

Abstract
Feline immunodeficiency virus (FIV) and the T cell-tropic strains of human immunodeficiency virus type 1 (HIV-1) share the use of the chemokine receptor CXCR4 for cell entry. To study this process further we developed a cell surface binding assay based on the expression of a soluble version of the FIV SU C-terminally tagged with the influenza virus hemagglutinin epitope (HA). The specificity of the assay was demonstrated by the following evidence: (1) the SU-HA protein bound to HeLa cells that express CXCR4 but not to MDCK cells that lack this chemokine receptor; and (2) binding of the SU-HA to HeLa cells was blocked by incubation with the CXCR4 antagonist AMD3100 as well as with the anti-CXCR4 monoclonal antibody (MAb) 12G5. Deletion of the V3 region from the FIV SU glycoprotein abolished its ability to bind CXCR4-expressing cells. Remarkably, substitution of the V3 domain of the FIV SU by the equivalent region of the HIV-1 NL4-3 isolate resulted in efficient cell surface binding of the chimeric SU protein to CXCR4. Moreover, transfection of MDCK cells with a plasmid encoding human CXCR4 allowed the association of the chimeric SU-HA glycoprotein to the transfected cells. Interestingly, while cell binding of the chimeric FIV-HIV SU was inhibited by an anti-HIV-1 V3 MAb, its association with CXCR4 was found to be resistant to AMD3100. Of note, the chimeric FIV-HIV Env glycoprotein was capable of promoting CXCR4-dependent cell-to-cell fusion.
Introduction
Feline immunodeficiency virus (FIV) induces in domestic cats an immunodeficiency syndrome similar to human AIDS.1 Therefore, FIV is not only an important cat pathogen but it is internationally recognized as a useful model for the study of human immunodeficiency virus type 1 (HIV-1) infections in humans.2–4
FIV infects a wide variety of feline cells such as CD4+ and CD8+ T lymphocytes, B lymphocytes, and macrophages.5–9 FIV, like the rest of the retroviruses, possesses a single envelope glycoprotein (Env) that, by interacting with specific receptors present at the surface of FIV target cells, mediates virus entry. Although FIV infects CD4+ T lymphocytes, it differs from the primate lentiviruses HIV-1 and simian immunodeficiency virus (SIV) in the sense that instead of using CD4 as primary receptor, it utilizes the CD134 molecule.10,11 However, FIV entry into its target cells also requires the binding of the Env glycoprotein to CXCR4, a chemokine receptor that is used as coreceptor by T cell-tropic (X4) HIV-1 isolates.12–14 Interestingly, FIV can use human CXCR4 as efficiently as its feline counterpart for Env-mediated cell fusion and viral entry.12 The FIV Env protein is initially synthesized as a precursor of 150 kDa, which after removal of an unusually long signal peptide yields a molecular species of 130 kDa that is further processed at the trans-Golgi generating the surface (SU, gp95) and transmembrane (TM, gp38) subunits of FIV Env.15
It is interesting to note that although the FIV SU protein specifically interacts with CXCR4, it exhibits a low degree of amino acid sequence homology with HIV-1 SU (gp120).16
In HIV-1, binding of gp120 to CD4 causes conformational changes in this Env subunit that lead to the formation of a domain, termed the bridging sheet, that is necessary for the interaction of gp120 with the coreceptors CCR5 or CXCR4.17 The bridging sheet consists of a four-stranded, antiparallel β sheet formed by the stems of the variable V1 and V2 loops as well as by residues of the conserved C4 region of gp120.17,18 The variable V3 loop of HIV-1 gp120 also contributes to the process of Env association with its cellular coreceptor. Indeed, since the bridging sheet region represents a chemokine binding domain highly conserved among the gp120 proteins of different HIV-1 isolates that interact with CCR5 or with CXCR4, it is the amino acid sequence of the V3 region that governs the coreceptor selectivity.19
While the biological properties of the gp120 proteins of HIV-1, HIV-2, and SIV have been the subject of numerous studies, little is known about the SU subunits of the rest of the lentiviruses. In this regard, the analysis of FIV entry is particularly interesting since FIV and the T cell-tropic HIV-1 strains share the use of CXCR4 as coreceptor. In addition, the study of the interaction between HIV-1 gp120 and CXCR4 has proven to be particularly difficult because the affinity of X4-tropic gp120 for this chemokine receptor is significantly lower (at least 20-fold) than that exhibited by R5-tropic gp120 proteins to CCR5.20–22 Therefore, given that all FIV isolates described so far exclusively use CXCR4 as coreceptor, it is likely that investigating the FIV SU-CXCR4 association will provide relevant information about the minimal structural requirements necessary for the interaction between lentiviral SU proteins and the chemokine receptor CXCR4.
Recently, it has been shown that FIV SU binding to CXCR4 specifically requires the V3 region of SU.23,24 We therefore decided to further investigate this process by using the SU of an FIV isolate that is independent of CD134 and requires only the presence on the cell surface of CXCR4 for cell entry.11,25 Considering that an FIV SU with such a biological property would be highly useful to directly evaluate the Env-CXCR4 association, we developed a cell surface SU binding assay that allowed us to readily detect the interaction between the FIV SU and CXCR4. This association was found to be specific since it was strictly dependent on the presence of CXCR4 on the cell surface and was inhibited by both the CXCR4 antagonist AMD3100 and the anti-CXCR4 monoclonal antibody (MAb) 12G5. Moreover, by means of this binding assay we demonstrated that deletion of the V3 domain of FIV Env abolishes the SU–CXCR4 interaction. Notably, we found that replacement in the FIV SU of its V3 region by the equivalent domain from an X4-tropic gp120 results in a chimeric SU protein that binds to CXCR4 as efficiently as wild-type FIV SU. In addition, the FIV glycoprotein carrying the HIV-1 V3 domain proved to be capable of mediating CXCR4-dependent cell fusion.
