

Abstract
Cationic, amphipathic host defense peptides represent a promising group of agents to be developed for anticancer applications. Poly-N-substituted glycines, or peptoids, are a class of biostable, peptidomimetic scaffold that can display a great diversity of side chains in highly tunable sequences via facile solid-phase synthesis. Herein, we present a library of anti-proliferative peptoids that mimics the cationic, amphipathic structural feature of the host defense peptides and explore the relationships between the structure, anticancer activity and selectivity of these peptoids. Several peptoids are found to be potent against a broad range of cancer cell lines at low-micromolar concentrations including cancer cells with multidrug resistance (MDR), causing cytotoxicity in a concentration-dependent manner. They can penetrate into cells, but their cytotoxicity primarily involves plasma membrane perturbations. Furthermore, peptoid 1, the most potent peptoid synthesized, significantly inhibited tumor growth in a human breast cancer xenotransplantation model without any noticeable acute adverse effects in mice. Taken together, our work provided important structural information for designing host defense peptides or their mimics for anticancer applications. Several cationic, amphipathic peptoids are very attractive for further development due to their high solubility, stability against protease degradation, their broad, potent cytotoxicity against cancer cells and their ability to overcome multidrug resistance.
Introduction
Chemotherapy remains a must-have treatment for many cancer patients, especially in those with advanced diseases [1]. Classic chemotherapeutical agents typically target the rapid proliferation of tumor cells by interrupting the synthesis or the functions of DNA, RNA or proteins. The lack of specificity of these drugs usually leads to many severe side effects. Moreover, it is very common for patients to develop multidrug resistance (MDR) through the efflux of drugs from cancer cells and become unresponsive to multiple chemotherapeutics [2]. In order to overcome drug resistance and provide more effective treatment, new molecular platforms with anti-proliferative activity employing a novel mechanism of action have been actively investigated.
Antimicrobial peptides, also known as host-defense peptides, represent a class of naturally occurring compounds that have recently been explored for their anticancer activity [3], [4], [5], [6], [7]. Natural products have been playing an important role in developing chemotherapeutics with a substantial amount of anticancer agents in use being either natural or derived from natural products from various sources [8]. Antimicrobial peptides are evolutionarily ancient weapons found throughout the animal and plant kingdoms [9]. A hallmark of this class is that the molecule can adopt a structure in which clusters of cationic and hydrophobic residues are spatially organized in discrete sectors, and this cationic, amphipathic structural feature is critical for their activity and selectivity [10]. Most host-defense peptides are believed to be membranolytic, with cationic residues selecting for anionic cellular membranes via electrostatic interactions and hydrophobic regions responsible for membrane permeation and disruption [11]. Magainin 2 and its analogs were first found in 1993 to display selective cytoxicity towards carcinoma cells in vitro and were proven to be as effective as doxorubicin in vivo via intraperitoneal delivery in ovarian cancer mouse models [12]. Over the last two decades, a growing number of studies have shown that some cationic, amphipathic peptides, including both natural host defense peptides and synthetic antimicrobial peptides, exhibit a broad spectrum of cytotoxic activity against cancer cells and are effective in reducing tumor burdens in several cancer animal models [3], [4], [5], [6], [7]. The selectivity of these peptides towards cancer cells is not well understood and is hypothesized to result from some altered membrane properties of cancer cells compared to normal tissue cells, e.g., more negative charges on outer membrane leaflets, more microvilli, higher transmembrane potentials, or higher membrane fluidity [3], [4], [5], [6], [7].
This class of cationic, amphipathic peptides possesses many features ideal for anticancer applications, including 1) high water solubility, 2) broad, potent cytotoxicity against cancer cells, and 3) the ability to overcome multidrug resistance developed in cancer cells [12], [13], [14]. However, the clinical use of peptide-based drugs has been limited due to their rapid degradation and clearance in vivo. Non-natural peptidomimetics can circumvent this proteolytic sensitivity while retaining the beneficial features of peptides [15]. A group of stable diastereomeric lytic peptides (containing both D- and L-forms of lysines and leucines) was developed based on antimicrobial model amphipathic peptides and investigated for anticancer applications [16], [17], [18]. Poly-N-substituted glycines, or peptoids, comprise another class of protease-resistant peptidomimetics, with side chains attached to the backbone nitrogen rather than to the α-carbon as in peptides [19], [20]. Peptoids can be readily synthesized on solid phase in a sequence-specific manner on an automated peptide synthesizer. Virtually any desired chemical functionality available as a primary amine can be incorporated into peptoids via a submonomer method, which provides great chemical “design diversity” in peptoid sequences [20].
Peptoids provide an ideal molecular platform to mimic the cationic, amphipathic structural feature of host-defense peptides. Though lacking backbone chirality and intrachain hydrogen bonding, peptoids can readily form helical structures with a periodic incorporation of bulky, α-chiral side chains, giving rise to polyproline type-I-like helices with approximately three residues per turn and a helical pitch of 6.0 – 6.7 Å [20], [21], [22]. This threefold periodicity of peptoids allows the cationic, amphipathic structure to be easily recapitulated in three-faced helices, simply using peptoids comprising trimer repeats, (X-Y-Z)n, which will display X, Y, and Z residues on separate faces. Accordingly, our group developed antimicrobial peptoids by mimicking magainin-2 [23]. A library of antimicrobial peptoids that typically adopt cationic, amphipathic helical structures was synthesized and studied for the relationships between their structures and their antimicrobial activity and selectivity [24], [25].
Here, inspired by anticancer peptides, we developed a new library of cationic, amphipathic peptoids and screened their anticancer activity and selectivity in vitro. We demonstrated for the first time that cationic, amphipathic peptoids can exhibit potent, fast cytotoxicity at low micromolar concentrations to a broad range of human cancer cell lines, and some peptoids were developed to show modest in vitro selectivity towards cancer cells. Moreover, actions of these peptoids were not influenced by multidrug resistance, killing primarily via plasma membrane disruptions. Finally, in vivo efficacy of the most potent peptoid derivative was validated in a preliminary study using a breast cancer xenotransplantation model established with human patient tumor cells.
