

Abstract
There has been an increasing interest to develop new types of stimuli-responsive drug delivery vehicles with high drug loading and controlled release properties for chemotherapeutics. An acid-labile, polyphosphoester-based degradable, polymeric paclitaxel (PTX) conjugate containing ultra-high levels of PTX loading has been improved significantly, in this second generation development, which involves connection of each PTX molecule to the polymer backbone via a pH-sensitive β-thiopropionate linkage. The results for this system indicate that it has great potential as an effective anti-cancer agent. Poly(ethylene oxide)-block-polyphosphoester-graft-PTX drug conjugate (PEO-b-PPE-g-PTX G2) was synthesized by organocatalyst-promoted ring-opening polymerization of 2-(but-3-en-1-yloxy)-1,3,2-dioxaphospholane-2-oxide from a PEO macroinitiator, followed by thermo-promoted thiolene click conjugation of a thiol-functionalized PTX prodrug to the pendant alkene groups of the block copolymer. The PEO-b-PPE-g-PTX G2 formed well-defined nanoparticles in aqueous solution, by direct dissolution into water, with a number-averaged hydrodynamic diameter of 114 ± 31 nm. The conjugate had PTX loading capacity as high as 53 wt%, and a maximum PTX concentration of 0.68 mg/mL in water (vs. 1.7 μg/mL for free PTX). Although the PTX concentration is ca. 10× less than for our first generation material, its accelerated release allowed for similar free PTX concentrations vs. time. The PEO-b-PPE-g-PTX G2 exhibited accelerated drug release under acidic conditions (~50 wt% PTX released in 8 d) compared to neutral conditions (~20 wt% PTX released in 8 d) and compared to the first generation analog that contained ester linkages between PTX and the polymer backbone (<5 wt% PTX released in 4 d), due to their acid-sensitive hydrolytically-labile β-thiopropionate linkages between PTX molecules and the polymer backbone. The positive cell-killing activity of PEO-b-PPE-g-PTX G2 against two cancer cell lines was demonstrated, and the presence of pendant reactive functionality provides a powerful platform for future work to involve conjugation of multiple numbers and/or types of targeting ligands, other drugs and imaging agents to achieve chemotherapy and bioimaging. Compared to our previously reported polyphosphoester-based PTX drug conjugates, PEO-b-PPE-g-PTX G1 without the β-thiopropionate linker, the PEO-b-PPE-g-PTX G2 showed pH-triggered drug release property and 5-to-8-fold enhanced in vitro cytotoxcity against two cancer cell lines.
Keywords: Polyphosphoester, pH-triggered release, polymer drug conjugates, paclitaxel, thiol-ene
1. Introduction
Created by the integration of nanotechnology with modern medicine, nanomedicines have been developed to possess unprecedented precision and efficacy.[1-8] Polymer-based nanomedicines have attracted an increasing interest in contemporary drug research because they have demonstrated the ability to serve as highly efficient drug delivery systems, in comparison with small molecule drugs alone.[9-17] Since Ringsdorf first proposed the concept of polymer-drug conjugates in the mid-1970s,[18] in which chemotherapeutic agents were covalently connected to a polymer, varieties of polymer-drug conjugate systems have been developed, and nearly a dozen of these systems have been advanced to different stages of clinical trials.[19-27] Compared to small molecule anticancer drugs, polymer-drug conjugates have shown several advantages, including, but not limited to, increased water solubility, decreased side effects, prolonged blood circulation time, and improved accumulation in tumors via the enhanced permeability and retention (EPR) effect and other targeting strategies.[28-31]
Paclitaxel (PTX), one of the most potent anticancer drugs widely used in breast and ovarian chemotherapies, is a particularly appropriate candidate for conjugation onto a polymer to form polymer-PTX conjugates to improve therapeutic efficacy.[32-35] Because the highly hydrophobic nature of PTX prevents intravenous administration of the drug, several studies were conducted to modify PTX to form water soluble prodrugs.[36, 37] Among the prodrugs developed, the C2' hydroxyl group of PTX is the most appropriate for derivatization, because it can be easily esterified without necessary protection of the C7 hydroxyl group.[38] During the past two decades, several polymer-PTX conjugates have been developed by the esterification of PTX C2'-OH with acid functionalities on polymers.[19-27] For instance, Greenwald et al. reported water-soluble poly(ethylene oxide) (PEO)-functionalized PTX drug conjugates with ~4 wt% PTX loading and increased PTX water solubility.[39] Ernsting et al. synthesized biocompatible carboxymethylcellulose-based docetaxel conjugates with a high (37 wt%) drug coupling that exhibited slow, controlled drug release kinetics.[40] Recently, Cheng's group developed well-defined brush polymer drug conjugates (BPDCs) via a grafting-through strategy by ring-opening metathesis copolymerization of norbornenefunctionalized PTX and PEO macromonomer.[41] These BPDCs had greatly improved PTX solubility (>10 mg/mL) and also achieved relatively high PTX loading (24 wt%). Among all reported polymer-PTX conjugates, poly(l-glutamic acid)-paclitaxel (PTX poliglumex, OPAXIO™, CT-2103, Xyotax®) is one of the most promising polymer-PTX conjugates, and has been advanced to Phase III clinical trials.[42-46] Although there have been significant advances, it still remains of broad interest to develop new water soluble and biocompatible polymer scaffolds that can serve as multi-functional platforms for PTX delivery, with triggered release and other functions.
