

Abstract
The inhaled corticosteroid fluticasone furoate (FF) and the long-acting β2 agonist vilanterol (VI) are in development as a combined once-daily therapy for asthma and chronic obstructive pulmonary disease. Our study objectives were to compare the efficacy and safety of once-daily FF/VI with FF alone and twice-daily fluticasone propionate (FP) in patients aged ≥12 years with moderate-to-severe persistent asthma.
Patients (n=586) received FF/VI 200/25 μg or FF 200 μg once-daily (evening dosing), or FP 500 μg twice-daily for 24 weeks. Co-primary end-points were change from baseline in trough forced expiratory volume in 1 s (FEV1) weighted mean (wm) 0–24 h serial FEV1. Secondary end-points included change from baseline in percentage of rescue-free 24-h periods, percentage of symptom-free 24-h periods and total Asthma Quality of Life Questionnaire (AQLQ). Safety assessments included adverse events, 24-h urinary cortisol excretion, vital signs and ECG.
FF/VI significantly improved trough FEV1 and wmFEV1 versus FF and FP. Significantly more rescue-free and symptom-free 24-h periods were reported with FF/VI versus FF. Treatment differences for AQLQ were not significant. Incidence of adverse events was similar across groups. No clinically significant differences were seen for 24-h urinary cortisol excretion, vital signs or ECG.
FF/VI resulted in statistically greater improvements in lung function and symptomatic end-points versus FF, and was well tolerated in this asthma population.
Short abstract
Fluticasone furoate (FF)/vilanterol improved lung function and symptomatic end-points compared with FF alone http://www.ow.ly/siK33
Introduction
Inhaled corticosteroids (ICSs) are the most effective anti-inflammatory treatments for all severities of persistent asthma [1]. The benefits of ICSs include improved control of asthma symptoms, improvement in lung function and fewer exacerbations [2]. A once-daily dosing of ICS can improve treatment adherence relative to twice-daily treatments [3], potentially improving asthma control. Fluticasone furoate (FF) is an ICS with 24-h activity approved for intranasal treatment of allergic rhinitis [4, 5] and is under development as a once-daily treatment for asthma. FF is structurally distinct from fluticasone propionate (FP) [6] and has both greater affinity for the glucocorticoid receptor and longer retention in respiratory tissues than FP [7]. FF has been shown to be effective in asthma in earlier trials, with daily doses of 100 μg and 200 μg having a favourable therapeutic index [8–11]. Additionally, noninferiority of FF 200 μg once-daily to FF 100 μg twice-daily has been shown, providing clinical evidence that FF is suitable for once-daily dosing [12].
The benefits of adding long-acting β2 agonists (LABAs) to an ICS in twice-daily treatments are well documented for lung function, symptom control, rescue medication use and asthma exacerbation frequency [13]. Vilanterol (VI) is a novel once-daily inhaled LABA shown to provide bronchodilation for at least 24 h; 25 μg is considered the optimal dose [14]. Once-daily FF/VI use is being investigated as a combination therapy for asthma at strengths of 100/25 μg and 200/25 μg [14].
The aims of this study were to compare the efficacy and safety of once-daily FF/VI 200/25 μg with once-daily FF 200 μg alone in patients aged ≥12 years with moderate-to-severe persistent asthma; twice-daily FP 500 μg was included as an active comparator. Additionally, the study sought to demonstrate noninferiority of FF 200 μg once-daily compared with FP 500 μg twice-daily. Preliminary results have been published in abstract form [15].
Methods
Patients
The study enrolled asthma patients [16] aged ≥12 years with documented use of ICS, with or without LABA, for ≥12 weeks with stable ICS dose (FP 500 μg twice daily (or equivalent) or mid-dose ICS/LABA (FP/salmeterol 250/50 μg twice daily or equivalent)) for ≥4 weeks. Eligible patients were required to demonstrate an evening pre-bronchodilator forced expiratory volume in 1 s (FEV1) of 40–90% of predicted normal and FEV1 reversibility of ≥12% and ≥200 mL on inhalation of albuterol/salbutamol. Patients were ineligible if they had a history of life-threatening asthma in the previous 10 years, an asthma exacerbation requiring overnight hospitalisation or emergency department attendance within 6 months of screening, and/or an asthma exacerbation requiring oral corticosteroids within 12 weeks of screening.