Materials and Methods
Peptoid synthesis and purification
Peptoids were synthesized using an ABI 433A peptide synthesizer (Applied Biosystems, Inc.) on Rink amide MBHA resin (EMD Biosciences, Gibbstown, NJ) using the submonomer protocol [20], [24]. Briefly, the amine on the nascent chain is bromoacetylated or chloroacetylated followed by SN2 displacement of bromide or chloride by a primary amine to form the side chain. Resin-bound peptoids were then exposed to a mixture of trifluoroacetic acid (TFA): triisopropylsilane: water (95∶2.5∶2.5, volume ratio) for 10 minutes to cleave peptoids from the resin. Crude peptoids were purified by reversed-phase high performance liquid chromatography (RP-HPLC) (Waters Corporation) using a C18 column and a linear acetonitrile/water gradient. A final purity >95% as measured by analytical RP-HPLC (Waters Corporation) was achieved, and the identity of each peptoid was confirmed using electrospray ionization mass spectrometry (ESI/MS). Pexiganan was synthesized by standard Fmoc chemistry on an ABI 433A peptide synthesizer (EMD Biosciences). Unless indicated otherwise, all reagents were purchased from Sigma Aldrich (St. Louis, MO). Among the submonomers used, Nspe was derived from (S)-N-(1-phenylethyl)amine; Npm from benzylamine (1-phenylethylamine); NHis from histamine (2-[4-imidazolyl]ethylamine); NLeu from isobutylamine; NLys from N-tert-butoxycarbonyl-1,4-butanediamine (CNH Technologies, MA). Guanidinylation of NLys was carried out according to the reported procedure [26]. When NHis was used in the peptoid sequence, chloroacetic acid was used [27]. 5(6)-Carboxyfluorescein was used to label the N-terminus of peptoid 1 [28].
Cell cultures
MRC-5 were purchased from American Type Culture Collection (ATCC) and were grown in media suggested by ATCC supplemented with 10% FBS (Hyclone, US sources) and antibiotics (Sigma). MCF-7, MCF-7/TxT50 and OVCAR-3 (available in ATCC) were kindly provided by Professor Branimir Sikic's lab at Stanford University [35], and were cultured in McCoy's 5A media (GIBCO) with 10% FBS and antibiotics. Primary dermal fibroblasts were gifts from Lifeline Cell Technology and were cultured <6 passages according to the protocol Lifeline provides. All the cells were cultured in cell incubators (Thermo Scientific) at 37°C with 5% CO2.
The MTS assay
This assay is a colorimetric method for determining the number of viable cells in proliferation. Aliquots of 100 µl media containing 1×104 cells were distributed into each well of a 96-well plate (BD Falcon). The following day, when cell density reaches about ∼40% confluency, the cell media were removed and replaced with serial dilutions of peptoid stocks in culturing media. For peptoid dilutions, peptoid stocks were initially diluted in media at 100 µM and then diluted by half in series using a multichannel pipette, and maintained in 100 µl media for each concentration with triple repeats. After peptoid solutions were transferred onto cells in 96-well plates, cells were incubated at 37°C for certain time periods. Then 20 µl of the CellTiter 96 Aqueous Non-Radioactive cell proliferation assay (Promega) reagent which contains a tetrazolium compound, [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS(a)], was added to each well and cells were further incubated for 2 h to metabolize. The absorbance of formazan products were measured at 490 nm in a microplate reader (Molecular Devices). Percentage of cell viability = (A -A testblank)/(A control -A blank)×100, where A is the absorbance of the test well and A control the average absorbance of wells with cells not treated with peptoids. A testblank (media, MTS, and diluted peptoids) and A blank (media and MTS) were background absorbances measured in the absence of cells.
The Guava Viacount assay
This assay measures cell viability by using two DNA-binding dyes, one membrane-permeant, the other membrane-impermeant. Briefly, cells were plated and peptoids were diluted as described in MTS assays. After peptoid treatments, media supernatant with floating cells were collected. Cells were washed once, trypsinized and neutralized with previously collected media supernatant with floating cells. Cells were centrifuged at 1800 rpm for 3 min, and cell pellets were resuspended in 180 µl of fresh media. 20 µl of Guava Viacount reagents (Millipore) was added to each cell suspension, gently mixed and incubated at room temperature (protected from light) for 5 min before being submitted to the Guava Easycyte Plus flow cytometry system (Millipore) to measure the cellular fluorescent signals stained with dyes in Viacount reagents and to quantify cell viability.
The LDH assay
This assay is used to measure the membrane integrity as a function of the amount of cytoplasmic LDH (lactic dehydrogenase) released into the medium. Experiments were carried out according to the protocol of the In Vitro Toxicology Assay Kit, Lactic Dehydrogenase (LDH) based (Sigma-Aldrich). Briefly, cells were plated as described before, and peptoids were diluted similarly but in culturing media without phenol red to reduce background signal. After peptoid treatments, media supernatant were collected and centrifuged to remove any cell debris, and analyzed for LDH activity in a 96-well plate using the kit, absorbance at 490 nm and 690 nm measured using a microplate reader. All the following absorbance difference = A 490nm –A 690nm. Percentage of LDH leakage = (A -A testblank)/(A lysis -A blank)×100, where A is the average absorbance difference of the test wells and A lysis the average absorbance difference of wells with cells treated with cell lysis solution provided by the kit for 45 min. A testblank (media, diluted peptoids and LDH measuring reagents) and A blank (media and LDH measuring reagents) were background absorbance differences measured in the absence of cells.
Confocal imaging
MCF-7 cells seeded in 35 mm glass-bottom dishes (MatTek Corporation) were treated with 8 µM carboxyfluorescein-peptoid1 (CF-peptoid 1) diluted in culturing media with 10% FBS for 1 h. Supernatants were removed, and cells were washed with PBS three times before being imaged via a Leica SP2 AOBS confocal laser scanning microscope.
Peptoid evaluation in a human breast cancer xenograft mice model
80,000 cells from a dissociated second generation metastatic breast cancer tumor were suspended in Medium 199 containing 25% Matrix Matrigel (Becton Dickinson 354248) and injected into the mammary fat pad of 4–8 week old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. After two weeks, 100 µl PBS containing 110 µM peptoid 1 or control peptide was injected into the mammary fat pad three times a week, roughly at a dose of 1 mg/kg. Tumor volumes were determined using direct caliper measurements following twelve weeks of treatment.
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by Stanford University Institutional Animal Care and Use Committee (IACUC) (Protocol Number: 10725).
Results
Peptoid Design and in vitro Screening
Peptoids were utilized herein as a peptidomimetic scaffold to capture the cationic, amphipathic nature of anticancer peptides, as well as to improve molecular stability and to increase chemical diversity. The design of anticancer peptoids were derived from previous antimicrobial peptoids and were further optimized hererin to improve the activity and selectivity of peptoids against anionic membranes [23], [24], [25]. Peptoid 1, [H-(NLys-Nspe-Nspe)4-NH2], composed of NLys (the peptoid analog of Lys) and Nspe (Figure 1), is the most active antimicrobial dodecamer we had previously developed yet it had some minor hemolytic effects at high concentrations (Figure 1A). Several novel variants were designed based on peptoid 1 but with different charges, amphipathicity, length, and helicity to change their activity or selectivity. The sequences of these variants, the solvent composition at RP-HPLC elution as a relative measure of molecular hydrophobicity, molecular net charges and their charge-to-length ratio are summarized in Table 1. Variants marked with an asterisk were previously reported to possess antimicrobial activity [24], [25]. Pexiganan, a clinically-relevant peptide analog of magainin 2 which was developed for topical treatment for diabetic foot infection [29], and LL-37, the only known human cathelicidin which is a non-selective antimicrobial peptide [30], were also tested for comparison.