Recently, our group developed a type of poly(ethylene oxide)-b-polyphosphoester (PEO-b-PPE)-based polymer drug conjugate, which is now defined as the first generation material, PEO-b-PPE-g-PTX G1, for PTX delivery (Scheme 1).[47] These highly water-soluble diblock copolymers contained alkyne functionalities on their polyphosphoester block that allowed for copper-catalyzed azide-alkyne cycloaddition (CuAAC) click conjugation of PTX onto the PPE segment of the block copolymer. Furthermore, these materials achieved ultra-high PTX loading capacity (up to 65 wt%), and after the spontaneous formation of amphiphilic nanoparticles, the PEO-b-PPE-g-PTX G1 with 55 wt% of PTX loading could achieve a maximum of 6.2 mg/mL PTX concentration in water. However, the release of free PTX from the PEO-b-PPE-g-PTX G1 polymer carrier required further optimization, because only 5 % of free PTX was detected by high-performance liquid chromatography (HPLC) 4 d after the incubation of PEO-b-PPE-g-PTX G1 nanoparticles in 20 mM acetate buffer at pH 6.0. This slow release accounted for the 8-to-63-fold lower cytotoxicity than the Taxol-mimicking formulation, as tested in OVCAR-3 and RAW 264.7 cell lines. The relative hydrolytic stability and added steric hindrance effects of the C2' PTX-ester packaged within the hydrophobic core of the polymer assemblies, which limits accessibility, each may have contributed to the slow release of PTX and the low cytotoxicity. Therefore, our recent efforts have focused on addressing the bioavailability of PTX, and, as reported here, its release from the polymer nanoparticles has been greatly improved by connection through an acid-sensitive hydrolytically-labile β-thiopropionate linkage.
Scheme 1.

The structure of polymer-PTX conjugates generation 1, with 55 wt% PTX loading giving the maximum PTX concentration packaged within block copolymer nanoparticles in water.
In order to achieve sustained and controlled PTX release from polymer carriers and further improve the cytotoxicity, incorporation of acid-labile linkages between the drug and the polymer scaffold was performed to enable the pH-triggered release of active drug from the carrier in the tumor tissue.[48-50] The mildly acidic pH in tumor tissues (pH ~ 6.8), as well as the acidic environments in the endosomal and lysosomal compartments of cancer cells (pH ~ 5-6) after endocytosis, provide ideal acid triggered drug release environments.[51, 52] Many polymeric anticancer drugs with pH-sensitive spacers bridging the drugs and polymers have demonstrated pH-dependent drug release profiles.[22, 53-55] Among all kinds of acid-labile linkages, the hydrazone bond and amide bond of a cis-aconityl containing linker have been widely used because of their facile, efficient and straightforward synthesis.[56-58] The structural complexity of PTX requires consideration in the design of synthetic routes to achieve acid-sensitive PTX prodrug conjugates without decrease of the therapeutic activity of PTX. In this paper, we report novel PEO-PPE-based polymeric PTX conjugates, PEO-b-PPE-g-PTX G2, with acid-sensitive β-thiopropionate linkages between PTX molecules and the polymer scaffold. Both the polymer backbone and the PTX prodrug are reported for the first time in this study, to the best of our knowledge. The polyphosphoesters with alkyne/alkene functionalities were synthesized based upon recently developed organocatalyst-promoted ring-opening polymerization (ROP).[59-61] The acid-labile PTX prodrug was obtained in high yield by a two-step organic synthesis. The pendant alkene functional groups on the polymer allowed for the efficient installation of thiol-functionalized PTX prodrug by click type thiolene reaction. The resulting PEO-b-PPE-g-PTX G2 assembled into well-defined nanoparticles with high drug loading (~53 wt%) and increased PTX water solubility (0.68 mg/mL vs. 1.7 μg/mL for free PTX). Three advantages of PEO-b-PPE-g-PTX G2 are demonstrated in this work, compared to our previously reported polymeric conjugate system: 1) The thermo-induced thiol-ene click conjugation eliminates the use of potentially toxic metal catalysts; 2) The acid labile linkage renders faster hydrolytic release kinetics of free PTX at acidic pH compared to neutral conditions; 3) The acid-labile linker promotes greater PTX release from assembled polymeric assemblies. As a result, the cell cytotoxicity studies of PEO-b-PPE-g-PTX G2 against two cell lines showed reduced IC50 values, compared to the previously reported PEO-b-PPE-g-PTX G1 system.
