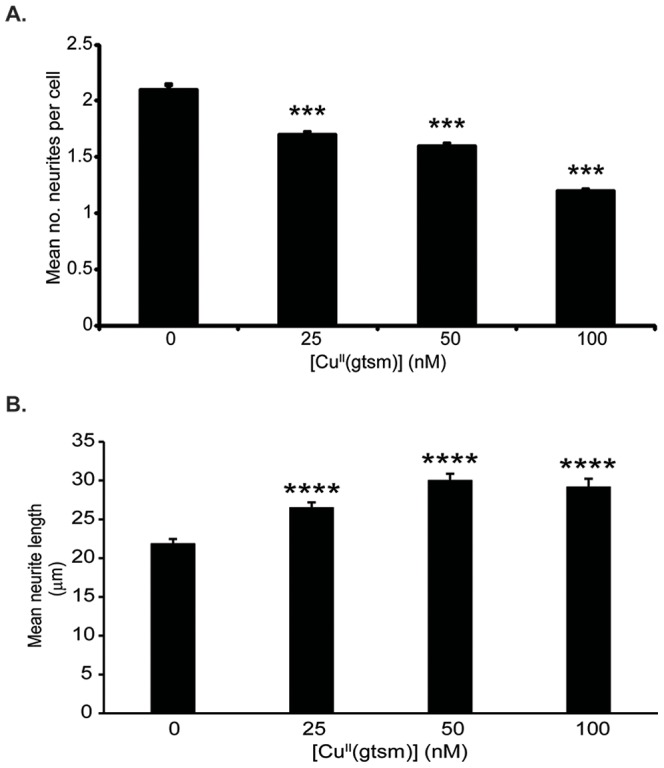
The effect of CuII(gtsm) (25, 50 and 100 nM, 18 hr) on total neurite numbers and neurite elongation was examined. (A) CuII(gtsm) reduced the mean number of neurites per cell in a dose dependent manner (n = 3/treatment). (B) CuII(gtsm) induced an increase in neurite elongation (assessed as mean neurite length) (n = 3/treatment). Values are mean ± SEM. **p<0.01, ***p<0.001.
Figure 3. CuII(gtsm) effects on neurite elongation of NGF-treated PC12 cells.

(A) The effect of CuCl2, (gtsm)H2, CuII(atsm) and CuII(gtsm) (25 and 50 nM, 18 hr) on neurite elongation (assessed as % neurites 2x or more than cell body width). CuII(gtsm) induced a significant increase in neurite elongation (at both concentrations used) whereas the other treatments had no effect (n = 3/treatment). (B) The effect of 50 nM CuII(gtsm) on neurite elongation was examined further by grouping neurites according to length (measured in µm). CuII(gtsm) induced a significant increase in the number of neurites in the 75–99 µm range. Values are mean ± SEM. *p<0.05, **p<0.01.
CuII(atsm) induced neurite elongation at higher concentrations than CuII(gtsm)
To determine how the effects seen with CuII(gtsm) related to Cu delivery, we analysed the ability of CuII(atsm) to modulate neurite elongation. CuII(atsm) is very similar in structure to CuII(gtsm) with only two methyl groups differentiating it from CuII(gtsm) [25]. We (and others) have shown that Cu is not readily released from CuII(atsm) in most cells examined under basal conditions [19], [26]. Limited Cu release from CuII(atsm) was supported here as treatment of PC12 cells with 50 and 500 nM CuII(atsm) induced little change to cell-associated Cu when compared to 50 nM CuII(gtsm) (Figure 4A), indicating that the CuII(atsm) complex was mostly effluxed from the cell as previously reported [24]. When neurite elongation was examined, we found that although 50 nM CuII(atsm) had no effect (Figure 3A), 500 nM CuII(atsm) was able to induce a significant increase in neurite elongation (+48%, p<0.05) similar to that induced by 50 nM CuII(gtsm) (Figure 4B). The ability of 500 nM CuII(atsm) to induce similar neurite elongation to 50 nM CuII(gtsm) but with little effect on Cu levels demonstrated that neurite elongation may not be directly related to Cu release by the Cu-complexes. Alternatively, the effects may be induced by significant alterations to specific sub-cellular pools or organellar localization of Cu that cannot be differentiated by ICP-MS and/or early transient changes to cellular Cu [24], [25], [27].
Figure 4. The effect of CuII(atsm) on Cu levels and neurite elongation.

(A) ICP-MS results showed that 18 hr exposure to 50 nM CuII(atsm) increased cellular Cu content slightly while 500 nM had no effect on cellular Cu levels (n = 6/treatment). (B) The effect of CuII(atsm) (500 nM, 18 hr) on neurite elongation (assessed as % neurites 2x or more than cell body width). CuII(atsm) at 500 nM concentration induced a significant increase in neurite elongation similar to that induced by 50 nM CuII(gtsm) (n = 4/treatment). Values are mean ± SEM. *p<0.05, **p<0.01.