Figure 1. The cationic, amphipathic structure of peptoids and monomers.
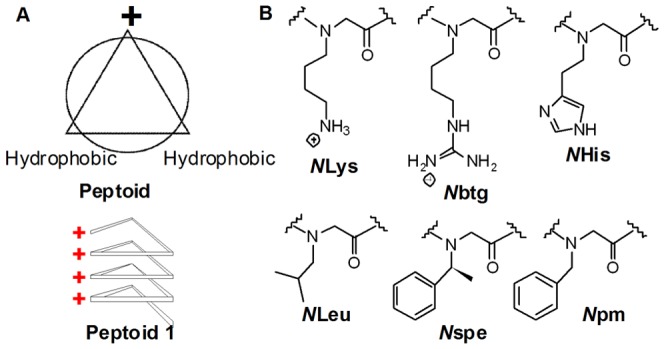
A, top-view of the cationic, amphipathic structure of peptoids (upper), and side-view of the helical structure of peptoid 1 (bottom); B, peptoid monomer side-chain structures with shorthand names.
Table 1. Sequences and molecular properties of peptoids and comparator peptides.
| Varient Category | Compound | Sequence | MW (Da) | HPLC elution | Net charge | CTLR |
| Basis | Peptoid 1 * | H-(NLys-Nspe-Nspe)4-NH2 | 1819 | 65.1 | 4 | 0.33 |
| Negative control | 1 11mer-NLeu | H-(NLys-NLeu-NLeu)3-NLys-NLeu-NH2 | 1321 | 51 | 4 | 0.36 |
| Cationic charge | 1-NLys5,11 * | H-(NLys-Nspe-Nspe-NLys-NLys-Nspe)2-NH2 | 1753 | 51.2 | 6 | 0.5 |
| 1-Nbtg | H-(Nbtg-Nspe-Nspe)4-NH2 | 1987 | 65 | 4 | 0.33 | |
| 1- NHis1,7 | H-(NHis-Nspe-Nspe-NLys-Nspe-Nspe)2-NH2 | 1767 | 62.5 | 2∼4** | 0.17∼0.33 | |
| Length | 1 13mer * | H-(NLys-Nspe-Nspe)4-NLys-NH2 | 1948 | 62.8 | 5 | 0.38 |
| 1 11mer * | H-(NLys-Nspe-Nspe)3-NLys-Nspe-NH2 | 1658 | 63.5 | 4 | 0.36 | |
| 1 9mer * | H-(NLys-Nspe-Nspe)3-NH2 | 1368 | 60 | 3 | 0.33 | |
| Achiral | 1 achiral * | H-(NLys-Npm-Npm)4-NH2 | 1701 | 59.8 | 4 | 0.33 |
| 1 achiral-Nspe2 * | H-NLys-Nspe-Npm-(NLys-Npm-Npm)3-NH2 | 1721 | 60.8 | 4 | 0.33 | |
| 1 achiral-Nspe2, 12 | H-NLys-Nspe-Npm-(NLys-Npm-Npm)2-NLys-Npm-Nspe-NH2 | 1735 | 62 | 4 | 0.33 | |
| 1-Npm2,3,8,9 * | H-(NLys-Npm-Npm-NLys-Nspe-Nspe)2-NH2 | 1763 | 63.3 | 4 | 0.33 | |
| Peptides | Pexiganan | GIGKFLKKAKKFGKAFVKILKK-NH2 | 2477 | 50.2 | 9 | 0.41 |
| LL 37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 4495 | 66 | 6 | 0.16 |
See Figure 1 for the structures of the peptoid monomers indicated in each sequence. HPLC elution is reported as the percentage of acetonitrile in water (% ACN) in the water/acetonitrile (0.1% trifluoroacetic acid) solvent in analytic HPLC. A linear water/acetonitrile (0.1% trifluoroacetic acid) gradient of 5%-95% acetonitrile over 30 min was run on a C18 column. Net charge indicates molecular charges at neutral pH. (“**”, NHis has 10% probability to be charged around neutral pH, so the net charge of 1- NHis1,7 is between +2 to +4). CTLR stands for charge-to-length ratio, which is defined as the ratio of the total number of cationic residues to the total number of residues in each sequence. Peptoids labeled with asterisk have been reported previously to possess antimicrobial activities [23], [24], [25].
The activity and selectivity of these peptoids against cancer cells were evaluated in vitro. Their cytotoxicity was tested in three cancer cell lines, MCF-7 (human breast cancer), LNCaP (human prostate cancer), and OVCAR-3 (human ovarian cancer). To estimate the selectivity of these peptoids in vitro, hemolytic activities of some peptoids were cited from previously published work (Table 2 **column), and the MRC-5 cell line (human fibroblasts derived from fetal lung tissues) and primary dermal fibroblasts were also tested as normal cell controls. Briefly, cells were treated with peptoids diluted in culturing media for certain time periods, and then cell viability was measured via MTS assays. These peptoids as well as pexiganan and LL-37 were found to be more potent in media with low serum concentrations (data not shown), but results reported here are all tested in media supplemented with 10% FBS.
Table 2. Peptoid Cytotoxicity in cancer cell lines and control cells.