2. Results and Discussion
The synthesis of PEO-b-PPE-g-PTX G2 by the post-functionalization of PTX prodrug onto the polymer backbone via thiol-ene click reaction is illustrated in Scheme 2. PEO, 1 (Mn ~ 2.0 kDa), was used as initiator in the ROP of 2-(but-3-en-1-yloxy)-1,3,2-dioxaphospholane 2-oxide, 2, with 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) as organocatalyst. After carefully screening the ROP conditions by tuning the initiator-to-monomer feed ratio, monomer concentration, reaction time and catalyst amount, well-defined diblock copolymers were synthesized with narrow molecular weight distributions and controlled molecular weights.[47, 59, 60] The diblock copolymer PEO45-b-PPE27, 3, was synthesized within 10 min at room temperature, purified by thrice precipitation from dichloromethane to ether, and dried into the form of a viscous liquid. The theoretical molecular weight calculated from the initiator-to-monomer feed ratio (1:2 = 1:30) and the monomer conversion (90%, obtained from 31P NMR spectroscopy) was in good agreement with the molecular weight (Mn = 6.5 kDa) determined by 1H NMR spectroscopy of the purified polymer product. The narrow molecular weight distribution was confirmed by gel permeation chromatography (GPC) (Mw/Mn = 1.16), which also revealed Mn = 16.8 kDa based on polystyrene standards (Figure 1). The full characterization of 3 is detailed in the supporting information.
Scheme 2.

Synthesis of PEO-b-PPE-g-PTX G2 6.
Figure 1.
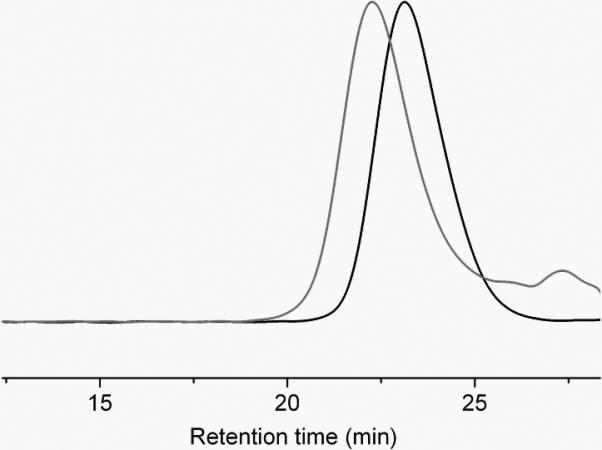
GPC traces of diblock copolymer 3 (black curve) and PEO-b-PPE-g-PTX G2 6 (gray curve).
To develop appropriate acid-labile PTX prodrug with a functional group, which could be further easily conjugated onto the alkene side chains of PPE polymer block, a β-thiopropionate linkage was installed within the PTX prodrug 5. The β-thiopropionate, a recently developed acid-sensitive covalent linker, has been employed in stimuli responsive micelles[62] and several biomedical materials.[11, 63, 64] The formation of a six-membered ring intermediate via intramolecular hydrogen bonding under acidic conditions, allows for rapid hydrolysis of the ester group of the linkage. The commercially-available 3,3'-disulfanediyldipropanoic acid was selected as the starting material to react with an excess amount of PTX and form diester 4 specifically at the C2' of PTX, because the most accessible hydroxyl group on C2' could be selectively esterified by using N,N'-dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) as condensation reagents. Automated high performance flash chromatography with prepacked fine spherical silica gel (20-40 μm) was used to purify 4 in 64% yield and a high purity. The selective esterification on C2' of PTX and the functionalization of 3,3'-disulfanediyldipropanoic acid at both ends were confirmed by 1H NMR spectroscopy (Figure 2). The downfield shift of the resonance frequency for the methine proton of C2′(CH) on PTX from 4.78 ppm to 5.53 ppm, together with the disappearance of the C2′-OH signal at 3.61 ppm, were evidence for its esterification. In contrast, proton resonances of C7(CH) at 4.40 ppm did not undergo change, indicating that the C7-OH was not esterified. The chemical shift at 2.70~2.85 ppm corresponded to the methylene protons adjacent to the disulfide bond (labeled as protons a in the structure of Figure 2) plus two protons of PTX (C6a(CH) and C7-OH). Comparison of the integration value for the one proton of PTX, C2′(CH) resonating at 5.53 ppm with the signal at 2.70~2.85 ppm, gave a ratio of 1 : 4, consistent with the stoichiometry of diester formation. The redox reagent dithiothreitol (DTT) was used to break the disulfide bond of 4, forming 5 in 72% yield.