At screening, eligible patients entering the study on ICS/LABA therapy were switched to the same ICS at the same dose contained in the ICS/LABA combination for the 4-week run-in period prior to randomisation. No LABA was taken on the day of screening. Patients on ICS alone continued ICS-only therapy during run-in. All patients had to be able to replace their current short-acting bronchodilator with albuterol/salbutamol and to withhold albuterol/salbutamol for at least 6 h prior to study visits. At randomisation, all eligible patients were required to have recorded asthma symptoms (equating to a score of ≥3 on the asthma symptom scale) and/or daily albuterol/salbutamol use on four or more of the seven preceding days. Electronic diary cards were used to capture daily measurements of peak expiratory flow (PEF), symptom scores and rescue use.
Written informed consent from each adult (≥18 years) patient was obtained prior to performing any study-specific procedures, as was assent and written parental consent for each adolescent (12–17 years) patient. The study was approved by local ethics review committees and was conducted in accordance with the Declaration of Helsinki [17], Good Clinical Practice guidelines [18] and all applicable regulatory requirements.
Study design and treatments
This phase III, randomised, multicentre, double-blind, double-dummy, parallel-group study (GlaxoSmithKline study number HZA106829; registered at www.clinicaltrials.gov with registration number NCT01134042) was conducted between June 10, 2010 and October 18, 2011 at 63 centres in six countries (Germany, Japan, Poland, Romania, Russia and the USA). Patients were randomised (1:1:1) to FF/VI 200/25 μg (representing an emitted dose of 184/22 μg), FF 200 μg once daily in the evening, or FP 500 μg twice daily (morning and evening) for 24 weeks.
FF/VI and FF were administered via a dry powder inhaler and FP was administered via Diskus/Accuhaler (GlaxoSmithKline, London, UK). Treatment compliance was assessed at each study visit after randomisation by reviewing the dose counters on the inhalers. Patients were randomised using a registration and medication ordering system (RAMOS; GlaxoSmithKline, Brentford, UK), an automated, interactive telephone-based system, following the generation of a central randomisation schedule generated by the sponsor using a validated computerised system (RandAll; GlaxoSmithKline, Brentford, UK).
Details of medication prohibited during the study are given in the online supplementary material.
Outcome measurements
The co-primary end-points were mean change from baseline in pre-dose (trough) FEV1 and weighted mean (wm)FEV1 (0–24 h post-dose) after 24 weeks of treatment. Secondary efficacy end-points were mean change from baseline in the percentage of rescue-free 24-h periods during the 24-week treatment period (a nominated powered end-point), change from baseline in the percentage of symptom-free 24-h periods during the 24-week treatment period, and change from baseline in Total Asthma Quality of Life Questionnaire (AQLQ+12) score after 12 and 24 weeks of treatment.
Other end-points included 12-h post-dose FEV1 at week 24, wmFEV1 (0–4 h post-dose) at week 24, mean change from baseline in daily morning and evening PEF over weeks 1–12 and 1–24, change from baseline in the Asthma Control Test (ACT) at weeks 12 and 24, number of withdrawals due to lack of efficacy over 24 weeks, and unscheduled healthcare resource utilisation for severe asthma exacerbations and other asthma-related healthcare. A post hoc analysis was performed to assess week 24 trough FEV1 as a percentage of the post-salbutamol screening value.
Safety
Safety and tolerability of study treatments were assessed by monitoring adverse events (AEs), serious AEs (SAEs), and incidence of severe asthma exacerbations (not recorded as an AE unless they met the definition of an SAE). AEs were coded using the Medical Dictionary for Regulatory Activities. Safety parameters, including vital signs, ECG measurements (recorded after 5 min rest before measurement of the clinic lung function tests), clinical chemistry and haematology, routine liver function assessments, and 24-h urinary cortisol (UC) excretion, were also assessed.
Statistical analysis
A sample size of 588 randomised patients (196 per treatment arm) had 95% power to detect a treatment difference of 150 mL in change from baseline in trough FEV1 between FF/VI and FF, 96% power to detect a treatment difference of 175 mL in 0–24-h serial wm FEV1 in the subset of patients performing serial FEV1 measurements, and >99% power to detect a treatment difference of 15% in change from baseline in percentage of rescue-free 24-h periods between FF/VI and FF. The overall power of the study to detect treatment differences for the co-primary end-points and the nominated powered secondary end-point was 92%. The sample size also provided 80% power to test for noninferiority on change from baseline in trough FEV1 only for the comparison of FF 200 μg to FP 500 μg with a noninferiority bound of -125 mL.
Co-primary and powered secondary end-points were analysed using an ANCOVA model with covariates for baseline, region, sex, age and treatment group. Imputation of missing data for the change from baseline in trough FEV1 end-point was carried out by last observation carried forward. Change from baseline in trough FEV1 was also analysed using a repeated measures model. wmFEV1 was calculated in a subset of patients (∼45% per arm) who performed serial FEV1 at week 24.