| Variant Category | Compounds | LC50 (µM) | HC10 (µM) ** | ||||
| MCF-7 (breast cancer) | LNCaP (prostate cancer) | OVCAR3 (ovarian cancer) | MRC-5 (fetal lung fibroblast) | Primary dermal fibroblast | |||
| Basis | Peptoid 1 * | 5 | 5 | 6 | 8 | 8 | 21 |
| Negative control | 1 11mer-NLeu | >100 | >100 | >100 | >100 | >100 | — |
| Cationic charge | 1-NLys5,11 * | 31 | 25 | 37 | 41 | 34 | >100 |
| 1-Nbtg | 5 | 8 | 8 | 10 | 8 | — | |
| 1- NHis1,7 | 19 | 23 | 30 | 30 | 30 | — | |
| 1 13mer * | 8 | 8 | 9 | 10 | 14 | 21 | |
| Length | 1 11mer * | 11 | 10 | 10 | 24 | 19 | 103 |
| 1 9mer * | 30 | 27 | 34 | 40 | 32 | 150 | |
| 1 achiral * | 13 | 11 | 16 | 30 | 39 | 183 | |
| Achiral | 1 achiral-Nspe2 * | 13 | 11 | 16 | 22 | 36 | 160 |
| 1 achiral-Nspe2, 12 | 12 | 10 | 16 | 21 | 32 | — | |
| 1-Npm2,3,8,9 * | 9 | 10 | 11 | 18 | 21 | 80 | |
| Peptides | Pexiganan | 8 | 6 | 13 | 21 | 19 | 70 |
| LL 37 | 21 | 27 | 24 | 24 | 26 | — | |
LC50 means lethal concentrations causing 50% of the cell death. The maximum peptoid and peptide concentration tested was 100 µM. Cell viability was measured via MTS assay in cells treated with compounds for 72 h, and LC50 of peptoids was derived from peptoids' viability curves. HC10 **, concentration causing 10% hemolysis of human red blood cells, all the numbers were cited from [25]. “—”, data not measured. Peptoids labeled with asterisk have been reported previously to possess antimicrobial activities [23], [24], [25].
These anticancer peptoids were found to exert fast killings in all tested cancer cell lines, and their activities were highly dependent on their structures and working concentrations. A majority of cell death occurred within only 4 h of peptoid treatments, and increased cytotoxicity was observed with longer treatments. The cytotoxicity curves of peptoid 1 in MCF-7 cells for different treatment times are shown in Figure 2A. Table 2 summarizes the compound activity for 72 h treatment to better correlate with cytotoxicity of apoptosis-inducing chemotherapeutics which are typically evaluated for 72 h treatment in vitro.
Figure 2. Cell viability curves of peptoids.
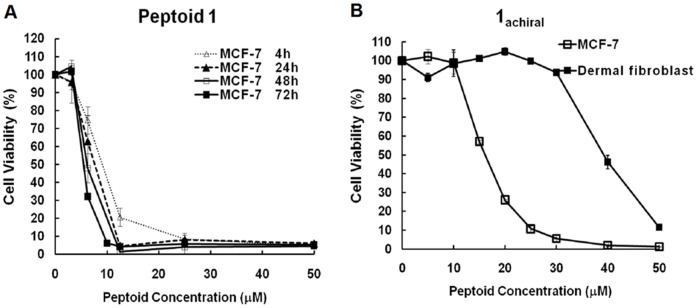
A, cell viability of MCF-7 cells treated with peptoid 1 for different time periods. Cell viability was measured with MTS assays; B, cell viability curve of 1achiral in MCF-7 and primary dermal fibroblast cells. 1achiral was more toxic to MCF-7 cells.
As shown in Table 2, LL-37, the non-selective antimicrobial peptide, showed little selectivity distinguishing cancer cell lines and normal control cells, while the clinically-relevant antimicrobial peptide, Pexiganan, exhibited modest in vitro selectivity towards cancer cell lines (indicated by higher LC50 in MRC-5 and primary dermal fibroblasts and higher HC10 against red blood cells than LC50 in cancer cell lines). We observed that the cytotoxicity of designed peptoids varied in different cancer cell lines, with LC50 in the low micromolar range. Some peptoids showed little selectivity, but several peptoids were found with modest in vitro selectivity towards cancer cells similar to Pexiganan, killing cancer cells efficiently while exhibiting less influence on MRC-5, primary dermal fibroblasts, and red blood cells in certain concentration ranges. How peptoid sequences could influence the cytotoxicity and selectivity will be discussed in the following structure-activity studies. The highest selectivity ratio (LC50 in primary dermal fibroblast divided by LC50 in cancer cells) we have observed for peptoids was ∼3 for 1 achiral (Figure 2B). We grouped the peptoid hits into two categories: (1) Peptoid 1 is the most potent peptoid with good water solubility, ease of synthesis and relatively low hemolytic activity, though it has similar cytotoxicity against cancer cells and fibroblasts cultured in vitro; (2) Similar to Pexiganan, 1 11mer, 1 achiral and 1 achiral-Nspe2,12 retain good potency and display modest selectivity in the in vitro screening.
Structure-Activity Relationship Studies
Cationic, amphipathic structure
We intended to study how hydrophobic and cationic residues as well as the structural amphipathicity influenced peptoid potency and selectivity (Table 2). Knowing that aromatic side chains are critical for the biological activities of cationic, amphipathic peptoids [23], an 11-mer with NLeu as hydrophobic residues was designed as a negative control and was confirmed to be inactive, with LC50 above 100 µM in all the cells tested. Furthermore, hydrophobicity and structural amphipathicity were found to be important for biological activity. Reduced potency against mammalian cells was observed with 1-NLys5,11 which had reduced hydrophobicity with only 6 Nspe residues per molecule and reduced amphipathicity with cationic residues present in hydrophobic faces [25]. Other cationic residues were also employed in peptoids. Guanidinium head groups have been previously reported to be critical for the cellular uptake of certain cell penetrating peptides [31]. Therefore, we synthesized 1-Nbtg, or [N-(4-butylguanidine) glycine], by guanidinylating NLys in peptoid 1. The activity of 1-Nbtg was similar to peptoid 1 without improvement on selectivity. Using histidines (pKa ∼6.1) as cationic residues has been reported to result in the pH-dependent activity of anticancer peptides with enhanced selectivity against tumor environments in vivo which are significantly more acidic than normal tissues [32]. 1-NHis1,7 was synthesized with NHis replacing NLys at position 1 and 7 in peptoid 1, which would reduce peptoid cationic charges in neutral pH. Decreased cytotoxicity against both cancer cell lines and fibroblasts was observed with 1-NHis1,7. We did not observe a pH-dependent activity of this NHis-NLys hybrid peptoid in vitro (data not shown), consistent with the previous result of histidine-containing peptide which only showed enhanced selectivity in vivo [32]. 1-NHis1,7 could exhibit better selectivity in vivo yet needs to be further validated.
Chain length variants
The chain length is believed to influence not only the activity but also modes of action of cationic, amphipathic peptides [33], [34]. In a previous study, increasing the length beyond a 12mer, such as peptoid 1 15mer, [H-(NLys-Nspe-Nspe)5-NH2], did not benefit antibacterial potency but resulted in substantially higher hemolytic activity, indicating that long chain lengths may lead to undesirable systemic toxicity of anticancer peptoids [23]. Thus we limited the chain length of peptoids to 13 residues at most, and studied 1 9mer which has the same charge-to-length ratio (CTLR) as peptoid 1 at 0.33, 1 11mer with CTLR at 0.36, and 1 13mer with CTLR at 0.38 [25]. As shown in Table 2, the relatively low toxicity of 19mer against mammalian cells indicated that peptoids have to reach certain chain lengths to gain high potency. 1 11mer was found to be more selective than peptoid 1 with reduced toxicity against MRC-5 and primary dermal fibroblasts and significantly reduced hemolytic activity, though its potency against cancer cells was slightly reduced as well. 1 13mer was slightly less active than peptoid 1 yet without noticeable improvements on its selectivity.