CuII(gtsm) inhibited kinases of the mitogen-activated protein kinase (MAPK) cell signaling pathway
In order to examine the mechanism(s) by which CuII(gtsm) modulated neurite elongation, the phosphorylation state of several key cell signaling kinases known to control neurite elongation was examined [28]–[30]. The basis for selecting these specific kinases was that CuII(gtsm) modulated these kinases both in cultured cells and in brain tissue of treated mice in previous studies [12], [18], [19]. The kinase inhibitors did not significantly alter LDH release compared to CuII(gtsm) treatment alone (Figure S1). Exposure of PC12 cells to CuII(gtsm) (50 nM, 18 hr as for neurite analysis) significantly decreased activation of ERK, JNK and p38 (−87%, −85% and −64% respectively with p<0.05 for all three) but had no effect on glycogen synthase kinase 3β (GSK3β) or protein kinase C (PKC) (Figure 5A and B). Akt was decreased but not significantly. This was surprising as we have previously shown that CuII(gtsm) can induce activation of these kinases, albeit at considerably higher doses [12], [31], although we have also reported that CuII(atsm) can inhibit these kinases in SH-SY5Y cells [16].
Figure 5. The effect of CuII(gtsm) on kinase activation in NGF-treated PC12 cells.

Activation of several cell signaling kinases was examined to determine which pathways might be involved in CuII(gtsm)-mediated neurite elongation. (A&B) CuII(gtsm) treatment decreased activation of ERK, JNK and p38 (n = 3/treatment). Values are mean ± SEM. *p<0.05
A time course analysis was then performed to determine if phosphorylation of ERK or JNK was affected at different time points up to 18 hr. While ERK activation showed a trend towards an increase within the first hour of treatment, this was not significant. ERK phosphorylation was significantly inhibited by CuII(gtsm) after 8 hr (−40% p<0.05) (Figure 6A). Similarly, JNK activation was also inhibited after 8 hr and also 18 hr treatment with CuII(gtsm) (−75% and −45% with p<0.05 for both time points) (Figure 6B). These findings suggest that altered cell signaling kinase activity by CuII(gtsm) (i.e., from altered Cu levels associated with CuII(gtsm) treatment) could be associated with the enhanced neurite elongation.
Figure 6. Timecourse of CuII(gtsm)-mediated ERK and JNK phosphorylation and the effect of specific kinase inhibition on neurite elongation in NGF-treated PC12 cells.
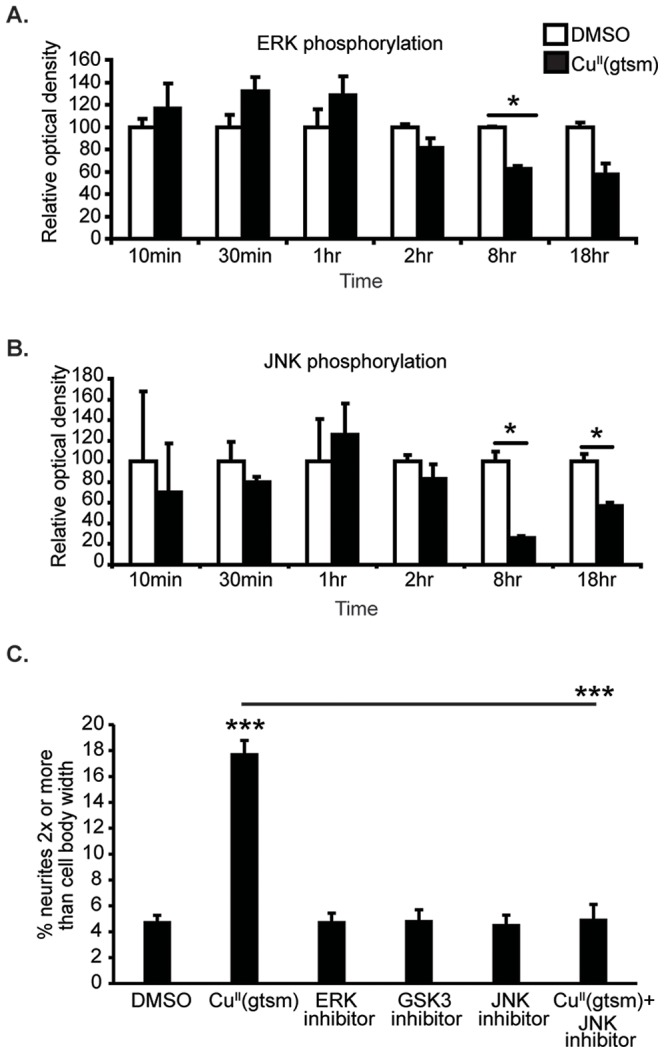
(A & B) CuII(gtsm) (18 hr, 50 nM) decreased ERK and JNK activation from the 8 hr timepoint. (C) Neither JNK, ERK nor GSK3 inhibition had an effect on neurite elongation. Values are mean ± SEM. *p<0.05, ***p<0.001.