The intent-to-treat (ITT) population comprised all patients randomised to treatment and who received at least one dose of study medication. The per protocol population comprised all patients in the ITT population who did not have any full protocol deviations. The UC population comprised all patients whose urinary samples did not have confounding factors considered to affect the interpretation of 24-h UC data. The ITT population was the primary population for all efficacy and safety analyses other than the UC analysis. The per protocol population was of equal importance to the ITT in the assessment of noninferiority of FF 200 μg once daily to FP 500 μg twice daily on change from baseline in trough FEV1.
To account for multiplicity across treatment comparisons and key end-points, a step-down, closed-testing procedure was applied whereby treatment comparisons for the co-primary end-points (trough and wmFEV1) were required to be significant at the 0.05 level in order to infer significance for key secondary end-points percentage of rescue-free and percentage of symptom-free 24-h periods and AQLQ+12 (online supplementary fig. S1). Significance at the 0.05 level was required for each secondary end-point in sequence to allow significance to be inferred for comparison of all remaining secondary and “other” end-points. If p>0.05 for any comparison in the hierarchy, then statistical significance could not be inferred for comparisons below that point in the hierarchy. The analysis of noninferiority for FF 200 μg once daily compared with FP 500 μg twice daily on the end-point of trough FEV1 was not part of this statistical procedure.
Following routine quality control surveillance by the study sponsor, data quality issues were identified for one trial site. A post hoc sensitivity analysis was therefore conducted after the blind was broken for the co-primary and powered secondary efficacy end-points that excluded all patients randomised at this site (n=48). The decision to keep the ITT population as the primary population for presentation of results was documented prior to conducting the sensitivity analysis.
Results
Study population
Of 1206 patients screened, 587 were randomised, 586 comprised the ITT population, 515 comprised the per protocol population and 476 (81%) completed the study (fig. 1). Patient demographics and baseline characteristics were well balanced across treatment groups (table 1). FP/salbutamol was the most common pre-study treatment (taken by 50–54% of patients). During run-in, FP was the most common ICS (taken by 59–64% of patients); mean daily doses of run-in FP ranged from 551.1 μg to 583.2 μg.
Figure 1–

Consort/patient flow diagram. FF: fluticasone furoate; FP: fluticasone propionate; ITT: intent-to-treat; VI: vilanterol. #: one patient was randomised to the FP 500 μg group in error, but did not receive study drug and was thus not included in the ITT population.
Table 1– Patient demographics and baseline characteristics (intent-to-treat population).
| FF/VI 200/25 μg once daily | FF 200 μg once daily | FP 500 μg twice daily | Total | |
| Subjects | 197 | 194 | 195 | 586 |
| Age years | 46.6±15.05 | 44.6±14.33 | 47.3±14.06 | 46.2±14.51 |
| Females | 116 (59) | 113 (58) | 116 (59) | 345 (59) |
| Duration of asthma years | 17.01±13.227 | 14.71±11.920 | 14.85±12.533 | 15.53±12.597 |
| Baseline pre-bronchodilator FEV1 L | 2.129±0.6539 | 2.190±0.6756 | 2.138±0.6725 | 2.153±0.6668 |
| Baseline FEV1 % predicted | 66.59±12.614 | 66.66±12.388 | 67.57±12.185 | 66.94±12.383 |
| Screening reversibility FEV1 % | 29.58±19.828 | 29.17±17.035 | 29.56±16.375 | 29.44±17.790 |
| Screening absolute reversibility FEV1 mL | 561.7±367.91 | 583.3±346.30 | 568.0±313.08 | 570.9±342.77 |
| Baseline rescue-free 24-h periods % | 7.6±19.22 | 7.8±20.68 | 6.3±18.03 | |
| Baseline symptom-free 24-h periods % | 5.1±15.20 | 4.7±16.06 | 2.7±9.83 | |
| Pre-study ICS regimen | ||||
| ICS alone | 47 (24) | 44 (23) | 49 (25) | 140 (24) |
| ICS + salmeterol | 106 (54) | 102 (53) | 98 (50) | 306 (52) |
| ICS + formoterol | 44 (22) | 48 (25) | 48 (25) | 140 (24) |
Data are presented as n, mean±sd or n (%). FF: fluticasone furoate; VI: vilanterol; FP: fluticasone propionate; FEV1: forced expiratory volume in 1 s; ICS: inhaled corticosteroid.