Figure 2.
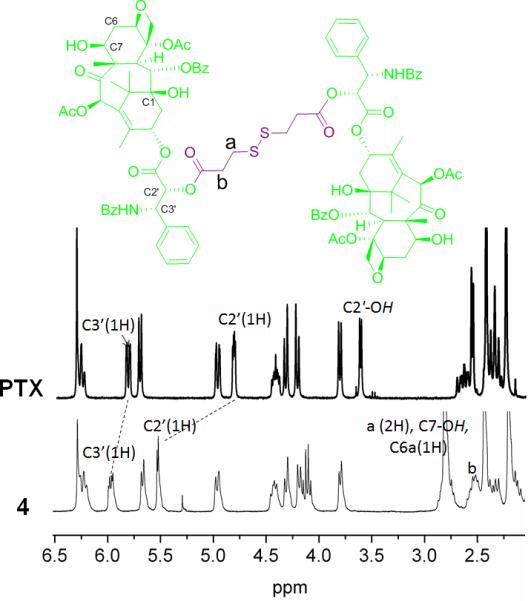
1H NMR spectra of PTX and 4.
The radical-mediated thiol-ene click reaction was employed to conjugate 5 onto the backbone of 3 to afford PEO-b-PPE-g-PTX G2 6. The conjugation of expensive drug moieties onto the polymer requires highly efficient reactions with minimal side reactions. For the efficient conjugation of highly sterically hindered 5 onto the polymer, thiol-ene click conditions were screened. UV irradiation-promoted thiol-ene click using photoinitiator 2,2-dimethoxy-2-phenylacetophenone (DMPA) was initially attempted. However, even with a high feed ratio of 20 equivalents of 5 to 1 equivalent of 3, and long UV irradiation time of >2 h, the conjugation efficiency reached <10% and by-products were observed. By using azobisisobutyronitrile (AIBN) as radical initiator, the thermo-promoted thiol-ene reaction was conducted at 60 °C for 16 h in N,N-dimethylformamide (DMF), to give high conjugation efficiency of 50% of 5 conjugated onto the polymer when the feeding ratio of 5 to 3 was 20 to 1. The resulting polymer-PTX conjugate 6 had PTX loading capacity of 53 wt% with relatively high effectiveness for PTX solubility in water (0.68 mg/mL), which was 400-fold higher than free PTX. Unreacted 5 was removed by repeated precipitation from DMF into diethyl ether three times, because PTX and 5 were well soluble in diethyl ether. Complete removal of 5 was confirmed by 1H NMR spectroscopy and HPLC analysis. The supernatant of the third precipitation showed no PTX signals, as confirmed by 1H NMR spectroscopy and HPLC. The HPLC analysis of 6 also indicated that no unreacted 5 remained in the polymer. The GPC trace of 6 shows the formation of well-defined polymer-drug conjugates (Mn = 26.6 kDa, Mw/Mn = 1.14) with obvious shorter retention time compared with the polymer precursor 3 (Figure 1).