Materials and Methods
Cell lines
293T, HeLa, and MDCK cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS, GIBCO). MAGI-CXCR4 cells were additionally maintained in medium containing 0.2 mg/ml G418, 0.1 mg/ml hygromycin B, and 1 μg/ml puromycin as described previously.25
Monoclonal antibodies
The wild-type and mutant FIV SU-HA glycoproteins were visualized using the rat MAb directed to the HA epitope conjugated to horseradish peroxidase (clone 3F10; Roche Applied Science). The following MAbs were obtained from the NIH AIDS Reagent Program: anti-FIV SU V3 (SUFc1-30), anti-CXCR4 12G5,26 and that directed against the V3 domain of the HIV-1 LAV strain (MAb 902).27 The anti-CXCR4 MAb clone 1F8 as well as the MAb directed to α-tubulin were purchased from Sigma-Aldrich. The MAb against the V4 region of the FIV SU (clone 2-3) was purchased from Santa Cruz Biotechnology. The FIV Env and TM glycoproteins were detected by using a mouse polyclonal antibody directed to the TM ectodomain prepared in our laboratory.25
DNA constructs and site-directed mutagenesis
All FIV constructs were derived from the infectious molecular clone FIV-14 of the Petaluma isolate.28 The FIV SU coding region corresponding to nucleotides (nt) 6266 to 8089 of the FIV genome (amino acids 1–608 of Env) was polymerase chain reaction (PCR) amplified with specific primers using the Elongase enzyme high-fidelity mix (Invitrogen). The 3′ oligonucleotide included the sequence for the influenza virus hemagglutinin (HA) epitope (YPYDVPDYA) followed by a stop codon at the SU-TM cleavage site.
The PCR fragment was cloned into the BamHI and EcoRV sites of the pcDNA3.1 vector. The SU V3 deletion mutant lacking amino acids 377–417 was generated by first amplifying two genomic regions: one coding for amino acids 1–376 (nt 6266–7393) and the other encoding residues 418–608 (nt 7517–8089). EcoRI sites were introduced at the 3′ and 5′ ends of the amplified DNAs encoding the upstream and downstream regions of the V3 domain, respectively. Both PCR products were digested with EcoRI and ligated. The EcoRI sequence provided the codons for N418 and S419, which are the first two residues after the end of the V3 domain. It should be noted that the EcoRI junction introduces a histidine to glutamine substitution at position 376. However, glutamine is frequently found at this position in many FIV SU proteins.
The resulting DNA fragment lacking the V3 coding region of the FIV SU was then ligated into the BamHI and EcoRV sites of pcDNA3.1. To create a plasmid encoding the SU-HA in which the V3 domain of FIV was replaced by its HIV-1 counterpart (SUHV3-HA), we derived the V3 loop from the pNL4-3 molecular clone, which is a chimeric DNA encoding the Gag and Pol polyproteins of the HIV-1 NY5 isolate and the Env glycoprotein from the LAV isolate.29 The genomic region (nt 7100–7207) of the HIV-1 NL4-3 strain that encodes the V3 loop (amino acids 296–331) was PCR amplified with primers that introduced PstI and EcoRI sites at the 5′ and 3′ ends of the DNA product, respectively. In parallel, the region coding for amino acids 1–376 of the FIV SU was PCR amplified using an antisense oligonucleotide that added a PstI site at the 3′ end of the DNA fragment. Both the HIV-1- and FIV-derived PCR products were digested with PstI and ligated. The ligation product was reamplified by PCR, cut with BamHI and EcoRI enzymes, and gel purified. This DNA fragment encoding the first 376 residues of the FIV SU joined to the HIV-1 V3 loop was used to replace the BamHI–EcoRI sequence in the SUΔV3-HA clone to generate the plasmid coding for the SUHV3-HA.
In this construct the PstI site introduces a conservative threonine-to-serine substitution at position 2 in the HIV-1 V3 domain. In addition, the EcoRI sequence linking the end of the HIV-1 loop to the FIV SU downstream sequence introduces an extra glycine immediately downstream of the C-terminal cysteine of the HIV-1 V3 loop.
For the cell fusion assays, we have constructed an FIV env expressing plasmid (pcDNA-FIVenvWT) by cloning into the BamHI and NotI sites of the pcDNA 3.1 vector the viral genomic region encompassing the env gene, the two rev exons, and the Rev-responsive element (nt 6266–9474).25 To generate the expression plasmids pcDNA-FIVenvΔV3 and pcDNA-FIVenvHV3, the BamHI–MfeI fragment (nt 6266–7759) of the pcDNA-FIVenvWT was replaced by the corresponding fragments derived from the SU ΔV3-HA and SUHV3-HA constructs, respectively.
All the FIV env-derived constructs were confirmed by DNA sequencing.
Expression of the SU-HA glycoproteins
Since the SU-HA, SUΔV3-HA, and SUHV3-HA DNA constructs were cloned in the pcDNA3.1 vector that contains the T7 RNA polymerase promoter, these glycoproteins were expressed in mammalian cells using the vaccinia T7 system essentially as we have previously reported.30 Briefly, confluent monolayers of 293T cells (grown in 60-mm-diameter dishes) were infected for 1 h at 37°C with the vTF7-3 recombinant vaccinia virus that directs the synthesis of the T7 RNA polymerase.31 After infection, the cells were washed twice with DMEM and then transfected with the SU-HA, SUΔV3-HA, and SUHV3-HA plasmids using Lipofectamine 2000 (Invitrogen) as previously described.25,32 Thirty hours postinfection, culture supernatants were filtered through 0.45-μm-pore-size filters (Corning) and the amounts of the three different SU-HA glycoproteins present in the culture media were normalized by western blotting using the anti-HA MAb.