Co-primary end-points
Trough FEV1 at week 24 was improved from baseline with all therapies. The differences between FF/VI and FF, and FF/VI and FP were both significant, while FF was noninferior to FP (table 2). Change from baseline in trough FEV1 by treatment showed sustained benefit with FF/VI over FF and FP at all study time-points (fig. 2).
Table 2– Mean change from baseline and treatment differences in trough forced expiratory volume in 1s (FEV1) (last observation carried forward) and 0–24 h wmFEV1 (subset of patients who performed serial FEV1) at week 24 (population).
| FF/VI 200/25 μg once daily | FF 200 μg once daily | FP 500 μg twice daily | |
| Subjects | 197 | 194 | 195 |
| Trough FEV1 (week 24) | |||
| Subjects | 187 | 186 | 190 |
| Least squares mean change from baseline mL | 394±30.2 | 201±30.3 | 183±30.0 |
| Treatment difference versus FF 200 μg once daily mL | 193 (108–277)*** | ||
| Treatment difference versus FP 500 μg twice daily mL | 210 (127–294)*** | 18 (-66–102)# | |
| 0–24-h wmFEV1 (week 24) | |||
| Subjects n | 89 | 83 | 86 |
| Least squares mean change from baseline mL | 464±47.0 | 328±49.3 | 258±48.3 |
| Treatment difference versus FF 200 μg once daily mL | 136 (1–270)¶ | ||
| Treatment difference versus FP 500 μg twice daily mL | 206 (73–339)+ |
Data are presented as n, mean±se or mean (95% confidence interval). The ANCOVA model with covariates for baseline, region, age, sex and treatment was used. FF: fluticasone furoate; FP: fluticasone propionate; VI: vilanterol; wm: weighted mean. #: noninferiority comparison, where noninferiority is shown as lower bound of comparison 95% confidence interval exceeded margin of -125 mL. ***: p<0.001; ¶: p=0.048; +: p=0.003.
Figure 2–

Repeated measures analysis for the primary end-point of change from baseline in trough forced expiratory volume in 1 s over 24 weeks of treatment (intent-to-treat population). Data are presented as least squares mean (95% CI). FF: fluticasone furoate; VI: vilanterol; FP: fluticasone propionate.
0–24-h wmFEV1 at 24 weeks was assessed in 89, 83 and 86 patients in the FF/VI, FF and FP arms, respectively. In all arms, 0–24-h wmFEV1 was improved at week 24 compared with baseline and there were statistically significant treatment differences between FF/VI and FF or FP (table 2). A 24-h time-course of FEV1 at week 24 is shown in figure 3.
Figure 3–
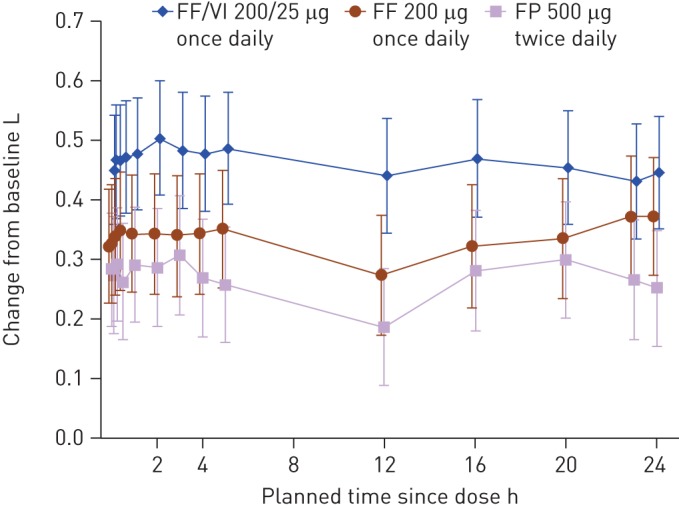
Adjusted mean change from baseline of individual serial forced expiratory volume in 1 s assessments following 24 weeks of treatment (intent-to-treat population). Data are presented as least squares mean (95% CI). FF: fluticasone furoate; VI: vilanterol; FP: fluticasone propionate.
Results for both co-primary end-points in the per protocol population supported those of the ITT population (online supplementary fig. S2).
Powered secondary end-points
The percentage of rescue-free 24-h periods increased over the study with all therapies. The difference in improvement was significant for the comparison of FF/VI with FF, but not for FF/VI compared with FP (table 3). The number of additional rescue-free 24-h periods per week compared with baseline was 2.7 with FF/VI, 1.9 with FF and 2.2 with FP.