Achiral monomer variants
The molecular chirality of peptoids is derived from the chirality of the side chains rather than that of the backbone [21]. 1 achiral with less hydrophobic, achiral Npm residues lost helical signals in circular dichroism (CD) spectroscopy, which suggests a lack of stable secondary structure, and increased replacement of Nspe with Npm in peptoid 1 decreased peptoid helical intensities in CD. The achiral monomer variants had significantly reduced hemolytic activities compared to peptoid 1 [25]. In our in vitro screening, 1 achiral, 1 achiral–Nspe2, 1 achiral–Nspe2,12, 1-Npm2,3,8,9 all displayed better selectivity than peptoid 1 with reduced toxicity to MRC-5 cells and primary dermal fibroblasts. 1 achiral was the most selective peptoid in this group with good potency against cancer cells (Figure 2B).
Anticancer peptoids overcome multidrug resistance developed in cancer cells
Several anticancer peptides, such as magainin 2 [12] and buforin IIb [13], [14], have been reported to overcome multidrug resistance developed in cancer cells. Interestingly, activities of our anticancer peptoids were also found to be unaffected by the multidrug resistance in cancer cells. The resistant MCF-7/TxT50 cell line which was selected by increasing exposure to docetaxel is resistant due to the high expression of the ABCB1/MDR1, P-glycoprotein [35]. As shown in Figure 3A, MCF-7/TxT50 cells were confirmed to be resistant to Docetaxel compared to wild-type MCF-7 cells. However, peptoid 1 and 1 achiral had similar activities in both MCF-7 and the resistant MCF-7/TxT50 cells (Figure 3B-C), indicating a different mode of action of peptoids from the hydrophobic small molecule chemotherapeutic agents.
Figure 3. Activity of docetaxel and peptoids in MCF-7 and MCF-7/TxT50 cells.
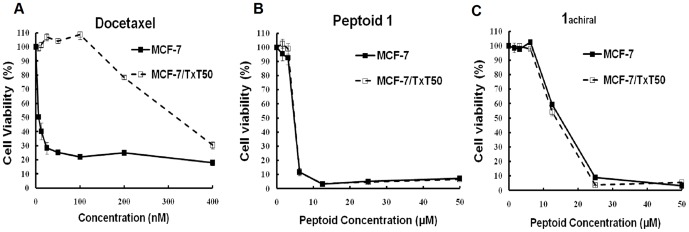
MCF-7/TxT50 cells were selected by docetaxel screening and are resistant due to overexpression of MDR1. Figures show the cell viability of MCF-7 and MCF-7/TxT50 cells treated with docetaxel (A), peptoid 1 (B) and 1achiral (C) for 72 h. Cell viability was measured with MTS assays.
Primary cytotoxicity of anticancer peptoids in cancer cells involves plasma membrane damage
These anticancer peptoids exert fast killing in cancer cells and are not affected by the multidrug resistance. To study how these peptoids interact with cells, we synthesized CF-peptoid 1 with fluorescent labeling on the N-terminus of peptoid 1. Via live cell confocal imaging, peptoid 1 was found to penetrate into cells efficiently with a dot-like cytoplasmic distribution, even at low concentrations without causing any noticeable cytotoxicity (Figure 4A), indicating that these peptoids can interact with plasma membranes and translocate into cells. To evaluate if plasma membranes are damaged upon peptoid interactions and to determine how much membrane damage accounts for the cell death caused by peptoid 1, cells were treated with peptoids at various concentrations for 5, 30 and 60 minutes. The cells were then either cultured in fresh media for another 48 h before testing via the MTS assay, which measures cell viability by tracking cellular metabolism (Figure 4B), or immediately tested via two cytotoxicity assays that evaluate plasma membrane intactness, the Guava Viacount assay (Figure 4C) and the lactate dehydrogenase (LDH) assay (Figure 4D). The Guava Viacount assay quantifies dead cells with a cell-impermeable DNA binding dye that can only stain cells with damaged membranes and cellular signals are measured by Guava flow cytometry. The LDH assay measures membrane damage by quantifying the leakage of the cytoplasmic enzyme LDH. Cell viability curves quantified by the Guava Viacount assay right after peptoid treatment were similar to those measured 48 h later with MTS assays, suggesting that plasma membranes were damaged upon peptoid treatments and most of the cell deaths caused by peptoids were due to plasma membrane damage. Moreover, in the LDH assay, a fast release of LDH into culturing media was also observed upon peptoid treatment. With LDH leakage caused by cell lysis solution as the 100% control, 50 µM of peptoid 1 (Figure 4D) and melittin (Figure S1) caused ∼50% of enzyme leakage. A linear correlation with r2 = 0.955 was observed between LDH leakage and cell viability quantified via the MTS assay (Figure 4E). Similar results were observed with melittin which is generally accepted to be lytic (Figure S1), further confirming the primary toxicity of peptoids being on plasma membranes. Moreover, typical apoptotic DNA ladders were not observed in cells after 24 h of peptoid 1 treatment (Figure S2). Taken together, these anticancer peptoids killed cancer cells primarily through plasma membrane damage. However, there could be intracellular targets as well, since peptoids can translocate into cells and are widely distributed in the cytoplasm, and we have observed increased cell death with longer peptoid treatment in some concentration ranges (Figure 2A).
Figure 4. Killing mechanisms.

A, live cell confocal images. MCF-7 cells were treated with 8 µM of CF-peptoid 1 for 1 h, and cells were imaged with ×63 oil lens. MCF-7 cells were treated with peptoids for indicated time and cell viability was measured with MTS assays after another 48 h incubation with fresh media (B), or measured via the Guava assay immediately after peptoid treatment (C), or quantified with LDH leakage immediately (D). E, correlation of LDH leakage and cell viability upon peptoid treatment measured with MTS assays, with r2 = 0.955.