The supramolecular assembly of polymer drug conjugate 6 was achieved by direct dissolution of dry polymer powder into nanopure water. Taking advantage of the high hydrophilicity of the polyphosphoester backbone, the direct dissolution self-assembly method provided an efficient way to construct well-defined nanoassemblies. Certain amounts of amphiphilic 6 were suspended into nanopure water (18 mΩ·cm) and then sonicated for 2 min followed by 2 h stirring at room temperature (r.t.). The purification was conducted by passing the solution through a 450 nm polypropylene filter to remove dust and large aggregates. The nanoparticle solution (concentration of 6 = 1.28 mg/mL) had a maximum PTX solubility at 0.68 mg/mL, and was stable at r.t. for one week without any observable precipitation. Dynamic light scattering (DLS) analysis showed the number-averaged hydrodynamic diameter (Dh)n of the nanoparticles to be 114 ± 31 nm (Figure 3). Transmission electron microscopy (TEM) images confirmed that the PEO-b-PPE-g-PTX G2 nanoassemblies were well-dispersed in water in the form of core-shell assemblies with Dav = 139 ± 25 nm. Zeta potential analysis indicated a negatively-charged feature of the polymeric assemblies (-17.1 mV), which is in accordance with the characteristic of typical polyphosphoester-based nanomaterials.[65, 66]
Figure 3.

(a) TEM image, (b) zeta potential Lorentzian peak and top view of frequency distribution across the cell during electrophoretic light scattering measurement, (c) DLS size distribution profile of polymeric assemblies of 6.
The acid-triggered hydrolytic PTX release from 6 (PEO-b-PPE-g-PTX G2) was analyzed by HPLC. Our recently published PEO-b-PPE-g-PTX G1 polymeric assemblies presented slow PTX release kinetics having only 5 wt% of PTX release after 4 d incubation in 20 mM acetate buffer at pH 6.0 (supporting information Figure S2). The low release of PTX was attributed to a combination of the inaccessibility of C-2′ PTX-ester in the densely loaded hydrophobic cores of polymeric assemblies and the relative stability of the ester linkage. In comparison, the PEO-b-PPE-g-PTX G2 with an acid-labile β-thiopropionate linkage between polymer and drug moieties allows for accelerated cleavage of PTX (Scheme 3). The studies of hydrolytic release of PTX from PEO-b-PPE-g-PTX G2 nanoparticles were conducted under sink conditions. The successful separation of PEO-b-PPE-g-PTX G2, PTX and PTX prodrugs by reverse phase HPLC was achieved with an eluent combination of 55% water and 45% acetonitrile. The PEO-b-PPE-g-PTX G2 had a retention time of ~3 min, while the prodrugs 5 and 4 and pure PTX had elution times of 7.9 min, 17.6 min and 9.3 min, respectively. The calibration curve of PTX for HPLC indicated that the method could measure the PTX concentration as low as 6.25 ng/mL (supporting information Figure S1). The solubilities of PTX in 10 mM PBS buffer at pH 7.4 and 5.5 were determined by HPLC to be 1.7 μg/mL and 1.8 μg/mL, respectively. The release of PTX from polymeric assemblies of 6 was first measured by a dialysis method, in which 1 mL micelle 6 solution (0.1 mg/mL) was injected into a dialysis cassette having a membrane molecular weight cut-off (MWCO) of 8000 Da. The dialysis cassette was submerged into 100 mL of 10 mM PBS buffer at pH 7.4 and stirred at 37 °C. In order to analyze the concentration of released PTX, 0.1 mL of released medium outside the cassette was taken out at different time points. However, during the long release period, precipitation in the dialysis cassette was observed which prevented an accurate measurement of released PTX. Therefore, direct dissolution of 6 (1.0 μg/mL) in 10 mM PBS buffer pH = 7.4 and pH = 5.5, respectively, without dialysis was conducted at 37 °C. As shown in Figure 4, the release of PTX from 6 was time and pH dependent. Under neutral conditions (pH = 7.4), the release of PTX was relatively slow, only 25 wt% of drug was released after 8 d. Due to the acid-sensitive linker, the PTX release was enhanced at acidic condition (pH = 5.5), and over 50 wt% of free PTX was detected in the release medium after 8 d, nearly twice as much PTX released than that under neutral conditions. The acid-labile polymer PTX conjugates provided for sustained PTX release kinetics without burst release effect, which is usually observed in drug delivery systems using physical loading methods.
Scheme 3.
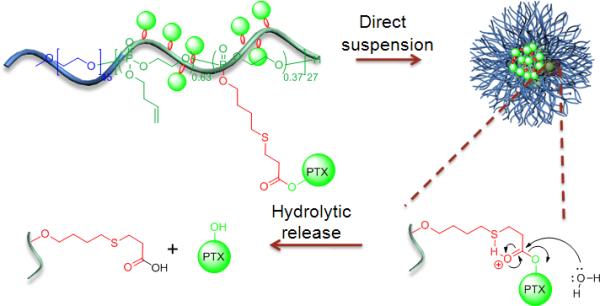
Self assembly of nanoparticles of 6 and acid-sensitive hydrolytic release of PTX.
Figure 4.