Cell surface binding assay
To examine the association of the FIV SU glycoprotein with CXCR4, we modified the cell surface binding assay that has been used to study the interaction between the gp120 of SIV and HIV-2 to the cellular coreceptors CCR5 and CXCR4, respectively.22,33 The secreted SU-HA, SUΔV3-HA, and SUHV3-HA proteins were generated as described above and the clarified supernatants containing these glycoproteins were directly overlaid on the receptor cells (HeLa or MDCK cells grown in 60-mm-diameter dishes) for 1 h at 37°C. Alternatively, receptor cells were first detached from the flasks, pelleted, and then resuspended and incubated with the SU-HA-containing supernatants to reduce the binding volume. This modification had no effect on the binding results. Cells were then washed three times with ice-cold phosphate-buffered saline (PBS) and lysed at 4°C in buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml aprotinin followed by a 5-min centrifugation at 16,000×g to remove cellular debris. Cell lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), blotted onto nitrocellulose membranes, and analyzed by western blotting using the MAb directed to the HA epitope. Western blots were developed with ECL Advance reagent (GE Life Sciences). Protein band signals were quantitated as previously described.25,30
Inhibition studies
HeLa cells (2×106 cells) were detached from flasks, pelleted, and resuspended in 100 μl of DMEM containing the potential inhibitors of the cell surface binding of SUWT-HA or SUHV3-HA and incubated for 30 min at 37°C. The culture supernatants (400 μl) containing the soluble SU-HA glycoproteins and the appropriate inhibitor final concentration were then added to the cell suspensions, which were further incubated for 1 h at 37°C. Cells were washed, lysed, and analyzed by SDS–PAGE and western blotting as described above. The inhibitors as well as their concentrations were as follows: AMD3100 (100 μg/ml) and anti-CXCR4 MAb 12G5 (10 μg/ml). When we tested the effect of the HIV-1 V3-specific MAb on the cell surface binding ability of the SU-HA glycoproteins, the receptor cells were incubated with 500 μl of SU-HA culture supernatants pretreated with the MAb (20 μg/ml) for 30 min at 25°C.
Cellular ELISA
To determine the levels at which the SUWT-HA and SUHV3-HA bind to the surface of HeLa cells, we also performed cellular enzyme-linked immunosorbent assays (cellular ELISA) similar to those previously described.32 Triplicate samples corresponding to 3×105 HeLa cells were first incubated at 37°C for 1 h with supernatants containing normalized amounts of the two different SU-HA glycoproteins, washed three times with ice-cold PBS, and resuspended in 50 μl of ELISA buffer (PBS containing 0.4% bovine serum albumin and 0.1% sodium azide). Cells were incubated for 1 h at 4°C with the anti-HA MAb conjugated to horseradish peroxidase followed by three washes with ice-cold ELISA buffer. The enzymatic reaction was performed using the 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonic acid) substrate. The resulting colored reaction signal was measured on a microtiter plate ELISA reader (Bio-Rad) at 405 nm (reference wavelength 490 nm) as previously described.30 HeLa cells successively incubated with the supernatant of mock-transfected 293T cells and the anti-HA antibody were used as a negative control.
Cloning of human CXCR4 cDNA and expression of the chemokine receptor in MDCK cells
Two micrograms of total RNA from HeLa cells was reverse-transcribed with SuperScript II reverse transcriptase (Invitrogen) in a 20-μl reaction mixture containing 50 mM Tris–HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 0.5 mM dNTPs, 40 units of recombinant RNasin ribonuclease inhibitor (Promega), and 500 ng oligo(dT)12–18. The mixture was incubated at 42°C for 50 min followed by heat inactivation at 70°C for 15 min. Four microliters of the first strand cDNA synthesis reaction was PCR amplified using the following specific primers for human CXCR4 gene: sense primer, 5′-TGGAAGCTTATGTCCATTCCTTTGCCT-3′ (the HindIII restriction site is underlined); antisense primer, 5′-TTAGCGGCCGCTTAGCTGGAGTGAAA-3″ (the NotI restriction site is underlined). The PCR product was digested with HindIII and NotI and cloned into pcDNA3.1 cut with the same restriction enzymes. The pcDNA-CXCR4 clone was subjected to DNA sequencing prior to its use in the transfection experiments. MDCK cells were transfected with this plasmid as described above following the manufacturer's (Invitrogen) recommendations for this cell line.
Cell-to-cell fusion assays
To examine the ability of the mutant ΔV3 and chimeric HV3 Env glycoproteins to mediate cell-to-cell fusion, 293T cells were transfected with the pcDNA-env/rev constructs together with an HIV-1 Tat-expressing plasmid created in our laboratory.25 Forty-eight hours posttransfection, the cells were dissociated and equivalent numbers of cells were added at a 1:5 ratio to 4×104 MAGI-CXCR4 cells in 24-well plates. Coculture was continued for 48 h, after which cells were stained for β-galactosidase activity and scored for syncytia formation as previously described.25,32 Cell-to-cell fusion was quantitated as the total number of blue syncytia per well by first counting their number in 20 nonoverlapping fields. The average number of syncytia per field was multiplied by the total number of fields per well and the result was referred to that obtained for the wild-type FIV Env protein.
Results
Expression of secreted FIV SU-derived glycoproteins
The mature form of the FIV Env glycoprotein is a heterodimer composed of the SU and TM subunits that remain associated by noncovalent interactions (Fig. 1A). The SU glycoprotein exhibits six variable domains (V1–V6) that encompass amino acid regions 197–205, 248–354, 377–417, 462–500, 534–563, and 586–611, respectively (Fig. 1A).34 Since it has recently been reported that the V3 domain plays a key role in the association of the FIV SU with CXCR4,23,24 we decided to study the SU–CXCR4 interaction using an Env protein that mediates virus entry into target cells independently of the primary receptor CD134.
FIG. 1.

(A) Schematic diagram of the feline immunodeficiency virus (FIV) Env glycoprotein showing the cleavage site between its surface (SU) and transmembrane (TM) subunits (arrow). The variable regions of the SU glycoprotein (V1–V6) are indicated with hatched boxes whereas the structural features of the TM glycoprotein: ectodomain, membrane-spanning domain, and cytoplasmic region are highlighted with black, gray, and open boxes, respectively. Amino acid numbering corresponds to the Env protein of the Petaluma FIV-14 isolate. (B) Amino acid sequence alignment of the V3 domains of FIV (Petaluma isolate) and HIV-1 (NL4-3 strain). The identical amino acids present at the same position in both sequences are indicated. Asterisks show the conservative amino acid changes between the FIV and HIV-1 V3 regions.
To this end, we first generated a plasmid construct encoding a secreted version of the FIV SU tagged at its C-terminus with the HA epitope. The supernatant of 293T cells expressing the SU-HA protein as well as the clarified culture medium of cells transfected with the FIV proviral DNA were subjected to SDS-PAGE and western blotting using the anti-FIV V3 (SUFc1-30) MAb. As shown in Fig. 2A, the soluble SU-HA exhibits the same electrophoretic mobility as that of the native SU, which is spontaneously shed into the culture medium after dissociating from the SU-TM complex anchored at the plasma membrane. This result indicates that the SU-HA is efficiently expressed and secreted into the medium, and that it exhibits a degree of glycosylation similar to that of the native FIV SU.