Peptoid 1 inhibits tumor growth in a clinically relevant orthotopic xenotransplantation model
As a preliminary study of the in vivo efficacy and safety of peptoids, the most potent peptoid, peptoid 1, was evaluated in an orthotopic xenograft mouse model. Human breast cancer cells were implanted in the mammary fat pad of immunocompromised mice. After two weeks, peptoid 1 or the inactive negative control peptoid 1 11mer-NLeu was injected into the mammary fat pad at a dose of ∼1 mg/kg three times a week. Peptoid 1 significantly inhibited the tumor growth compared to the control peptoid (Figure 5). In addition, the applied dosages of peptoids did not cause any noticeable acute toxicity in mice. These results indicate that peptoid derivatives may be effective therapeutic agents for the treatment of human tumors, but their in vivo efficacy, toxicity, and proper local delivery methods will need to be thoroughly investigated in future.
Figure 5. Reduction of tumor volumes in a human breast cancer xenograft mouse model treated with peptoid 1.
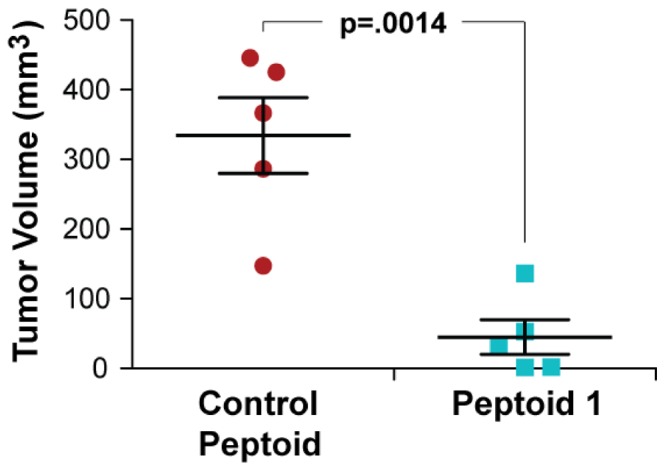
Cancer cells (8×104 cells) isolated from the second generation xenografts of human breast cancer tissues were implanted s.c. into the lower left mammary fat pads of 5–6-week-old immunocompromised NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) female mice. Two weeks after cell implantation, 100 µl of peptoid 1 and control peptoids were injected into xenografts at 110 µM in PBS (∼1 mg/kg) three times a week, up to 8 weeks. Tumor sizes were measured by a caliper. P = 0.0014, t test.
Discussion
Several cationic, amphipathic antimicrobial peptides or their derivatives have recently been reported with anticancer activity, displaying selective cytotoxicity against cancer cells in vitro and being effective in several in vivo xenograft models [3], [4], [7]. Moreover, a recent report indicated a naturally occurring anticancer role of a host defense peptide, cathelicidin, in natural killer cells' antitumor functions in mice, most strongly supported by the experiment that cathelicidin knockout (Camp -/-) mice permitted faster tumor growth than wild type controls [36]. All these findings suggest that these cationic, amphipathic peptides, which are used by nature in host defense, represent a very interesting and promising group of candidates to be developed for anticancer applications.
In this study, we exploited the protease resistance and the propensity for helix formation of certain peptoids to develop stable anticancer peptidomimetics, expanding available monomer chemistry and capturing the cationic, amphipathic structural feature. Several anticancer peptoids were developed successfully with potent cytotoxicity toward a broad range of human cancer cell lines and their killing was not affected by multidrug resistance. Moreover, the most potent hit, peptoid 1, greatly reduced tumor growth via intratumor injections in a human breast cancer xenograft mouse model without causing any obvious acute side effects in mice, suggesting the in vivo anticancer efficacy and good local tolerance of peptoids.
Our structure-activity studies revealed that it was critical for peptoids to have the cationic, amphipathic structure and to reach a certain chain length to obtain good potency against cancer cell lines. Peptoids needed to retain certain proportions of hydrophobic residues in their structures, namely a certain size of the hydrophobic arc in order to be potent. A typical ratio of cationic and hydrophobic residues per molecule was ∼1∶2 in peptoids with good potency. Increasing cationic residues (as in 1-NLys5,11 with 1∶1 cationic to hydrophobic ratio) or reducing cationic charges (as in 1-NHis1,7, with ∼90% of NHis base in uncharged form around neutral pH) both weakened their activity. Moreover, using Nbtg which has a guanidinium head group instead of NLys in peptoid 1 did not increase peptoid activity significantly. Instead, bulkier aromatic side chains in hydrophobic residues are found to be critical for peptoid potency. The cytotoxicity of 1 11mer-NLeu was low and was used as the inactive control. Taken together, the results with our peptoids support current explanations for the actions of cationic, amphipathic peptides: cationic residues select for anionic cellular membranes via electrostatic interactions; hydrophobic regions lead to membrane disruptions.
To evaluate peptoid selectivity in vitro, MRC-5 and primary dermal fibroblasts were tested as normal control cells, and hemolytic activities of several peptoids were also cited from previous work [25]. Though there are many reports about changed membrane properties of cells once they become cancerous [3], [7], [33], [37], it could be challenging to develop selective cationic, amphipathic peptides or peptoids to distinguish the slight differences. In our study, we found several peptoids with modest selectivity towards cancer cells, killing several cancer cell lines efficiently while showing little influence on normal control cells and red blood cells in some concentration ranges, such as achiral monomer peptoid variants with less stable secondary structures. The hemolytic activities of peptoids are generally quite low. These results prove the successful mimicry of anticancer peptides using non-natural peptoids.
The selectivity ratio of peptoids in our in vitro screening system was not strikingly high. Besides, given the membrane perturbation mechanism of these cationic, amphipathic molecules, the peptoids may only be suitable for certain anticancer applications, and local delivery or prodrug methods may mitigate any potential side effects. We understand that in vitro systems have limitations in mimicking in vivo environments; therefore, further investigation on the in vivo efficacy and toxicity as well as proper administration routes of these peptoids are required in a rigorous preclinical setting to establish their potential as cancer therapeutics.
There are several methods to further improve selectivity or reduce toxicity. First, we can engineer the peptoids at the molecular level by attaching “tumor homing” moieties to peptoids to enhance their accumulation in tumors and reduce their nonspecific interactions [38], [39], [40], or by developing peptoid-based prodrugs, the full activity of which need to be activated by tumor related enzymes [41], [42]. Second, systemic toxicity of peptoids can be tuned by the careful choice of delivery methods. Mitomycin C, a DNA crosslinking agent with antitumor antibiotic activity is administered as a single instillation within 6 hours of bladder tumor resection to minimize its toxicity and is proven to be effective in reducing recurrence [43]. The nonselective melittin was incorporated into nanocarriers with favorable pharmacokinetics, named “nanobees”, and was selectively accumulated in multiple tumor targets, dramatically reducing tumor growth without causing any apparent signs of toxicity [44]. Thus, a proper delivery method can be explored to further improve the in vivo behaviors of these peptoids.