The hydrolytic release profile of PTX from assemblies of PEO-b-PPE-g-PTX G2, evaluated in PBS buffer at pH values of 5.0 and 7.4.
The in vitro cytotoxic effect of PEO-b-PPE-g-PTX G2 against two cell lines was studied. The IC50 values were measured after incubating the formulations for 72 h at 37 °C and compared against that of PTX in Cremophor-EL/ethanol (1:1 v/v) (PTX free drug) and the control polymer 3, which had the same polymer backbone as 6. The control polymer 3 was not cytotoxic over the range of tested concentrations (33.5 ng/mL - 33.5 μg/mL). As shown in Table 1, the polymeric-PTX conjugates 6 had IC50 values (PTX moieties concentrations) of 0.022 μM against OVCAR-3 and 0.38 μM against RAW 264.7 cell lines. PEO-b-PPE-g-PTX G2 showed 3-to-10-fold lower cytotoxic than the PTX free drug. Compared with the PEO-b-(PPE-g-PTX) G1, PEO-b-PPE-g-PTX G2 had 5-to-8-fold enhanced cytotoxicity. The improved in vitro cytotoxicities of PEO-b-PPE-g-PTX G2 to PEO-b-PPE-g-PTX G1 might be due to faster PTX release kinetics. By introducing the β-thiopropionate linkage, PTX molecules were released in a pH controlled manner. The lower cytotoxicity of PEO-b-PPE-g-PTX G2 compared to free PTX might provide better pharmacokinetics in vivo and, by controlling the chemistry of the backbone and conjugation, the release rate and cytotoxicity of nanoparticles utilized for the delivery of chemotherapeutics can be tailored.
Table 1.
Comparison of the IC50 values of PTX (as a Taxol®-mimicking formulation; Cremophor-EL and ethanol, 1:1 v/v), PEO-b-PPE-g-PTX G1 and PEO-b-PPE-g-PTX G2 6 in OVCAR-3 and RAW 264.7 cells incubated for 72 h.
| Formulation | IC5o(μM) | |
|---|---|---|
| OVCAR-3 | RAW 264.7 | |
| PTX | 0.007 | 0.04 |
| PEO-b -(PPE-g-PTX) G1 | 0.119 | 2.83 |
| PEO-b -(PPE-g-PTX) G2 6 | 0.022 | 0.38 |
3. Conclusions
We have designed and synthesized an advanced type of amphiphilic polyphosphoester-based polymeric drug conjugate with acid-labile linkages between the polymer backbone and the drug moieties, which undergoes supramolecular assembly into well-defined nanoscopic PTX-loaded polymer drug delivery agents. Organocatalyst-promoted ring-opening polymerization of an alkenyl-functionalized phospholane monomer from a PEO initiator provided a well-defined highly water soluble PEO-b-polyphosphoester backbone with pendant alkene functionalities for further post modification along one segment of the block copolymer. By using thermo-induced thiol-ene click chemistry, a thiol-functionalized PTX prodrug was reacted with the alkene groups on the polyphosphoester side chains and yielded the PTX polymer drug conjugates with 53 wt% PTX loading, covalently through a β-thiopropionate linkage. The combination of organocatalyzed ROP and metal-free thiol-ene reaction diminished the metal induced toxicity in biomedical use, while also having the potential for the rapid construction of drug-conjugated polymeric nanomaterials. Compared to the reported PEO-b-PPE-g-PTX G1 polymeric PTX delivery system, the PEO-b-PPE-g-PTX G2 with β-thiopropionate linkage released more PTX over a given time period and exhibited acid-promoted PTX release kinetics. The increased cytotoxicity against two cell lines, compared to PEO-b-PPE-g-PTX G1, demonstrated the advantages of PEO-b-PPE-g-PTX G2 in vitro. The accelerated PTX release profile at acidic pH provides for a promising application in chemotherapy. The PEO-b-PPE-g-PTX G2 conjugates are expected to provide stability during blood circulation to accumulate in tumor tissues via the EPR effect. Moreover, the residual alkenyl functionalities offer opportunities for conjugation of imaging labels, other drugs, etc., to advance toward theranostic systems. The high concentration of polymeric assemblies and the accelerated release in acidic tumor tissues might be advantageous for in vivo applications. With the promising initial in vitro results, in vivo evaluation of these complex nanoscopic drug-polymerconjugates is our next major challenge.
Supplementary Material
Acknowledgements
We gratefully acknowledge financial support from the Covidien Inc., the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C) and the National Science Foundation under grant numbers DMR-0906815 and DMR-1105304. The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry, Grant No. A-0001.