FIG. 2.

Expression in human cells of wild-type (WT), mutant (ΔV3), and chimeric (HV3) SU glycoproteins C-terminally tagged with the HA epitope. (A) 293T cells were transfected with the FIV proviral DNA and 48 h posttransfection the culture medium was recovered and clarified as described in Materials and Methods. In parallel, 293T cells were first infected with the recombinant vaccinia virus expressing the T7 RNA polymerase and then transfected with the SU-HA-expressing construct. Thirty hours postinfection, the cell-free supernatant was harvested. Aliquots of both cell culture supernatants were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the anti-FIV V3 monoclonal antibody (MAb) so as to detect in these samples the native FIV SU and the wild-type SU glycoprotein carrying the HA epitope at its C-terminus. (B) The wild-type (WT) and mutant (ΔV3) SU-HA glycoproteins were expressed in 293T cells as explained in Materials and Methods. The SU proteins secreted into the cell culture media were visualized by western blotting using the anti-HA MAb. (C) The clarified supernatants from 293T cells expressing either the wild-type (WT) or the chimeric (HV3) SU-HA glycoproteins were resolved by SDS-PAGE and immunoblotted with MAbs directed to the FIV V3, the HIV-1 V3, or the HA epitope. The positions of the molecular weight standards (in kDa) are indicated.
We then analyzed the expression of the SUΔV3-HA protein in which the V3 domain of the FIV SU was removed without altering the protein reading frame. Comparative analysis of SUΔV3-HA and SUWT-HA by western blotting revealed that both proteins were efficiently detected in the cell-free media of transfected cells with the anti-HA antibody and that the SUΔV3-HA glycoprotein migrates more rapidly than its wild-type counterpart, which is in agreement with the fact that the SUΔV3-HA protein has a 41-amino-acid deletion (Fig. 2B).
To investigate whether the V3 domain of the FIV SU could be functionally replaced with the equivalent region of a T cell-tropic (X4) HIV-1 strain, we created a construct coding for the SUHV3-HA glycoprotein. Of note, alignment of the V3 domain of the FIV SU with that of an X4 HIV-1 strain reveals that these V3 regions share 44% similarity at the amino acid level (Fig. 1B). As shown in Fig. 2C, expression of SUHV3-HA in 293T cells resulted in secretion of this protein with an efficiency similar to that of SUWT-HA. In addition, the SUHV3-HA is readily detected by western blotting using the anti-HA and anti-HIV-1 V3 MAbs but, as expected, it is not recognized by the anti-FIV V3 antibody (Fig. 2C).
Cell surface binding of SU-HA glycoproteins
We next examined the ability of the SUWT-HA, SUΔV3-HA, and SUHV3-HA glycoproteins to bind to the surface of cells expressing CXCR4. To this end, the supernatants of 293T cells containing the soluble SU-HA proteins were incubated either with HeLa cells that express CXCR435 or with MDCK cells that lack this chemokine receptor at their cell surface.36 After the incubation step, cells were washed and lysed, and the presence in the cell lysates of the different SU-HA glycoproteins was detected by western blotting using the anti-HA MAb as described in Materials and Methods. As shown in Fig. 3A, the SUWT-HA binds to HeLa cells but not to MDCK cells. By contrast, deletion of the V3 domain from the FIV SU abolishes cell surface binding to HeLa cells. Notably, SUHV3-HA associates with CXCR4-expressing cells as efficiently as SUWT-HA (Fig. 3A).
FIG. 3.

Cell surface binding of SUWT-HA, SUΔV3-HA, and SUHV3-HA glycoproteins. (A) Samples of culture supernatants containing similar amounts of the SU-HA glycoproteins (inputs) were incubated with HeLa or MDCK cells as described in Materials and Methods. Cells were washed and lysed and the presence in the cell lysates of each SU-HA glycoprotein was detected by western blotting using the anti-HA MAb. Inputs: As control, one-tenth of the total amount of SU-HA used in each binding reaction was loaded on the gel and immunoblotted with the HA-specific MAb. (B) Quantitation of the cell surface binding of the SUWT-HA and SUHV3-HA glycoproteins by cellular ELISA. HeLa cells were incubated with equivalent amounts of each soluble SU-HA protein and the surface levels of the wild-type and chimeric (HV3) SU-HA glycoproteins were quantitated by cellular ELISA as described in Materials and Methods. The values (mean±SD, three independent assays) correspond to the absorbance of each sample at 405 nm. As negative control (–), cells were incubated with the culture supernatant of mock-transfected cells.
To confirm and quantitate the cell surface binding of the SUWT-HA and SUHV3-HA glycoproteins we used the cellular ELISA as explained in Materials and Methods. This technique is highly specific and as sensitive as (or even more sensitive than) flow cytometry for the quantitative analysis of molecules expressed on the cell surface.37,38 Moreover, we have shown that cellular ELISA can successfully be used to determine the relative levels at the cell surface of wild-type and mutant FIV Env proteins.32 HeLa cells were incubated with SUWT-HA, SUHV3-HA, or culture medium from mock-transfected cells as negative control, washed, and incubated with the anti-HA MAb conjugated to peroxidase. After washing the cells, color was developed and quantitated as described in Materials and Methods. The cellular ELISA experiments demonstrated that SUWT-HA and SUHV3-HA specifically bind to HeLa cells, confirming the results obtained with the other methodology based on the immunoblotting of cell lysates after cell surface binding of the SU-HA proteins (Fig. 3B).
Cell surface binding to MDCK expressing CXCR4
To determine that cell surface binding of SUHV3-HA is dependent on the presence of CXCR4, we transfected MDCK cells with the plasmid encoding human CXCR4 constructed in our laboratory (see Materials and Methods). Transfected and mock-transfected MDCK cells were incubated with the supernatants containing the SUHV3-HA protein, washed, lysed, and subjected to western blotting using the anti-HA MAb. Figure 4A shows that the chimeric FIV SU-HA carrying the V3 loop of HIV-1 NL4-3 exclusively binds to MDCK cells expressing CXCR4. Expression of CXCR4 in transfected MDCK cells was verified by the detection of this chemokine receptor using the anti-CXCR4 MAb 1F8 that is suitable for western blot analyses (Fig. 4B). Moreover, probing the blots for the constitutive cellular protein α-tubulin lets us confirm that similar protein levels of transfected and mock-transfected MDCK cell lysates were compared in this experiment (Fig. 4B).