To test this, cells were treated with inhibitors of JNK (SP600125) 10 µM and ERK (PD98059) 10 µM for 18 hr to determine if inhibition of these kinases resulted in neurite elongation (these concentrations provide robust inhibition of JNK and ERK [32]). Neither inhibitor induced a significant change to neurite outgrowth, demonstrating that decreased JNK or ERK phosphorylation by CuII(gtsm) was unlikely to be directly related to CuII(gtsm)-mediated neurite elongation (Figure 6C). However to examine this further, we exposed cells to the JNK inhibitor (SP600125) in combination with CuII(gtsm). Interestingly, SP600125 abrogated CuII(gtsm)-mediated neurite outgrowth compared to CuII(gtsm) alone (p<0.001) (Figure 6C), despite both compounds having analogous effects on JNK phosphorylation (inhibition of phosphorylation). The reason for this apparently conflicting result is not known. It is possible that SP600125 may target other kinases that are crucial to CuII(gtsm)-mediated neurite elongation. However, examination of SP600125 action on casein kinase 1 (a known SP600125 target) did not reveal any inhibitory effect (data not shown), suggesting that the concentration used was unlikely to inhibit off-target kinases.
An alternative hypothesis is that CuII(gtsm) may have induced differential effects on JNK phosphorylation within the neurites and this could explain the neurite elongation observed with CuII(gtsm) treatment. This possibility is supported by studies showing that JNK activity is required for neurite outgrowth [33], [34]. Examination of JNK phosphorylation by immunofluorescence was performed to observe localized changes in response to CuII(gtsm). Immunofluorescence analysis demonstrated that neurites of longer length (2x cell body width) contained ‘nodes’ of phosphorylated JNK (Figure 7A). Quantification of the percentage of longer neurites containing these nodes revealed that there was no significant change in response to CuII(gtsm) exposure (Figure 7B). This does not necessarily mean that phosphorylated JNK is not involved in neurite elongation, as basal levels could be required for neurite elongation to occur; however, CuII(gtsm)-mediated elongation did not alter the formation of these phosphorylated JNK nodes.
Figure 7. The localised effect of CuII(gtsm) on JNK phosphorylation (pJNK) in NGF-treated PC12 cells.

CuII(gtsm)-mediated effect on pJNK was examined using immunofluorescence with rabbit anti-phospho-JNK primary antibody (Cat. # 4668, CST) and the percentage of longer neurites that have a pJNK node was quantified. (A) Images of pJNK immunofluorescence (B) The percentage of neurites that are two times or more than cell body width were increased by CuII(gtsm) and the percentage of these longer neurites that have a pJNK node was found to be unchanged (n = 4/treatment). Values are mean ± SEM. **p<0.01.
CuII(gtsm) inhibited cellular phosphatase activity
As we observed no direct relationship between phosphorylation state of JNK or ERK and CuII(gtsm)-mediated neurite elongation, we examined the relationship between phosphatase activity and neurite elongation in CuII(gtsm)-treated cells. Phosphatases are the main effectors of kinase dephosphorylation and since CuII(gtsm) inhibited several kinases we examined whether this was due to an up-regulation of phosphatase activity. As before, NGF-treated PC12 cells were exposed to CuII(gtsm) (50 nM, 18 hr) and then assayed for broad spectrum phosphatase activity. Interestingly, CuII(gtsm)-treated cells showed a 28% decrease in phosphatase activity (p<0.01) (Figure 8A). The phosphatase assay we used contained FDP, which is a highly sensitive fluorogenic substrate for most phosphatases, i.e., alkaline phosphatases, protein tyrosine phosphatases and serine/threonine phosphatases. Calcineurin inhibition by FK506 has previously been shown to enhance NGF-mediated neurite elongation in vitro [35] and improve Aβ-induced spine loss in APP/PS1 mice [36]. Cells were therefore assayed for calcineurin using a specific calcineurin (PP2B) activity assay. CuII(gtsm) treatment was found to inhibit total cellular calcineurin activity by 45% when compared to controls (p<0.05) (Figure 8B). In contrast, no effect of CuII(gtsm) was seen on protein tyrosine phosphatases at 50 nM (data not shown).
Figure 8. Effect of CuII(gtsm) on phosphatase activity and effect on calcineurin.