Footnotes
Supporting Information
Supporting Information is available online from the Wiley Online Library or from the author.
Contributor Information
Jiong Zou, Departments of Chemistry and Chemical Engineering, Laboratory for Synthetic-Biologic Interactions, Texas A&M University, P.O. Box 30012, 3255 TAMU, College Station, TX 77842, USA.
Fuwu Zhang, Departments of Chemistry and Chemical Engineering, Laboratory for Synthetic-Biologic Interactions, Texas A&M University, P.O. Box 30012, 3255 TAMU, College Station, TX 77842, USA.
Shiyi Zhang, Departments of Chemistry and Chemical Engineering, Laboratory for Synthetic-Biologic Interactions, Texas A&M University, P.O. Box 30012, 3255 TAMU, College Station, TX 77842, USA.
Stephanie F. Pollack, Departments of Chemistry and Chemical Engineering, Laboratory for Synthetic-Biologic Interactions, Texas A&M University, P.O. Box 30012, 3255 TAMU, College Station, TX 77842, USA
Mahmoud Elsabahy, Departments of Chemistry and Chemical Engineering, Laboratory for Synthetic-Biologic Interactions, Texas A&M University, P.O. Box 30012, 3255 TAMU, College Station, TX 77842, USA; Department of Pharmaceutics, Faculty of Pharmacy, Assiut University, Assiut, Egypt.
Jingwei Fan, Departments of Chemistry and Chemical Engineering, Laboratory for Synthetic-Biologic Interactions, Texas A&M University, P.O. Box 30012, 3255 TAMU, College Station, TX 77842, USA.
Karen L. Wooley, Departments of Chemistry and Chemical Engineering, Laboratory for Synthetic-Biologic Interactions, Texas A&M University, P.O. Box 30012, 3255 TAMU, College Station, TX 77842, USA.
References
- 1.Zhang LF, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. Acs Nano. 2008;2:1696. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Clin. Pharmacol. Ther. 2008;83:761. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 3.Wagner V, Dullaart A, Bock AK, Zweck A. Nat. Biotechnol. 2006;24:1211. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- 4.Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Angew. Chem. Int. Ed. 2009;48:872. doi: 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganta S, Devalapally H, Shahiwala A, Amiji M. J. Control. Release. 2008;126:187. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 6.Naahidi S, Jafari M, Edalat F, Raymond K, Khademhosseini A, Chen P. J. Control. Release. 2013;166:182. doi: 10.1016/j.jconrel.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Bauer M, Schroeder S, Tauhardt L, Kempe K, Schubert US, Fischer D. J. Polym. Sci. Pol. Chem. 2013;51:1816. [Google Scholar]
- 8.Chen CK, Law WC, Aalinkeel R, Nair B, Kopwitthaya A, Mahajan SD, Reynolds JL, Zou J, Schwartz SA, Prasad PN, Cheng C. Adv. Healthcare Mater. 2012;1:751. doi: 10.1002/adhm.201200094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elsabahy M, Wooley KL. Chem. Soc. Rev. 2012;41:2545. doi: 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elsabahy M, Wooley KL. J. Polym. Sci. Pol. Chem. 2012;50:1869. doi: 10.1002/pola.25955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee Y, Kataoka K. Soft Matter. 2009;5:3810. [Google Scholar]
- 12.Duncan R, Izzo L. Adv. Drug. Deliver. Rev. 2005;57:2215. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 13.Tong R, Cheng JJ. Polym. Rev. 2007;47:345. [Google Scholar]
- 14.Matsumura Y, Kataoka K. Cancer Sci. 2009;100:572. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurtoglu YE, Mishra MK, Kannan S, Kannan RM. Int J Pharm. 2010;384:189. doi: 10.1016/j.ijpharm.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 16.Regnier-Delplace C, du Boullay OT, Siepmann F, Martin-Vaca B, Demonchaux P, Jentzer O, Danede F, Descamps M, Siepmann J, Bourissou D. J. Control. Release. 2013;166:256. doi: 10.1016/j.jconrel.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Gao GH, Lee DS. Adv. Healthc. Mater. 2013;2:388. doi: 10.1002/adhm.201200313. [DOI] [PubMed] [Google Scholar]