FIG. 4.
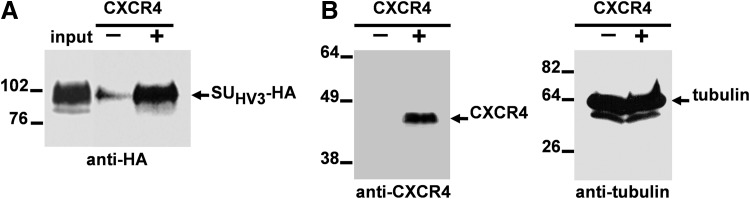
Cell surface binding of SUHV3-HA to MDCK cells expressing CXCR4. (A) Equivalent amounts of soluble SUHV3-HA were incubated with MDCK cells that were either transfected with a plasmid expressing human CXCR4 (+) or mock-transfected (–). Binding of SUHV3-HA to the surface of MDCK cells was detected by western blotting using the anti-HA MAb. Input: One-tenth of the total amount of SUHV3-HA used in each binding reaction was loaded on the gel. (B) The expression of CXCR4 in MDCK-transfected cells was verified by immunoblotting with the 1F8 MAb directed to human CXCR4. Normalization of protein levels in transfected and mock-transfected MDCK cells was performed by testing the cell lysates for the presence of α-tubulin as described in Materials and Methods. The positions of the molecular weight standards (in kDa) are indicated.
Inhibition studies
At this point our results indicated that the SUWT-HA and SUHV3-HA glycoproteins bind efficiently to CXCR4. We therefore sought to establish whether viral entry inhibitors of X4-tropic HIV strains also interfere with the cell surface binding of FIV-derived SU-HA proteins. We tested the CXCR4 antagonist AMD310039 and the anti-CXCR4 MAb 12G526 that binds to an epitope located in the CXCR4 extracellular loop (ECL) 2.40 As shown in Fig. 5A, we found that binding of SUWT-HA to the surface of HeLa cells was inhibited by both AMD3100 and the anti-CXCR4 MAb 12G5. Interestingly, AMD3100 had no effect on the cell surface binding capacity of SUHV3-HA (Fig. 5B). However, preincubation of the SUHV3-HA glycoprotein with the anti-HIV-1LAV V3 MAb prior to its addition to HeLa cells abrogated binding of this glycoprotein to the receptor cells (Fig. 5C).
FIG. 5.
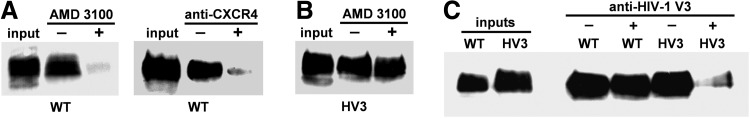
Inhibition studies of cell surface binding of SUWT-HA and SUHV3-HA glycoproteins. (A) Cell culture supernatants containing the soluble SUWT-HA protein were incubated with HeLa cells pretreated with either 100 μg/ml AMD3100 or 10 μg/ml anti-CXCR4 12G5 MAb. (B) The soluble SUHV3-HA was incubated with HeLa cells treated with 100 μg/ml AMD3100. (C) To test the ability of the anti-HIV-1 V3 MAb to interfere with the cell surface binding of the SU-HA glycoproteins, the SUWT-HA and SUHV3-HA proteins were first incubated with this MAb and then used in the binding assays as described in Materials and Methods. Input: One-tenth of the total amount of SUWT-HA or SUHV3-HA used in each binding reaction was immunoblotted with the anti-HA MAb.
Fusogenic capacity of the mutant ΔV3 and chimeric HV3 Env glycoproteins
We have previously developed for the Env glycoprotein of the Petaluma isolate a sensitive and quantitative assay that evaluates fusion between FIV Env-expressing cells and human CXCR4 cells.25 This method is based on the fact that the FIV Petaluma strain is independent of the primary receptor CD134 for cell entry.11 293T cells expressing wild-type, mutant ΔV3, or HV3 Env glycoproteins together with HIV-1 Tat were added to and cocultured with MAGI-CXCR4 indicator cells (HeLa-CD4-LTR-β-galactosidase)41 as described in Materials and Methods. If Env-mediated CXCR4-specific fusion between 293T and MAGI cells occurs, the HIV-1 Tat activates the LTR in the syncytia thereby promoting β-galactosidase synthesis, which is detected with the chromogenic substrate X-Gal.25
Using this assay, 293T cells expressing wild-type FIV Env yielded 925±65 blue syncytia per well (average of three independent experiments±standard deviation). By contrast, only 0.5% of blue syncytia were obtained with FIV EnvΔV3 when compared to the wild-type value, which is in line with the inability of this mutant Env glycoprotein to bind to CXCR4 (Table 1). Interestingly, the chimeric FIV EnvHV3 glycoprotein was capable of promoting substantial cell-to-cell fusion. Indeed, this chimeric protein exhibited a fusion activity representing approximately 60% of the wild-type value (Table 1). Efficient expression and processing of the wild-type, ΔV3, and HV3 Env glycoproteins were monitored by immunoblotting of a fraction of the transfected 293T cells with a polyclonal antibody specific for the FIV TM (Fig. 6A). Moreover, comparable levels of these glycoproteins were present at the cell surface as determined by means of cellular ELISA using the anti-FIV SU MAb 2-3 (Fig. 6B).
Table 1.
Cell-to-Cell Fusion Mediated by Feline Immunodeficiency Virus Env Glycoproteins
Blue foci were counted in 20 independent fields and the mean number of syncytia per field was multiplied by the total number of fields per well. The resulting values were referred to that obtained with wild-type Env,±standard deviations (three independent experiments).
293T cells expressing only HIV-1 Tat.
No blue syncytia were detected.
293T cells expressing the wild-type, the mutant ΔV3, or the chimeric HV3 Env glycoproteins together with the HIV-1 Tat protein were dissociated and equivalent numbers of cells were added at a 1:5 ratio to 4×104 MAGI-CXCR4 cells. Coculture was continued for 48 h. Cells were then stained for β-galactosidase and scored for syncytia formation.