Table 3– Mean change from baseline and treatment differences for powered secondary and other efficacy end-points (intent-to-treat population).
| FF/VI 200/25 μg once daily | FF 200 μg once daily | FP 500 μg twice daily | |
| Subjects | 197 | 193 | 194 |
| Rescue-free 24-h periods % (weeks 1–24) | |||
| Least squares mean change from baseline | 38.2±2.42 | 26.6±2.45 | 31.9±2.45 |
| Treatment difference versus FF 200 μg once daily | 11.7 (4.9–18.4)*** | ||
| Treatment difference versus FP 500 μg twice daily | 6.3 (-0.4–13.1)# | ||
| Symptom-free 24-h periods % (weeks 1–24) | |||
| Least squares mean change from baseline | 29.3±2.29 | 21.0±2.32 | 24.5±2.31 |
| Treatment difference versus FF 200 μg once daily | 8.4 (2.0–14.8)¶ | ||
| Treatment difference versus FP 500 μg twice daily | 4.9 (-1.6–11.3)+ | ||
| AQLQ+12 (week 24) | |||
| Least squares mean change from baseline | 0.93±0.065 | 0.88±0.071 | 0.90±0.068 |
| Treatment difference versus FF 200 μg once daily | 0.05 (-0.14–0.24)§ƒ | ||
| Treatment difference versus FP 500 μg twice daily | 0.03 (-0.16–0.21)## | ||
| 12-h post-dose FEV1 (week 24) L | |||
| Least squares mean | 2.645±0.0489 | 2.477±0.0513 | 2.390±0.0497 |
| Treatment difference versus FF 200 μg once daily | 0.167 (0.027–0.307) | ||
| Treatment difference versus FP 500 μg twice daily | 0.254 (0.117–0.392) | ||
| 0–4 h wmFEV1 (week 24) L | |||
| Least squares mean change from baseline | 0.484±0.0469 | 0.342±0.0492 | 0.285±0.0483 |
| Treatment difference versuss FF 200 μg once daily | 0.142 (0.008–0.276) | ||
| Treatment difference versus FP 500 μg twice daily | 0.198 (0.066–0.331) | ||
| Morning PEF (weeks 1–24) L·min−1 | |||
| Least squares mean change from baseline | 51.8±2.94 | 18.2±2.97 | 18.8±2.95 |
| Treatment difference versus FF 200 μg once daily | 33.5 (25.3–41.7) | ||
| Treatment difference versus FP 500 μg twice daily | 32.9 (24.8–41.1) | ||
| Evening PEF (weeks 1–24) L·min−1 | |||
| Least squares mean change from baseline | 39.8±2.93 | 9.1±2.98 | 13.6±2.96 |
| Treatment difference versus FF 200 μg once daily | 30.7 (22.5–38.9) | ||
| Treatment difference versus FP 500 μg twice daily | 26.2 (18.0–34.3) | ||
| ACT (week 24) | |||
| Least squares mean change from baseline | 5.5±0.28 | 5.2±0.30 | 4.7±0.29 |
| Treatment difference versus FF 200 μg once daily | 0.3 (-0.5–1.1) | ||
| Treatment difference versus FP 500 μg twice daily | 0.7 (-0.1–1.5) |
Data are presented as n, mean±se and mean (95% confidence interval). The ANCOVA model with covariates for baseline, region, age, sex and treatment was used. FF: fluticasone furoate; VI: vilanterol; FP: fluticasone propionate; AQLQ+12: total Asthma Quality of Life Questionnaire; FEV1: forced expiratory volume in 1 s; wm: weighted mean; PEF: peak expiratory flow; ACT: Asthma Control Test. #: p=0.067; ¶: p=0.010; +: p=0.137; §: p=0.587; ƒ: failure to achieve p<0.05 for the FF/VI versus FF comparison on the AQLQ+12 end-point in the intent-to-treat population analysis meant that significance could not be inferred for any subsequent comparisons using the closed step-down statistical hierarchy employed, as such, differences and associated 95% confidence inerval values are shown only for subsequent comparisons; ##: p=0.786; ***: p<0.001.
Sensitivity analysis
The results for the sensitivity analysis excluding the site with quality issues, gave results consistent with the full ITT population for trough FEV1 and for 24-h rescue-free periods (online supplementary table S1 and fig. S2). For 0–24-h wmFEV1 at 24 weeks, the treatment difference for FF/VI versus FF was smaller than that observed in the ITT population and was no longer statistically significant; the treatment difference for FF/VI versus FP remained statistically significant (online supplementary table 1 and fig. S2).