Following exposure to CuII(gtsm) (50 nM, 18 hr) cells were assayed to determine overall phosphatase activity and also specifically for calcineurin activity. Cells were also exposed to the specific calcineurin inhibitor FK506 at 5 and 10 nM concentrations (18 hr) with and without CuII(gtsm) (50 nM) and neurite elongation was assessed. (A) CuII(gtsm) inhibited total phosphatase activity by 28% (n = 3/treatment). (B) CuII(gtsm) inhibited calcineurin activity by 45% (n = /treatment). (C) CuII(gtsm) and 5 nM FK506 each enhanced neurite elongation but when combined, their effects were blocked. At 10 nM concentration the FK506 effect on neurite elongation was not as strong, and again, when combined with CuII(gtsm) the enhanced elongation was blocked (n = 4/treatment). Values are mean ± SEM. *p<0.05, **p<0.01.
The inhibitory effects of CuII(gtsm) on phosphatase activity were contrary to our initial expectation (due to the decreased phosphorylation of JNK and ERK). Therefore, we examined whether this inhibitory effect on phosphatase activity could be associated with neurite elongation. Since total cellular calcineurin activity was decreased by 45% in CuII(gtsm)-treated cells, it is possible that localized changes to calcineurin e.g. in neurites, were responsible for the altered neurite elongation (it is not feasible to measure this using current calcineurin assays). Therefore, we examined the effect of treating cells with 5 nM calcineurin inhibitor, FK506 [37], [38]. FK506 (5 nM) did not affect cell LDH release when added alone or with CuII(gtsm) (Figure S1B). This concentration of FK506 was found to decrease overall phosphatase activity by 18% and decreased calcineurin activity by 50% (Figure 8A and B). This treatment increased neurite elongation to a level equivalent to 50 nM CuII(gtsm), supporting a potential relationship between phosphatase inhibition and neurite elongation (Figure 8C). We also examined the effect of 5 nM FK506 combined with CuII(gtsm) as well as 10 nM FK506 with and without CuII(gtsm). Alone, 10 nM FK506 enhanced neurite elongation but to lesser extent than the 5 nM treatment. When either 5 or 10 nM FK506 was combined with CuII(gtsm) the effect on neurite elongation was abolished suggesting that there is an optimal level of calcineurin inhibition for this process. We also examined whether lower concentrations of CuII(gtsm) and FK506 resulted in synergistic effects on neurite elongation. However, treatment of cells with 10 nM CuII(gtsm) and 1 nM FK506 had no effect on neurite elongation. The lack of improved elongation with combined CuII(gtsm) and FK506 was unlikely to be due to increased cellular toxicity as no change in LDH activity was observed (Figure S1B). The results suggest that interaction of these agents negates the positive effects of each alone but the mechanism is unknown.
We examined whether other inhibitors of Ser/Thr phosphatases (okadaic acid and phenylarsine oxide) also induced elongation. However, the concentrations that inhibited phosphatase activity to the level induced by CuII(gtsm) treatment were also partially toxic and therefore compromised the neurite elongation assay. Our data indicates that calcineurin inhibition may be involved in CuII(gtsm)-mediated neurogenerative effects.
Discussion
8-hydroxyquinoline-based compounds such as CQ and PBT2 have established the therapeutic potential of metal-modulation for the treatment of AD and have led to testing the effectiveness of other Cu-complexing compounds, such as bis(thiosemicarbazones) [6], [10], [39]. CuII(gtsm) is a compound that enables delivery of Cu across the blood-brain barrier, making it potentially bioavailable to neurons. Treatment of APP/PS1 mice with CuII(gtsm) resulted in cognitive improvements accompanied by decreased soluble oligomeric Aβ levels [12]. However, whether the cognition-enhancing activity of CuII(gtsm) is related to altered Aβ levels or alternative effects induced by modulation of Cu homeostasis is not known. Recent studies demonstrated that PBT2, an 8-hydroxyquinoline metal chelator, reduced loss of apical spine density in Tg2576 mice, and parallel cell culture studies suggested a link between these neuroprotective effects and enhanced neurite elongation, which was dependent on the presence of extracellular metals (Zn and/or Cu) [17]. However, these studies did not clearly establish whether the delivery of metals into cells as preformed metal-complexes is the driving factor in the beneficial outcomes observed in treated neurons.
In the present study we used the stable bis(thiosemicarbazonato) metal complex CuII(gtsm) which allows tighter control over metal delivery. We examined the effects of CuII(gtsm) on neurogeneration-related end points using a PC12 cell culture model of neurite formation and elongation. We determined that while CuII(gtsm) reduced the total number of neurites per cell compared to control cultures, it enhanced neurite elongation in a dose-dependent manner and this effect was dependent on the delivery/bioavailability of Cu. Increased neurite length in vivo can be accompanied by a decrease in the total number of neurites as the cell shifts resources from de novo neurite formation to increased length of major projections [40]. The effects of CuII(gtsm) could be related to interaction of the complex with cell surface receptors. However, we have reported analogous actions using the structurally unrelated 8-hydroxyquinoline compound, PBT2, suggesting that it is more likely the delivery of metal, than complex-receptor interactions, that trigger the effects on neurites, but this remains to be confirmed.