FIG. 6.

(A) Synthesis and processing of the FIV Env glycoproteins. 293T cells expressing the wild-type (WT), ΔV3, or HV3 glycoproteins were lysed and viral proteins were resolved by SDS-PAGE and immunoblotted with the polyclonal serum against the FIV TM. The mobilities of the Env precursor (Pre) and TM subunit are indicated as are those of the molecular weight markers (in kDa). (B) Relative cell surface levels of mutant ΔV3 and chimeric HV3 glycoproteins as percentages of those of wild-type Env (WT) considered as 100%. The surface levels in transfected 293T cells of wild-type, ΔV3, or HV3 glycoproteins were quantitated by cellular ELISA as described in Materials and Methods.
Discussion
After interacting with the primary receptor CD134, FIV binds to the chemokine receptor CXCR4.42 Interestingly, despite the extensive amino acid sequence divergence exhibited by the SU proteins of FIV and T cell-tropic HIV-1 strains, these glycoproteins share the use of CXCR4 as coreceptor.14
In this article, we developed a cell surface CXCR4 binding assay based on the use of a soluble CD134-independent FIV SU glycoprotein tagged at its C-terminus with the HA epitope. This system offers two advantages. First, the fact that the SU glycoprotein derives from the Petaluma isolate that requires only CXCR4 for cell entry ensures that the results of the binding experiments will reflect only the Env association with CXCR4 since Petaluma SU does not require any previous interaction with CD134 to undergo the conformational changes necessary for binding to CXCR4. Second, expression of the FIV SU glycoprotein with the HA epitope as a C-terminal extension allows the detection by means of the anti-HA MAb of cell-surface bound SU not only in the case of the wild-type SU but also of any modified or mutated version of this glycoprotein.
The specificity of our cell surface FIV SU-CXCR4 association assay was demonstrated by the following evidence: (1) the SU-HA glycoprotein binds only to cells expressing CXCR4 such as the HeLa cell line, and (2) the interaction between the SU-HA glycoprotein and CXCR4 is inhibited by both the CXCR4 antagonist AMD3100 and the anti-CXCR4 12G5 MAb, which, by binding to the CXCR4 ECL2 domain, blocks the FIV SU binding site on CXCR4.43,44
It has previously been reported that the V3 region of the SU glycoprotein from the FIV PPR isolate is required for binding of SU-Fc immunoadhesins (soluble SU fused to the Fc of human IgG1) to the surface of 3201 cells, a lymphoma cell line that expresses CXCR4 but not CD134.23,24 Testing the ability of a set of V3 peptides to inhibit binding of PPR SU-Fc to 3201 cells together with site-directed mutagenesis of V3 led to the identification within this region of nine contiguous amino acids (SSWKQRNRW; Env residues 392–400) that are critical for FIV SU binding to CXCR4-expressing cells.23,24 In agreement with these previous reports, we demonstrate here that deletion of the V3 domain from a CD134-independent FIV SU abrogates binding to the surface of CXCR4-expressing cells. It should be pointed out, however, that despite the key role of the V3 region in SU-CXCR4 association, other as yet unidentified SU domains are likely to contribute to this process.
In HIV-1 the V3 loop of gp120 is the primary determinant of coreceptor usage. Indeed, the so-called “11/25” rule stated that if a positive charge was present at either of these positions in V3 (considering the first V3 cysteine as position 1), the virus was predicted to be X4-tropic.45,46 However, this rule proved to be more accurate for R5 HIV-1 isolates than for X4 viruses.47 This prediction uncertainty was probably the result of using virus populations instead of cloned viruses and the lack in many cases of the experimental determination of the virus tropism for CXCR4 or CCR5. In this regard, Cardozo et al.,19 by analyzing a large number of cloned viruses whose V3 loop sequences were compared and their coreceptor usage experimentally established, proposed the “11/24/25” rule that states that the presence of positively charged amino acids at positions 11, 24, or 25 indicates X4 tropism whereas any other residue at these positions defines R5 viruses. Indeed, this rule has been shown to have 94% accuracy in predicting the coreceptor usage of 217 viruses.19
An interesting outcome of our studies is that replacement of the V3 region of the FIV SU by the equivalent domain of an X4-tropic HIV-1 gp120 results in a chimeric SU protein (SUHV3) that binds to CXCR4 with an efficiency similar to that of wild-type FIV SU.
The dependence on the presence of CXCR4 for the cell surface binding of SUHV3-HA was demonstrated by the experiments showing that the chimeric glycoprotein does not bind to MDCK cells that lack CXCR4 whereas transfection of this cell line with a plasmid encoding human CXCR4 allows cell surface association of the SUHV3-HA glycoprotein. Moreover, it should be stressed that we have clearly demonstrated that the HIV-1 V3 loop in the SUHV3-HA protein is essential for binding of this chimeric protein to CXCR4. Indeed, the association of SUHV3-HA with the surface of CXCR4-expressing cells is completely inhibited by the MAb directed to the V3 region of the HIV-1LAV gp120. Of note, the association of SUHV3-HA with CXCR4 is not inhibited by the CXCR4 antagonist AMD3100. This coreceptor inhibitor interacts with negatively charged amino acids in membrane-proximal domains of the ECLs.48,49 However, AMD3100 appears to act through an allosteric mechanism that modifies the conformational state of the chemokine receptor thereby preventing a functional interaction with HIV gp120.50 In this regard, resistance to AMD3100 is thought to result from an acquired ability of the gp120 glycoprotein to interact with conformationally modified versions of CXCR4 without switching to another chemokine receptor.50,51 In support of this notion, it has been shown that an X4-tropic HIV-1 that became resistant to AMD3100 in vitro exhibited several mutations that were not found in the wild-type virus.52 Analysis of the coding region for the gp120 of this AMD3100-resistant virus revealed the presence of eight amino acid substitutions: one in V2, four in V3, two in C4, and one in V5 that disrupted a potential N-glycosylation site.52 In addition, this gp120 had a deletion of four residues in V4 that also eliminated a glycosylation site.52
Based on this precedent, it is most likely that in the case of the SUHV3 the insertion of the V3 loop of the X4-tropic HIV-1 gp120 in the context of the FIV SU amino acid sequence confers to the chimeric glycoprotein the capacity to associate with CXCR4 molecules that have been previously treated with AMD3100. Although identification of the specific FIV SU residues that contribute to the AMD3100-resistant phenotype of SUHV3-HA is a laborious task, further studies of this chimeric glycoprotein may provide additional information regarding SU-CXCR4 interaction and its sensitivity to AMD3100.