Cationic, amphipathic peptides are generally believed to be membranolytic in vitro, accumulating on lipid membranes and subsequently disrupting the membrane structural integrity, either by forming pores in the membrane or acting like detergents and dissolving the membrane altogether [33]. We have observed membrane damage upon peptoid treatments, indicated by the Guava viacount assay and LDH assay, and there was a good correlation between immediate membrane damage and the cytotoxicity caused by peptoids. Interestingly, these peptoids can also penetrate into cells even at low concentrations. We even observed some co-localization of peptoids with mitochondria in the cytoplasm (data not shown). Therefore, we do not exclude potential intracellular targets of these peptoids, which is backed by the observation that longer peptoid treatments resulted in increased cell death at some concentration ranges. Our data support that these cationic, amphipathic peptoids interact with cells in a concentration dependent manner. At low concentrations, peptoids penetrate into cells in a way that does not disrupt plasma membranes or from which cells can recover, and they may disrupt intracellular organelles depending on their intracellular concentrations. At high concentrations, the way peptoids interact with plasma membranes causes membrane damage, which contributes to peptoids' observed, fast cytotoxicity.
Our preliminary in vivo experiment with peptoid 1 indicated promising anticancer potentials of these peptoids. Future research directions would include evaluating systemic toxicity of these peptoids in mouse models and testing their anticancer efficacy in clinical-relevant bladder cancer and ovarian cancer mouse models. This group of cationic, amphipathic peptoids, because of their broad spectrum of anticancer activity, unique modes of action, resistance to MDR and stability towards protease degradation, has a great potential to complement existing chemotherapy.
Associated Content
Electronic Supplementary Information includes the evaluation data of melittin as a comparison in the mechanism study, the DNA ladder assay for peptoid treatment, and ESI-MS data of peptoids (Figure S1-3).
Supporting Information
Evaluation of melittin as a comparison. MCF-7 cells were treated with melittin for indicated time and cell viability was measured with MTS assays after another 48 h incubation with fresh media (A), or measured via the Guava assay immediately after treatment (B), or quantified with LDH leakage immediately (C). D, correlation of LDH leakage and cell viability upon melittin treatment measured with MTS assays, with r2 = 0.726.
(TIF)
DNA ladder assay. MCF-7 cells were treated with control peptoid and peptoid 1 at the indicated concentrations for 24 h. Cells and floated cells were collected and combined for each sample. DNA was extracted using the apoptotic DNA ladder Kit (Roche), stained with GelStar (Lonza) and was run in 1% agarose gel. Experiments were done according to the kit, and the +DNA ladder control used the “lyophilized apoptotic U397 cells” sample provided in the kit as a positive control.
(TIF)
ESI-MS data of peptoids. Construct molecular weight (MW) and the corresponding peaks were indicated in the mass spectra. Previously reported peptoids in Table 1 are not listed here.
(TIF)
Acknowledgments
We thank Professor Branimir Sikic and Dr. George Duran for providing the MCF-7 and MCF-7/TxT50 cells, Lifeline Cell Technology for gifting the primary dermal fibroblasts and their culturing kits, Dr. Modi Wetzler for manuscript revisions, Professor Christopher Contag, Professor Marcus Covert, and Dr. Niv Papo for insightful discussions.
Funding Statement
This work was supported by the National Institutes of Health (5 R01 AI072666-04 to A.E.B.) and the National Research Foundation of Korea (2011-0014890 to J.S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G (2008) Immunological aspects of cancer chemotherapy. Nature Reviews Immunology 8: 59–73. [DOI] [PubMed] [Google Scholar]
- 2. Fletcher JI, Haber M, Henderson MJ, Norris MD (2010) ABC transporters in cancer: more than just drug efflux pumps. Nature Reviews Cancer 10: 147–156. [DOI] [PubMed] [Google Scholar]
- 3. Bhutia SK, Maiti TK (2008) Targeting tumors with peptides from natural sources. Trends Biotechnol 26: 210–217. [DOI] [PubMed] [Google Scholar]
- 4. Hoskin DW, Ramamoorthy A (2008) Studies on anticancer activities of antimicrobial peptides. Biochimica Et Biophysica Acta-Biomembranes 1778: 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leuschner C, Hansel W (2004) Membrane disrupting lytic peptides for cancer treatments. Curr Pharm Des 10: 2299–2310. [DOI] [PubMed] [Google Scholar]
- 6. Dennison SR, Whittaker M, Harris F, Phoenix DA (2006) Anticancer alpha-helical peptides and structure/function relationships underpinning their interactions with tumour cell membranes. Current Protein & Peptide Science 7: 487–499. [DOI] [PubMed] [Google Scholar]
- 7. Papo N, Shai Y (2005) Host defense peptides as new weapons in cancer treatment. Cell Mol Life Sci 62: 784–790. [DOI] [PubMed] [Google Scholar]
- 8. Cragg GM, Grothaus PG, Newman DJ (2009) Impact of Natural Products on Developing New Anti-Cancer Agents. Chem Rev 109: 3012–3043. [DOI] [PubMed] [Google Scholar]
- 9. McPhee JB, Hancock REW (2005) Function and therapeutic potential of host defence peptides. Journal of Peptide Science 11: 677–687. [DOI] [PubMed] [Google Scholar]
- 10. Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415: 389–395. [DOI] [PubMed] [Google Scholar]
- 11. Melo MN, Ferre R, Castanho MARB (2009) OPINION Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nature Reviews Microbiology 7: 245–250. [DOI] [PubMed] [Google Scholar]
- 12. Baker MA, Maloy WL, Zasloff M, Jacob LS (1993) Anticancer Efficacy of Magainin2 and Analog Peptides. Cancer Res 53: 3052–3057. [PubMed] [Google Scholar]
- 13. Park CB, Kim HS, Kim SC (1998) Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun 244: 253–257. [DOI] [PubMed] [Google Scholar]
- 14. Park CB, Yi KS, Matsuzaki K, Kim MS, Kim SC (2000) Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin II. Proc Natl Acad Sci U S A 97: 8245–8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Czyzewski AM, Barron AE (2008) Protein and peptide biomimicry: Gold-mining inspiration from nature's ingenuity. Aiche Journal 54: 2–8. [Google Scholar]
- 16. Papo N, Shahar M, Eisenbach L, Shai Y (2003) A novel lytic peptide composed of DL-Amino acids selectively kills cancer cells in culture and in mice. J Biol Chem 278: 21018–21023. [DOI] [PubMed] [Google Scholar]
- 17. Papo N, Shai Y (2003) New lytic peptides based on the D,L-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry (Mosc) 42: 9346–9354. [DOI] [PubMed] [Google Scholar]
- 18. Papo N, Braunstein A, Eshhar Z, Shai Y (2004) Suppression of human prostate tumor growth in mice by a cytolytic D-, L-amino acid peptide: Membrane lysis, increased necrosis, and inhibition of prostate-specific antigen secretion. Cancer Res 64: 5779–5786. [DOI] [PubMed] [Google Scholar]
- 19. Zuckermann RN, Kerr JM, Siani MA, Banville SC, Santi DV (1992) Identification of Highest-Affinity Ligands by Affinity Selection from Equimolar Peptide Mixtures Generated by Robotic Synthesis. Proc Natl Acad Sci U S A 89: 4505–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, et al. (1998) Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc Natl Acad Sci U S A 95: 4303–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu CW, Sanborn TJ, Huang K, Zuckermann RN, Barron AE (2001) Peptoid oligomers with alpha-chiral, aromatic side chains: Sequence requirements for the formation of stable peptoid helices. J Am Chem Soc 123: 6778–6784. [DOI] [PubMed] [Google Scholar]
- 22. Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, et al. (2003) Structural and spectroscopic studies of peptoid oligomers with alpha-chiral aliphatic side chains. J Am Chem Soc 125: 13525–13530. [DOI] [PubMed] [Google Scholar]
- 23. Patch JA, Barron AE (2003) Helical peptoid mimics of magainin-2 amide. J Am Chem Soc 125: 12092–12093. [DOI] [PubMed] [Google Scholar]
- 24. Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, et al. (2008) Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci U S A 105: 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czyzewski AM, Kapoor R, Chongsiriwatana NP, Dohm MT, Vakulenko S, et al. (In review) Antimicrobial peptoids with improved selectivity exhibit broad-spectrum actvity against multi-drug resistant bacterial strains. Journal of antimicrobial agents and chemotherapy.