CuII(gtsm) can inhibit the activity of several kinases known to be involved in neural differentiation and plasticity [41], but we found no evidence that this change was related to the effects of CuII(gtsm) on neurite elongation. Despite this, CuII(gtsm) could inhibit the activity of phosphatases with a significant inhibitory effect specifically on calcineurin. Furthermore, inhibition of calcineurin with FK506 mimicked the effect of CuII(gtsm) on neurite elongation potentially implicating a role for calcineurin and/or additional phosphatases in the neurogenerative effects observed.
The NGF-treated PC12 cell line provides a valuable model for investigating neurogenerative effects of potential therapeutic agents for the treatment of AD [42], [43]. However, unlike the in vivo studies involving mouse models of AD treated with CuII(gstm), these experiments were performed on non-transfected cells that did not overexpress the amyloid precursor protein (APP) and therefore Aβ. The fact that CuII(gtsm) induced neurite elongation in these cells (albeit within a narrow length range of neurites) is consistent with a broad neuroprotective action of bis(thiosemicarbazonato) complexes in multiple animal models of neurodegeneration [12], [14], [15], rather than a specific effect on Aβ production. However, it is not clear how such a specific change in neurites (within the 75–99 µm length range) could equate to broad neuroprotection. Instead, these results should be taken as an in vitro marker of neurogenerative activity of the Cu-complex rather than an indicator of its cognition enhancing mechanism.
Neuronal differentiation and maturation in vivo involve axon pruning (reviewed in [44]); however, this occurs mainly during development. In the healthy mature brain, most plasticity events involve changes (retraction and sprouting in different locations) in dendritic spine morphology and are accompanied by synapse elimination and formation respectively [45]. In neurodegenerative diseases such as AD, PD and Huntington's disease there is significant axon degeneration [46]. Whether the neurite elongation-enhancing property of metal-complexes such as CuII(gtsm) could be beneficial is not known.
A relationship between Cu bioavailability and neurite elongation was reported by Birkaya et. al. [47] who found that NGF-mediated differentiation of PC12 cells caused accumulation of Cu, while decreasing cellular Cu (using tetraethylene pentamine) impaired NGF-mediated neurite elongation. Our results are consistent with a role for bioavailable Cu in essential neurogenerative processes. Several studies have reported an inverse relationship between bioavailable Cu levels and cognition (reviewed [48]). Moreover, the Cu transporter ATP7a has a critical role in synaptogenesis as El Meskini et al. demonstrated that ATP7A expression levels are developmentally regulated and shifted from the cell bodies of developing neurons to the extending axons, peaking prior to synaptogenesis or during injury-stimulated neurogenesis [49]. These studies provide strong support for Cu (and ATP7A) as a mediator of neurogenerative pathways. Should bioavailable Cu in specific brain regions be diminished in AD, as reported in a recent meta-analysis [2], compounds such as CuII(gtsm) have the potential to enhance Cu-associated neuronal functions.
The effects of CuII(atsm) were also examined. CuII(atsm) was selected because of its relative resistance to cellular reductants, resulting in decreased Cu release inside the cell. Although 50 nM CuII(atsm) had no effect on neurite elongation, MTT reduction or cellular Cu content, the 500 nM CuII(atsm) dose increased neurite elongation significantly. This did not correlate with apparent cellular Cu levels as they were unchanged at 18 hr. However, this does not take into account either dynamic changes to Cu levels over the preceding 18 hr, as supported by our previous studies [24], [25], [27] or altered sub-cellular Cu pools. Indeed, we have shown previously that uptake by M17 (human neuroblastoma cells) of fluorescently tagged CuII(atsm) resulted in colocalisation with lysosomal and autophagic structures, as well as endoplasmic reticulum [24], [25], [27]. Interestingly, the analogous neurite elongation-enhancing effects of CuII(atsm) and CuII(gtsm) shown here provide support for potentially similar mechanisms of action for these compounds in vivo [14], [15].
MAPK signaling is associated with neurite formation and elongation [50]–[53]. We examined activation of several MAPKs that could potentially be involved in CuII(gtsm)-mediated enhanced cognition. ERK, JNK and p38 were potently inhibited while GSK3 and PKC were unaffected. Akt was moderately changed. However, when ERK or JNK were inhibited by pharmacological inhibitors, there was no change in neurite elongation suggesting that these kinases were not mediating the observed effects.