In summary, we have confirmed using a CD134-independent and soluble FIV SU glycoprotein that the integrity of the V3 region is essential for CXCR4 binding and have demonstrated that substitution of the V3 loop of an X4-tropic HIV-1 gp120 for the FIV SU V3 region creates a chimeric glycoprotein that retains its full ability to interact with CXCR4 and is capable of promoting cell-to-cell fusion.
Acknowledgments
This work was supported by the National Agency for the Promotion of Science and Technology (ANPCyT, Argentina) grant 641 to J.L.A. S.A.G. and J.L.A. are career investigators of the National Research Council of Argentina (CONICET).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Pedersen NC, Ho EW, Brown ML, and Yamamoto JK: Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science 1987;235:790–793 [DOI] [PubMed] [Google Scholar]
- 2.Bukhard MJ. and Dean GA: Transmission and immunopathogenesis of FIV in cats as a model for HIV. Curr HIV Res 2003;1:15–29 [DOI] [PubMed] [Google Scholar]
- 3.Elder JH, Lin YC, Fink E, and Grant CK: Feline immunodeficiency virus (FIV) as a model for study of lentivirus infections: Parallels with HIV. Curr HIV Res 2010;8:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamamoto JK, Sanou MP, Abbott JR, and Coleman JK: Feline immunodeficiency virus model for designing HIV/AIDS vaccines. Curr HIV Res 2010;8:14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown WC, Bissey L, Logan KS, Pedersen NC, Elder JH, and Collisson EW: Feline immunodeficiency virus infects both CD4+and CD8+T lymphocytes. J Virol 1991;65:3359–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunner D. and Pedersen NC: Infection of peritoneal macrophages in vitro and in vivo with feline immunodeficiency virus. J Virol 1989;63:5483–5488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dean GA, Himathongkham S, and Sparger EE: Differential cell tropism of feline immunodeficiency virus molecular clones in vivo. J Virol 1999;73:2596–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dow SW, Mathiason CK, and Hoover EA: In vivo monocyte tropism of pathogenic feline immunodeficiency viruses. J Virol 1999;73:6852–6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.English RV, Johnson CM, Gebhard DH, and Tompkins MB: In vivo lymphocyte tropism of feline immunodeficiency virus. J Virol 1993;67:5175–5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Parseval A, Chatterji U, Sun P, and Elder JH: Feline immunodeficiency virus targets activated CD4+T cells by using CD134 as a binding receptor. Proc Natl Acad Sci USA 2004;101:13044–13049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimojima M, Miyasawa T, Ikeda Y, McMonagle EL, Haining H, Akashi H, Takeuchi Y, Hosie MJ, and Willett BJ: Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science 2004;303:1192–1195 [DOI] [PubMed] [Google Scholar]
- 12.Poeschla EM. and Looney DJ: CXCR4 is required by a nonprimate lentivirus: Heterologous expression of feline immunodeficiency virus in human, rodent, and feline cells. J Virol 1998;72:6858–6866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson J, Pancino G, Merat R, Leste-Lasserre T, Moraillon A, Schneider-Mergener J, Alizon M, Sonigo P, and Heveker N: Shared usage of the chemokine receptor CXCR4 by primary and laboratory-adapted strains of feline immunodeficiency virus. J Virol 1999;73:3661–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willett BJ, Picard L, Hosie MJ, Turner JD, Adema K, and Clapham PR: Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J Virol 1997;71:6407–6415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verschoor EJ, Hulskotte EGJ, Ederveen J, Koolen MJM, Horzinek MC, and Rottier PJM: Post-translational processing of the feline immunodeficiency virus envelope precursor protein. Virology 1993;193:433–438 [DOI] [PubMed] [Google Scholar]
- 16.Pancino G, Fossati I, Chappey C, Castelot S, Hurtrel B, Moraillon A, Klatzmann D, and Sonigo P: Structure and variations of feline immunodeficiency virus envelope glycoproteins. Virology 1993;192:659–662 [DOI] [PubMed] [Google Scholar]
- 17.Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, and Sodroski J: A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 1998;280:1949–1953 [DOI] [PubMed] [Google Scholar]
- 18.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, and Hendrickson WA: Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998;393:648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardozo T, Kimura T, Philppot S, Weiser B, Burger H, and Zolla-Pazner S: Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res Hum Retroviruses 2007;23:415–426 [DOI] [PubMed] [Google Scholar]
- 20.Babcock GJ, Mirzabekov T, Wojtowicz W, and Sodroski J: Ligand binding characteristics of CXCR4 incorporated into paramagnetic proteoliposomes. J Biol Chem 2001;276:38433–38440 [DOI] [PubMed] [Google Scholar]
- 21.Doranz BJ, Baik SS, and Doms RW: Use of a gp120 binding assay to dissect the requirements and kinetics of human immunodeficiency virus fusion events. J Virol 1999;73:10346–10358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin G, Baribaud F, Romano J, Doms RW, and Hoxie JA: Identification of gp120 binding sites on CXCR4 by using CD4-independent human immunodeficiency virus type 2 Env proteins. J Virol 2003;77:931–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Q.Y, Fink E, Hong Y, Wang C, Grant CK, and Elder JH: Fine definition of the CXCR4-binding region on the V3 loop of feline immunodeficiency virus surface glycoprotein. PLoS One 2010;5:e10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundstrom M, White RL, de Parseval A, Sastry KJ, Morris G, Grant CK, and Elder JH: Mapping of the CXCR4 binding site within variable region 3 of the feline immunodeficiency virus surface glycoprotein. J Virol 2008;82:9134–9142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Celma CCP, Paladino MG, González SA, and Affranchino JL: Importance of the short cytoplasmic domain of feline immunodeficiency virus transmembrane glycoprotein for fusion activity and envelope glycoprotein incorporation into virions. Virology 2007;366:405–414 [DOI] [PubMed] [Google Scholar]