- 26. Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, et al. (2000) The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc Natl Acad Sci U S A 97: 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burkoth TS, Fafarman AT, Charych DH, Connolly MD, Zuckermann RN (2003) Incorporation of unprotected heterocyclic side chains into peptoid oligomers via solid-phase submonomer synthesis. J Am Chem Soc 125: 8841–8845. [DOI] [PubMed] [Google Scholar]
- 28. Fischer R, Kohler K, Fotin-Mleczek M, Brock R (2004) A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J Biol Chem 279: 12625–12635. [DOI] [PubMed] [Google Scholar]
- 29. Gottler LM, Ramamoorthy A (2009) Structure, membrane orientation, mechanism, and function of pexiganan - A highly potent antimicrobial peptide designed from magainin. Biochimica Et Biophysica Acta-Biomembranes 1788: 1680–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barlow PG, Beaumont PE, Cosseau C, Mackellar A, Wilkinson TS, et al. (2010) The Human Cathelicidin LL-37 Preferentially Promotes Apoptosis of Infected Airway Epithelium. Am J Respir Cell Mol Biol 43: 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA (2004) Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc 126: 9506–9507. [DOI] [PubMed] [Google Scholar]
- 32. Makovitzki A, Fink A, Shai Y (2009) Suppression of Human Solid Tumor Growth in Mice by Intratumor and Systemic Inoculation of Histidine-Rich and pH-Dependent Host Defense-like Lytic Peptides. Cancer Res 69: 3458–3463. [DOI] [PubMed] [Google Scholar]
- 33. Shai Y (2002) Mode of action of membrane active antimicrobial peptides. Biopolymers 66: 236–248. [DOI] [PubMed] [Google Scholar]
- 34. Oren Z, Shai Y (1998) Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 47: 451–463. [DOI] [PubMed] [Google Scholar]
- 35. Spicakova T, O'Brien MM, Duran GE, Sweet-Cordero A, Sikic BI (2010) Expression and Silencing of the Microtubule-Associated Protein Tau in Breast Cancer Cells. Molecular Cancer Therapeutics 9: 2970–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buchau AS, Morizane S, Trowbridge J, Schauber J, Kotol P, et al. (2010) The Host Defense Peptide Cathelicidin Is Required for NK Cell-Mediated Suppression of Tumor Growth. J Immunol 184: 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sok M, Sentjurc M, Schara M (1999) Membrane fluidity characteristics of human lung cancer. Cancer Lett 139: 215–220. [DOI] [PubMed] [Google Scholar]
- 38. Rege K, Patel SJ, Megeed Z, Yarmush ML (2007) Amphipathic peptide-based fusion peptides and immunoconjugates for the targeted ablation of prostate cancer cells. Cancer Res 67: 6368–6375. [DOI] [PubMed] [Google Scholar]
- 39. Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, et al. (1999) Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med 5: 1032–1038. [DOI] [PubMed] [Google Scholar]
- 40. Dharap SS, Wang Y, Chandna P, Khandare JJ, Qiu B, et al. (2005) Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci U S A 102: 12962–12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DeFeo-Jones D, Garsky VM, Wong BK, Feng DM, Bolyar T, et al. (2000) A peptide-doxorubicin ‘prodrug’ activated by prostate-specific antigen selectively kills prostate tumor cells positive for prostate-specific antigen in vivo. Nat Med 6: 1248–1252. [DOI] [PubMed] [Google Scholar]
- 42.Boudreault PL, Arseneault M, Otis F, Voyer N (2008) Nanoscale tools to selectively destroy cancer cells. Chemical Communications: 2118–2120. [DOI] [PubMed]
- 43. Tolley DA, Parmar MKB, Grigor KM, Lallemand G, Beynon LL, et al. (1996) The effect of intravesical mitomycin C on recurrence of newly diagnosed superficial bladder cancer: A further report with 7 years of followup - Reply. J Urol 155: 1238–1238. [PubMed] [Google Scholar]
- 44. Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, et al. (2009) Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest 119: 2830–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Evaluation of melittin as a comparison. MCF-7 cells were treated with melittin for indicated time and cell viability was measured with MTS assays after another 48 h incubation with fresh media (A), or measured via the Guava assay immediately after treatment (B), or quantified with LDH leakage immediately (C). D, correlation of LDH leakage and cell viability upon melittin treatment measured with MTS assays, with r2 = 0.726.
(TIF)
DNA ladder assay. MCF-7 cells were treated with control peptoid and peptoid 1 at the indicated concentrations for 24 h. Cells and floated cells were collected and combined for each sample. DNA was extracted using the apoptotic DNA ladder Kit (Roche), stained with GelStar (Lonza) and was run in 1% agarose gel. Experiments were done according to the kit, and the +DNA ladder control used the “lyophilized apoptotic U397 cells” sample provided in the kit as a positive control.
(TIF)
ESI-MS data of peptoids. Construct molecular weight (MW) and the corresponding peaks were indicated in the mass spectra. Previously reported peptoids in Table 1 are not listed here.
(TIF)