We found that CuII(gtsm) treatment resulted in diminished phosphatase activity (Figure 8B), and that the calcineurin-specific inhibitor FK506 mimicked the effect of CuII(gtsm) on neurite elongation (Figure 8C). This suggested calcineurin could be involved in the observed neurogenerative effects of CuII(gtsm). Calcineurin regulates neuronal plasticity [54], [55] and its inhibition improves synaptic function [56]. Furthermore, calcineurin is a therapeutic target for AD as its inhibition can ameliorate dendritic spine density deficits in a mouse model of AD [36]. The cellular mechanism of action of PBT2 possibly involved inhibition of calcineurin in a metal-dependent manner [9]. It is possible that there may be localized changes in calcineurin activity that are critical for neurite elongation. However, this would be difficult to determine as the calcineurin assays require large numbers of cells to measure activity and this technique could not be feasibly applied to neurites alone. Alternatively, calcineurin acts in concert with other phosphatases to modulate neurite elongation as supported by the 28% inhibition of broad spectrum phosphatases by CuII(gtsm) in our assays (Figure 8A).
Whether the neurogenerative effects of CuII(gtsm) are related specifically to Cu, Zn or both metals is difficult to decipher. An increase in cellular Cu may in fact result in increased Zn bioavailability via displacement of Zn ions that are bound to metallothionein (MT) as MT has a higher affinity for Cu than Zn [57]. This would explain why increasing Cu, by CuII(gtsm) treatment or Zn by PBT2-Zn treatment, has the same inhibitory effect on calcineurin. Although a decrease in phosphatase activity should result in an increase in phosphorylation of various kinases, we did not see increased phosphorylation of ERK, JNK or GSK3. This suggests that the altered phosphatase activity may be compartmentalized and only affect a subset of kinases in CuII(gtsm)-treated cells.
In summary, our study suggests that the Cu-delivery compounds, CuII(gtsm) and CuII(atsm) have the potential to enhance neurogenerative processes by increased metal bioavailability. This protective action could account for some of the neurotherapeutic effects of these complexes in animal models of neurodegeneration. Regardless, the results provide a valuable in vitro measure of neurogenic activity of this class of compound [14], [15]. Inhibition of calcineurin, and additional phosphatases, may be involved; however, the sub-cellular localization of this effect and the modulation of downstream kinases needs to be determined. Our studies demonstrated that low, physiological, concentrations of a Cu-complex (25–50 nM) can be beneficial and promote neurogenerative outcomes in neuron-related cells. This study provides valuable support for the therapeutic potential of CuII(gtsm) and PBT2 in the treatment of AD and other disorders where metal dyshomeostasis is involved.
Supporting Information
The effect of kinase inhibitors and calcineurin inhibitor on LDH release. The effects of (ERK inhibitor), SP600125 (JNK inhibitor), GSK3 inhibitor VII and calcineurin inhibitor (FK506) on NGF-treated PC12 cells was assessed. (A) LDH analysis of cell cultures treated with ERK, JNK or GSK3 inhibitors indicates that the 10 µM concentration can be used with no significant increase in LDH release (n = 5/treatment). (B) LDH analysis of cell cultures treated with 1, 5 or 10 nM FK506 demonstrated that these treatments had no effect on LDH release (n = 5/treatment). Values are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001.
(TIF)
Funding Statement
This work was supported by grants from the ANZ Trustees Wicking Foundation and Mason Trust, the Brain Foundation of Australia, Australian Research Council (ARC) and National Health and Medical Research Council of Australia (NHMRC). ARW is a recipient of an ARC Future Fellowship. RC is an NHMRC Senior Research Fellow. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Barnham KJ, Bush AI (2008) Metals in Alzheimer's and Parkinson's diseases. Curr Opin Chem Biol 12: 222–228. [DOI] [PubMed] [Google Scholar]
- 2. Schrag M, Mueller C, Oyoyo U, Smith MA, Kirsch WM (2011) Iron, zinc and copper in the Alzheimer's disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog Neurobiol 94: 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tamano H, Takeda A (2011) Dynamic action of neurometals at the synapse. Metallomics 3: 656–661. [DOI] [PubMed] [Google Scholar]
- 4.Gaier ED, Eipper BA, Mains RE (2012) Copper signaling in the mammalian nervous system: Synaptic effects. J Neurosci Res. [DOI] [PMC free article] [PubMed]
- 5. Kessler H, Pajonk FG, Supprian T, Falkai P, Multhaup G, et al. (2005) [The role of copper in the pathophysiology of Alzheimer's disease]. Nervenarzt 76: 581–585. [DOI] [PubMed] [Google Scholar]
- 6. Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, et al. (2010) PBT2 rapidly improves cognition in Alzheimer's Disease: additional phase II analyses. J Alzheimers Dis 20: 509–516. [DOI] [PubMed] [Google Scholar]