- 26.Endres MJ, Clapham PR, Marsh M, Ahuja M, Davis Turner J, McKnight A, Thomas JF, Stoebenau-Haggarty B, Choe S, Vance PJ, Wells TNC, Power CA, Sutterwala SS, Doms RW, Landau NR, and Hoxie JA: CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell 1996;87:745–756 [DOI] [PubMed] [Google Scholar]
- 27.Chesebro B. and Wehrly K: Development of a sensitive quantitative focal assay for human immunodeficiency virus infectivity. J Virol 1988;62:3779–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olmsted RA, Barnes AK, Yamamoto JK, Hirsch VM, Purcell RH, and Johnson PR: Molecular cloning of feline immunodeficiency virus. Proc Natl Acad Sci USA 1989;86:2448–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, and Martin MA: Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 1986;59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rauddi ML, Mac Donald CL, Affranchino JL, and González SA: Mapping of the self-interaction domains in the simian immunodeficiency virus Gag polyprotein. AIDS Res Human Retroviruses 2011;27:303–316 [DOI] [PubMed] [Google Scholar]
- 31.Fuerst TR, Earl PL, and Moss B: Use of a hybrid vaccinia virus-T7 RNA polymerase system for expression of target genes. Mol Cell Biol 1987;7:2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.González SA, Paladino MG, and Affranchino JL: Palmitoylation of the feline immunodeficiency virus envelope glycoprotein and its effect on fusion activity and envelope incorporation into virions. Virology 2012;428:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edinger AI, Blanpain C, Kunstman KJ, Wolinsky SM, Parmentier M, and Doms RW: Functional dissection of CCR5 coreceptor function through the use of CD4-independent simian immunodeficiency virus strains. J Virol 1999;73:4062–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu QY, Fink E, Happer M, and Elder JH: Identification of amino acid residues important for heparan sulfate proteoglycan interaction within variable region 3 of the feline immunodeficiency virus surface glycoprotein. J Virol 2011;85:7108–7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng Y, Broder CC, Kennedy PE, and Berger EA: HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996;272:872–877 [DOI] [PubMed] [Google Scholar]
- 36.Cervantes-Acosta G, Welman M, Freund F, Cohen EA, and Lemay G: CD4/CXCR4 co-expression allows productive HIV-1 infection in canine kidney MDCK cells. Virus Res 2006;120:138–145 [DOI] [PubMed] [Google Scholar]
- 37.Józefowski S, Czerkies M, Sobota A, and Kwiatkowska K: Determination of cell surface expression of Toll-like receptor 4 by cellular enzyme-linked immunosorbent assay and radiolabeling. Anal Biochem 2011;413:185–191 [DOI] [PubMed] [Google Scholar]
- 38.Lourenço EV. and Roque-Barreira MC: Immunoenzymatic quantitative analysis of antigens expressed on the cell surface (cell-ELISA). Methods Mol Biol 2010;588:301–309 [DOI] [PubMed] [Google Scholar]
- 39.Donzella GA, Schols D, Lin SW, Este JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, de Clercq E, and Moore JP: AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med 1998;4:72–77 [DOI] [PubMed] [Google Scholar]
- 40.Baribaud F, Edwards TG, Sharron M, Brelot A, Heveker N, Price K, Mortari F, Alizon M, Tsang M, and Doms RW: Antigenically distinct conformations of CXCR4. J Virol 2001;75:8957–8967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimpton J. and Emerman E: Detection of replication competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J Virol 1992;66:2232–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Parseval A, Grant CK, Sastry KJ, and Elder JH: Sequential CD134-CXCR4 interactions in feline immunodeficiency virus (FIV): Soluble CD134 activates FIV Env for CXCR4-dependent entry and reveals a cryptic neutralization epitope. J Virol 2006;80:3088–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brelot A, Heveker N, Adema K, Hosie MJ, Willett B, and Alizon M: Effect of mutations in the second extracellular loop of CXCR4 on its utilization by human and feline immunodeficiency viruses. J Virol 1999;73:2576–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willett BJ, Adema K, Heveker N, Brelot A, Picard L, Alizon M, Turner JD, Hoxie JA, Peiper S, Neil JC, and Hosie MJ: The second extracellular loop of CXCR4 determines its function as a receptor for feline immunodeficiency virus. J Virol 1998;72:6475–6481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jong JJ, De Ronde A, Keulen W, Tersmette M, and Goudsmit J: Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: Analysis by single amino acid substitution. J Virol 1992;66:6777–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fouchier RAM, Groenink M, Kootstra NA, Tersmette M, Huisman HG, Midema F, and Schuitemaker H: Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J Virol 1992;66:3183–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Resch W, Hoffman N, and Swanstrom R: Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology 2001;288:51–62 [DOI] [PubMed] [Google Scholar]
- 48.Gerlach LO, Skerlj RT, Bridger GJ, and Schwartz TW: Molecular interactions of cyclam and bicyclam non-peptide antagonists with CXCR4 chemokine receptor. J Biol Chem 2001;276:14153–14160 [DOI] [PubMed] [Google Scholar]
- 49.Hatse S, Princen K, Gerlach LO, Bridger G, Henson G, de Clercq E, Schwartz TW, and Schols D: Mutation of Asp (171) and Asp (262) of the chemokine receptor CXCR4 impairs its coreceptor activity for human immunodeficiency virus 1 entry and abrogates the antagonistic activity of AMD3100. Mol Pharmacol 2001;60:164–173 [DOI] [PubMed] [Google Scholar]
- 50.Schols D: HIV co-receptors inhibitors as novel class of anti-HIV drugs. Antiviral Res 2006;71:216–226 [DOI] [PubMed] [Google Scholar]
- 51.Schols D, Este JA, Cabrera C, and de Clercq E: T-cell-line-tropic human immunodeficiency virus type 1 that is made resistant to stromal cell-derived factor 1 alpha contains mutations in the envelope gp120 but does not show a switch in coreceptor use. J Virol 1998;72:4032–4037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Vreese K, Kofler-Mongold V, Leutgeb C, Weber V, Vermeire K, Schacht S, Anné J, de Clercq E, Datema R, and Werner G: The molecular target of bicyclams, potent inhibitors of human immunodeficiency virus replication. J Virol 1996;70:689–696 [DOI] [PMC free article] [PubMed] [Google Scholar]