Secondary and other end-points
The percentage of symptom-free 24-h periods increased over the course of the study and a significant effect of FF/VI over FF, but not over FP, was observed (table 3). Patients experienced an additional 2.1 symptom-free 24-h periods per week in the FF/VI arm; these values were 1.5 for FF and 1.7 for FP.
Improvements from baseline in the AQLQ+12 score were seen in all treatment groups at week 24. The improvements were similar in each arm and were not statistically significant (table 3). As statistical significance was not achieved on this end-point, statistical inference could not be drawn on any of the other efficacy end-points (table 3 and online supplementary fig. S3 for morning PEF data). At baseline, the proportion of patients rating themselves as controlled (ACT ≥20) was 2% (three out of 197), 6% (12 out of 194) and 8% (15 out of 195) in the FF/VI, FF and FP arms, respectively. At week 24, this proportion increased to 50% (85 out of 170), 51% (75 out of 147) and 48% (77 out of 162) in the FF/VI, FF and FP groups, respectively.
Over the 24-week treatment period, fewer patients withdrew due to lack of efficacy in the FF/VI group (3%) compared with the FF (11%) or FP (9%) groups (fig. 4).
Figure 4–

Cumulative incidence curves for withdrawals due to lack of efficacy over the 24-week treatment period (intent-to-treat population). FF: fluticasone furoate; VI: vilanterol; FP: fluticasone propionate.
In post hoc analysis, the mean percentage of trough FEV1 at week 24 relative to post-salbutamol FEV1 (screening) was 96.1% (FF/VI), 85.7% (FF) and 85.6% (FP) (online supplementary table S2 and fig. S4).
Safety assessment
On-treatment AEs were similar across treatment groups (46–50%). Nasopharyngitis (13–20%) and headache (6–8%) occurred most frequently (online supplementary table S3). More patients withdrew from the study due to an AE in the FF/VI group (seven; 4%) compared with FF (three; 2%) and FP (two; 1%). The incidence of treatment-related AEs was greater with FF/VI (9%) and FP (8%) than FF (4%). The incidence of on-treatment SAEs was greater for FF/VI (3%) than FF (<1%) or FP (1%). Two SAEs (atrial fibrillation in the FF/VI group and haemoptysis in the FP group) were considered treatment related. There were no statistically significant differences at week 24 in systolic or diastolic blood pressure or pulse rate, and there were no clinically significant differences for ECG measurements (QTcF or QTcB). No deaths occurred during the study.
Eight patients reported severe asthma exacerbations during treatment: six (3%) in the FF group, none in the FF/VI group and two (1%) in the FP group. All were treated with systemic/oral corticosteroids; only one patient in the FF group was hospitalised due to a severe asthma exacerbation.
The ratio to baseline for 24-h UC excretion at week 24 was slightly lower for FP (0.84) than FF (0.91) or FF/VI (0.98); treatment differences were not statistically significant for any comparison (online supplementary fig. S5).
Discussion
This study demonstrates the benefit of combining the new once-daily LABA VI with the once-daily ICS FF, versus once-daily FF or twice-daily FP alone, in patients with moderate-to-severe asthma. The main aims of this study, involving patients with moderate-to-severe asthma [1], were to compare the efficacy and safety of once-daily FF/VI 200/25 μg to once-daily FF 200 μg alone. An additional comparison of once-daily FF 200 μg to twice-daily FP 500 μg assessed the noninferiority of the two treatments on trough FEV1. A significantly greater improvement in lung function was observed with FF/VI versus FF for both co-primary end-points and the powered secondary end-point of change in percentage of rescue-free 24-h periods, as well as versus FP for both co-primary end-points. Additionally, FF 200 μg once daily was shown to be noninferior to FP 500 μg twice daily. All treatments were generally well-tolerated and no safety signals were observed.
The inclusion criteria in this study selected for patients whose asthma was uncontrolled on high-dose ICS or medium-dose ICS/LABA therapy. FF 200 μg has previously been shown to be effective for patients with moderate-to-severe persistent asthma, with higher doses not offering further benefits [10]. The active control (FP 500 μg twice daily) represents the maximum dose of FP recommended for use in patients with moderate-to-severe persistent asthma [1]. Therefore, doses of FF/VI, FF and FP used were appropriate for the patient population assessed.