- 7. Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, et al. (2008) Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol 7: 779–786. [DOI] [PubMed] [Google Scholar]
- 8. White AR, Du T, Laughton KM, Volitakis I, Sharples RA, et al. (2006) Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem 281: 17670–17680. [DOI] [PubMed] [Google Scholar]
- 9. Crouch PJ, Savva MS, Hung LW, Donnelly PS, Mot AI, et al. (2011) The Alzheimer's therapeutic PBT2 promotes amyloid-beta degradation and GSK3 phosphorylation via a metal chaperone activity. J Neurochem 119: 220–230. [DOI] [PubMed] [Google Scholar]
- 10. Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, et al. (2008) Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59: 43–55. [DOI] [PubMed] [Google Scholar]
- 11. Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, et al. (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30: 665–676. [DOI] [PubMed] [Google Scholar]
- 12. Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, et al. (2009) Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc Natl Acad Sci U S A 106: 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malm TM, Iivonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, et al. (2007) Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J Neurosci 27: 3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hung LW, Villemagne VL, Cheng L, Sherratt NA, Ayton S, et al. (2012) The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson's disease. J Exp Med 209: 837–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soon CP, Donnelly PS, Turner BJ, Hung LW, Crouch PJ, et al. (2011) Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J Biol Chem 286: 44035–44044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parker SJ, Meyerowitz J, James JL, Liddell JR, Nonaka T, et al. (2012) Inhibition of TDP-43 accumulation by bis(thiosemicarbazonato)-copper complexes. PLoS One 7: e42277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adlard PA, Bica L, White AR, Nurjono M, Filiz G, et al. (2011) Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer's disease. PLoS One 6: e17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, et al. (2008) Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J Biol Chem 283: 4568–4577. [DOI] [PubMed] [Google Scholar]
- 19. Donnelly PS, Liddell JR, Lim S, Paterson BM, Cater MA, et al. (2012) An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copperII. Proc Natl Acad Sci U S A 109: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beraldo H, Boyd L, West D (1997) Copper(II) and nickel(II) complexes of glyoxaldehyde bis{N(3)-substituted thiosemicarbazones}. Transition Metal Chemistry 23: 67–71. [Google Scholar]
- 21. Bottomley G, Clark I, Creaser I, Engelhardt L, Geue R, et al. (1994) The Synthesis and Structure of Encapsulating Ligands: Properties of Bicyclic Hexamines. Australian Journal of Chemistry 47: 143–179. [Google Scholar]
- 22. Gingras BA, Suprunchuk T, Bayley CH (1962) The preparation of some thiosemicarbazones and their copper complexes: part iii. Canadian Journal of Chemistry 40: 1053–1059. [Google Scholar]
- 23. Blower PJ, Castle TC, Cowley AR, Dilworth JR, Donnelly PS, et al. (2003) Structural trends in copper(ii) bis(thiosemicarbazone) radiopharmaceuticals. Dalton Transactions 0: 4416–4425. [Google Scholar]
- 24. Price KA, Crouch PJ, Volitakis I, Paterson BM, Lim S, et al. (2011) Mechanisms controlling the cellular accumulation of copper bis(thiosemicarbazonato) complexes. Inorg Chem 50: 9594–9605. [DOI] [PubMed] [Google Scholar]
- 25. Price KA, Crouch PJ, Lim S, Paterson BM, Liddell JR, et al. (2011) Subcellular localization of a fluorescent derivative of CuII(atsm) offers insight into the neuroprotective action of CuII(atsm). Metallomics 3: 1280–1290. [DOI] [PubMed] [Google Scholar]
- 26. Yoshii Y, Furukawa T, Kiyono Y, Watanabe R, Mori T, et al. (2011) Internal radiotherapy with copper-64-diacetyl-bis (N4-methylthiosemicarbazone) reduces CD133+ highly tumorigenic cells and metastatic ability of mouse colon carcinoma. Nucl Med Biol 38: 151–157. [DOI] [PubMed] [Google Scholar]
- 27. Price KA (2012) The challenges of using a copper fluorescent sensor (CS1) to track intracellular distributions of copper in neuronal and glial cells. Chemical Science 3: 2748–2759. [Google Scholar]
- 28. Cowley S, Paterson H, Kemp P, Marshall CJ (1994) Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77: 841–852. [DOI] [PubMed] [Google Scholar]
- 29. Pang L, Sawada T, Decker SJ, Saltiel AR (1995) Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem 270: 13585–13588. [DOI] [PubMed] [Google Scholar]
- 30. Morooka T, Nishida E (1998) Requirement of p38 mitogen-activated protein kinase for neuronal differentiation in PC12 cells. J Biol Chem 273: 24285–24288. [DOI] [PubMed] [Google Scholar]
- 31. Bica L, Meyerowitz J, Parker SJ, Caragounis A, Du T, et al. (2011) Cell cycle arrest in cultured neuroblastoma cells exposed to a bis(thiosemicarbazonato) metal complex. Biometals 24: 117–133. [DOI] [PubMed] [Google Scholar]