FF/VI was statistically significantly better than FF and FP for both co-primary FEV1 end-points, with FF being noninferior to FP on trough FEV1. The improvements in trough FEV1 with FF/VI over FF (193 mL) and FP (210 mL) were in the range considered to be clinically relevant. These findings indicate that good efficacy is achievable with less frequent administration and, together with previous findings that VI produces prolonged bronchodilation for at least a 24-h duration at an optimal dose of 25 μg [13], this suggests the FF/VI is suitable for once-daily dosing, thereby offering greater convenience to patients. This is an important consideration for a maintenance therapy in asthma; poor adherence is associated with inadequate control and occurrence of severe asthma exacerbations [19], often due to poor patient motivation [20]. Simpler dosing regimens with reduced frequency of dosing have been shown to improve adherence for a variety of treatments [21].
Adding VI to FF significantly increased the percentage of rescue-free and symptom-free 24-h periods compared with FF alone. These findings are consistent with those of two large Cochrane meta-analyses that compared twice-daily ICS/LABA with ICS-only therapies in ICS-experienced [12] or ICS-naïve [22] patients with persistent asthma. In addition, there were numerical improvements in these outcomes for FF/VI compared with FP. The AQLQ+12 score was improved with FF/VI compared with FF and FP, but the differences were not statistically significant. There was a numerically greater benefit with FF/VI versus FF or FP for 12 h FEV1, wmFEV1 0–4 h post-dose, morning and evening PEF, and the ACT (for which all treatments achieved the minimally clinically important difference of three compared with baseline [23]). Fewer withdrawals due to lack of efficacy were seen with the combination and no severe asthma exacerbations were reported for patients on FF/VI compared with FF (n=6) and FP (n=2).
The study was conducted in a clinically appropriate cohort of patients for the dose of FF/VI used and was sufficiently long to allow clinically meaningful conclusions to be drawn from the data. An additional strength of the study was that its power was sufficient to show noninferiority of once-daily FF to the maximum recommended dose of FP (500 μg twice daily), thereby providing a robust active treatment against which the effects of FF/VI and FF could be assessed. The use of electronic daily diaries provided a standardised method of capturing accurate date- and time-stamped information. The statistical hierarchy methodology represents both a strength and potential limitation; while it accounts for multiple comparisons and multiplicity, it also means that statistical significance could not be inferred for treatment differences for any comparison following AQLQ+12. The sponsor’s quality control processes identified a study site with data quality issues and a sensitivity analysis of the co-primary and powered secondary end-points was subsequently performed. The results of the sensitivity analysis did not affect the overall study conclusions.
In conclusion, the findings of this study demonstrate the efficacy of FF/VI in this patient population. Additionally, noninferiority (on trough FEV1) of once-daily FF 200 μg to twice-daily FP 500 μg was shown. Treatment with once-daily FF/VI was associated with statistically greater improvements in lung function and symptomatic end-points versus FF, showing that the addition of the LABA VI confers significant clinical benefit in this patient population relative to high-dose ICS monotherapy. The incidence of treatment-related AEs was low and there were no apparent treatment effects on UC levels or safety parameters.
Acknowledgments
We thank all patients and investigators involved in the study. Editorial support in the form of development of a draft outline in consultation with the authors, development of a manuscript first draft in consultation with the authors, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, fact checking, referencing and graphic services was provided by T. Gallagher and G. Weller (Gardiner-Caldwell Communications, Macclesfield, UK) and was funded by GlaxoSmithKline.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Clinical trial: The study is registered at www.clinicaltrials.gov with identifier number NCT01134042, and GlaxoSmithKline study number HZA106829.
Support statement: This study was funded by GlaxoSmithKline (study number HZA106829). The open access and colour figure reproduction charges were paid for by GlaxoSmithKline, and editorial support was funded by GlaxoSmithKline.