- 32. Meyerowitz J, Parker SJ, Vella LJ, Ng D, Price KA, et al. (2011) C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol Neurodegener 6: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waetzig V, Herdegen T (2003) The concerted signaling of ERK1/2 and JNKs is essential for PC12 cell neuritogenesis and converges at the level of target proteins. Mol Cell Neurosci 24: 238–249. [DOI] [PubMed] [Google Scholar]
- 34. Eom DS, Choi WS, Oh YJ (2004) Bcl-2 enhances neurite extension via activation of c-Jun N-terminal kinase. Biochem Biophys Res Commun 314: 377–381. [DOI] [PubMed] [Google Scholar]
- 35. Lyons WE, George EB, Dawson TM, Steiner JP, Snyder SH (1994) Immunosuppressant FK506 promotes neurite outgrowth in cultures of PC12 cells and sensory ganglia. Proc Natl Acad Sci U S A 91: 3191–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rozkalne A, Hyman BT, Spires-Jones TL (2011) Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol Dis 41: 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu J, Farmer JD Jr, Lane WS, Friedman J, Weissman I, et al. (1991) Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66: 807–815. [DOI] [PubMed] [Google Scholar]
- 38. Fruman DA, Klee CB, Bierer BE, Burakoff SJ (1992) Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A. 89: 3686–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Duncan C, White AR (2012) Copper complexes as therapeutic agents. Metallomics 4: 127–138. [DOI] [PubMed] [Google Scholar]
- 40. Kiddie G, McLean D, Van Ooyen A, Graham B (2005) Biologically plausible models of neurite outgrowth. Prog Brain Res 147: 67–80. [DOI] [PubMed] [Google Scholar]
- 41. Xiao J, Liu Y (2003) Differential roles of ERK and JNK in early and late stages of neuritogenesis: a study in a novel PC12 model system. J Neurochem 86: 1516–1523. [DOI] [PubMed] [Google Scholar]
- 42. Jeon CY, Jin JK, Koh YH, Chun W, Choi IG, et al. (2010) Neurites from PC12 cells are connected to each other by synapse-like structures. Synapse 64: 765–772. [DOI] [PubMed] [Google Scholar]
- 43. Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73: 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Low LK, Cheng HJ (2006) Axon pruning: an essential step underlying the developmental plasticity of neuronal connections. Philos Trans R Soc Lond B Biol Sci 361: 1531–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, et al. (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420: 788–794. [DOI] [PubMed] [Google Scholar]
- 46. Coleman MP, Perry VH (2002) Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 25: 532–537. [DOI] [PubMed] [Google Scholar]
- 47. Birkaya B, Aletta JM (2005) NGF promotes copper accumulation required for optimum neurite outgrowth and protein methylation. J Neurobiol 63: 49–61. [DOI] [PubMed] [Google Scholar]
- 48. Kaden D, Bush AI, Danzeisen R, Bayer TA, Multhaup G (2011) Disturbed copper bioavailability in Alzheimer's disease. Int J Alzheimers Dis 2011: 345614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. El Meskini R, Cline LB, Eipper BA, Ronnett GV (2005) The developmentally regulated expression of Menkes protein ATP7A suggests a role in axon extension and synaptogenesis. Dev Neurosci 27: 333–348. [DOI] [PubMed] [Google Scholar]
- 50. Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030–1038. [DOI] [PubMed] [Google Scholar]
- 51. Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1–10. [DOI] [PubMed] [Google Scholar]
- 52. Sweatt JD (2004) Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14: 311–317. [DOI] [PubMed] [Google Scholar]
- 53. Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5: 173–183. [DOI] [PubMed] [Google Scholar]
- 54. Groth RD, Dunbar RL, Mermelstein PG (2003) Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun 311: 1159–1171. [DOI] [PubMed] [Google Scholar]
- 55. Jurado S, Biou V, Malenka RC (2010) A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci 13: 1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jouvenceau A, Dutar P (2006) A role for the protein phosphatase 2B in altered hippocampal synaptic plasticity in the aged rat. J Physiol Paris 99: 154–161. [DOI] [PubMed] [Google Scholar]
- 57. Hidalgo J, Aschner M, Zatta P, Vasak M (2001) Roles of the metallothionein family of proteins in the central nervous system. Brain Res Bull 55: 133–145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effect of kinase inhibitors and calcineurin inhibitor on LDH release. The effects of (ERK inhibitor), SP600125 (JNK inhibitor), GSK3 inhibitor VII and calcineurin inhibitor (FK506) on NGF-treated PC12 cells was assessed. (A) LDH analysis of cell cultures treated with ERK, JNK or GSK3 inhibitors indicates that the 10 µM concentration can be used with no significant increase in LDH release (n = 5/treatment). (B) LDH analysis of cell cultures treated with 1, 5 or 10 nM FK506 demonstrated that these treatments had no effect on LDH release (n = 5/treatment). Values are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001.
(TIF)