Conflict of interest: Disclosures can be found alongside the online version of this article at www.erj.ersjournals.com
References
- 1.British Thoracic Society and Scottish Intercollegiate Guidelines Network British guideline on the management of asthma: a national clinical guideline. Thorax 2008; 63: Suppl. 4, iv1–iv121 [DOI] [PubMed] [Google Scholar]
- 2.Global Initiative for Asthma Global Strategy for Asthma Management and Prevention, 2012. www.ginasthma.org/documents/4 Date last updated: December, 2012. Date last accessed: October 9, 2013
- 3.Fanta CH. Drug therapy: asthma. N Engl J Med 2009; 360: 1002–1014 [DOI] [PubMed] [Google Scholar]
- 4.Price D, Robertson A, Bullen K, et al. Improved adherence with once-daily versus twice-daily dosing of mometasone furoate administered via a dry powder inhaler: a randomized open-label study. BMC Pulm Med 2010; 10: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.US prescribing information for veramyst (fluticasone furoate) nasal spray Indicated for treatment of symptoms of seasonal and perennial allergic rhinitis in adults and children ≥2 years. www.accessdata.fda.gov/drugsatfda_docs/label/2011/022051s007lbl.pdf Date last updated: October, 2011. Date last accessed: October 9, 2013 [Google Scholar]
- 6.European Medicines Agency authorisation codes EU/1/07/434/001; EU/1/07/434/002; EU/1/07/434/003 Avamys (fluticasone furoate) indicated for the treatment of symptoms of allergic rhinitis: summary of product characteristics. www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000770/WC500028814.pdf Date last accessed: October 9, 2013
- 7.Biggadike K, Bledsoe RK, Hassell AM, et al. X-ray crystal structure of the novel enhanced-affinity glucocorticoid agonist fluticasone furoate in the glucocorticoid receptor-ligand binding domain. J Med Chem 2008; 51: 3349–3352 [DOI] [PubMed] [Google Scholar]
- 8.Salter M, Biggadike K, Matthews JL, et al. Pharmacological properties of the enhanced-affinity glucocorticoid fluticasone furoate in vitro and in an in vivo model of respiratory inflammatory disease. Am J Physiol Lung Cell Mol Physiol 2007; 293: 660–667 [DOI] [PubMed] [Google Scholar]
- 9.Bateman ED, Bleecker ER, Lötvall J, et al. Dose effect of once-daily fluticasone furoate in persistent asthma: a randomized trial. Respir Med 2012; 106: 642–650 [DOI] [PubMed] [Google Scholar]
- 10.Bleecker ER, Bateman ED, Busse WW, et al. Once-daily fluticasone furoate is efficacious in patients with symptomatic asthma on low-dose inhaled corticosteroids. Ann Allergy Asthma Immunol 2012; 109: 353–358 [DOI] [PubMed] [Google Scholar]
- 11.Busse WW, Bleecker ER, Bateman ED, et al. Fluticasone furoate demonstrates efficacy in patients with asthma symptomatic on medium doses of inhaled corticosteroid therapy: an 8-week, randomised, placebo-controlled trial. Thorax 2012; 67: 35–41 [DOI] [PubMed] [Google Scholar]
- 12.Woodcock A, Bleecker ER, Busse WW, et al. Fluticasone furoate: once-daily evening treatment versus twice-daily treatment in moderate asthma. Respir Res 2011; 12: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ducharme FM, Ni Chroinin M, Greenstone I, et al. Addition of long-acting β2-agonists to inhaled corticosteroids versus same dose inhaled corticosteroids for chronic asthma in adults and children. Cochrane Database Syst Rev 2010; 5: CD005535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lötvall J, Bateman ED, Bleecker ER, et al. 24 h duration of the novel LABA vilanterol trifenatate in asthma patients treated with ICSs. Eur Respir J 2012; 40: 570–579 [DOI] [PubMed] [Google Scholar]
- 15.O’Byrne PM, Bleecker ER, Bateman ED, et al. Efficacy and safety of fluticasone furoate (FF)/vilanterol (VI) once daily (OD) for 24 weeks in persistent asthma. Eur Respir J 2012; 40: Suppl. 56, S313 [Google Scholar]
- 16.National Institutes for Health Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/index.htm Date last updated: August, 2007. Date last accessed: October 9, 2013 [Google Scholar]
- 17.World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. www.wma.net/en/30publications/10policies/b3/17c.pdf Date last updated: 2008. Date last accessed: October 9, 2013
- 18.International Conference on Harmonisation Tripartite Guideline Guidance for Good Clinical Practice E6 (R1). 1996. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf Date last accessed: October 9, 2013
- 19.Williams LK, Peterson EL, Wells K, et al. Quantifying the proportion of severe asthma exacerbations attributable to inhaled corticosteroid nonadherence. J Allergy Clin Immunol 2011; 128: 1185–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bender BG. Overcoming barriers to nonadherence in asthma treatment. J Allergy Clin Immunol 2002; 109: Suppl. 6, S554–S559 [DOI] [PubMed] [Google Scholar]
- 21.Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001; 23: 1296–1310 [DOI] [PubMed] [Google Scholar]
- 22.Ni Chroinin M, Greenstone I, Lasserson TJ, et al. Addition of inhaled long-acting β2-agonists to inhaled steroids as first line therapy for persistent asthma in steroid-naive adults and children. Cochrane Database Syst Rev 2009; 7: CD005307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cloutier MM, Schatz M, Castro M, et al. Asthma outcomes: composite scores of asthma control. J Allergy Clin Immunol 2012; 129: S24–S33 [DOI] [PMC free article] [PubMed] [Google Scholar]