- 18.Ringsdorf H. J. Polym. Sci. Pol. Sym. 1975:135. [Google Scholar]
- 19.Li C, Wallace S. Adv. Drug. Deliver. Rev. 2008;60:886. doi: 10.1016/j.addr.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greco F, Vicent MJ. Adv. Drug. Deliver. Rev. 2009;61:1203. doi: 10.1016/j.addr.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Duncan R. Nat. Rev. Cancer. 2006;6:688. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 22.Ulbrich K, Subr V. Adv. Drug. Deliver. Rev. 2004;56:1023. doi: 10.1016/j.addr.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 23.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Frechet JMJ, Dy EE, Szoka FC. Proc. Natl. Acad. Sci. USA. 2006;103:16649. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markovsky E, Baabur-Cohen H, Eldar-Boock A, Omer L, Tiram G, Ferber S, Ofek P, Polyak D, Scomparin A, Satchi-Fainaro R. J. Control. Release. 2012;161:446. doi: 10.1016/j.jconrel.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 25.Tong R, Cheng JJ. J. Am. Chem. Soc. 2009;131:4744. doi: 10.1021/ja8084675. [DOI] [PubMed] [Google Scholar]
- 26.Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Poyner R, Boivin C, Hesslewood S, Twelves C, Blackie R, Schatzlein A, Jodrell D, Bissett D, Calvert H, Lind M, Robbins A, Burtles S, Duncan R, Cassidy J. Int. J. Oncol. 2009;34:1629. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- 27.Yurkovetskiy AV, Fram RJ. Adv. Drug. Deliver. Rev. 2009;61:1193. doi: 10.1016/j.addr.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 28.Maeda H. Adv. Enzyme Regul. 2001;41:189. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 29.Seymour LW, Miyamoto Y, Maeda H, Brereton M, Strohalm J, Ulbrich K, Duncan R. Eur. J. Cancer. 1995;31A:766. doi: 10.1016/0959-8049(94)00514-6. [DOI] [PubMed] [Google Scholar]
- 30.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J. Control. Release. 2000;65:271. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 31.Taurin S, Nehoff H, Greish K. J. Control. Release. 2012;164:265. doi: 10.1016/j.jconrel.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 32.Khandare JJ, Jayant S, Singh A, Chandna P, Wang Y, Vorsa N, Minko T. Bioconjugate Chem. 2006;17:1464. doi: 10.1021/bc060240p. [DOI] [PubMed] [Google Scholar]
- 33.Majoros IJ, Myc A, Thomas T, Mehta CB, Baker JR. Biomacromolecules. 2006;7:572. doi: 10.1021/bm0506142. [DOI] [PubMed] [Google Scholar]
- 34.Yu Y, Zou J, Yu L, Jo W, Li YK, Law WC, Cheng C. Macromolecules. 2011;44:4793. [Google Scholar]
- 35.Yu Y, Chen CK, Law WC, Mok J, Zou J, Prasad PN, Cheng C. Mol. Pharmaceut. 2013;10:867. doi: 10.1021/mp3004868. [DOI] [PubMed] [Google Scholar]
- 36.Skwarczynski M, Hayashi Y, Kiso Y. J. Med. Chem. 2006;49:7253. doi: 10.1021/jm0602155. [DOI] [PubMed] [Google Scholar]
- 37.Kingston DGI. J. Nat. Prod. 2000;63:726. doi: 10.1021/np000064n. [DOI] [PubMed] [Google Scholar]
- 38.Damen EWP, Wiegerinck PHG, Braamer L, Sperling D, de Vos D, Scheeren HW. Bioorg. Med. Chem. 2000;8:427. doi: 10.1016/s0968-0896(99)00301-6. [DOI] [PubMed] [Google Scholar]
- 39.Greenwald RB, Gilbert CW, Pendri A, Conover CD, Xia J, Martinez A. J. Med. Chem. 1996;39:424. doi: 10.1021/jm950475e. [DOI] [PubMed] [Google Scholar]
- 40.Ernsting MJ, Tang WL, MacCallum N, Li SD. Bioconjugate Chem. 2011;22:2474. doi: 10.1021/bc200284b. [DOI] [PubMed] [Google Scholar]
- 41.Zou J, Jafr G, Themistou E, Yap Y, Wintrob ZAP, Alexandridis P, Ceacareanu AC, Cheng C. Chem. Commun. 2011;47:4493. doi: 10.1039/c0cc05531j. [DOI] [PubMed] [Google Scholar]
- 42.Ke S, Milas L, Charnsangavej C, Wallace S, Li C. J. Control. Release. 2001;74:237. doi: 10.1016/s0168-3659(01)00322-4. [DOI] [PubMed] [Google Scholar]
- 43.Eldar-Boock A, Miller K, Sanchis J, Lupu R, Vicent MJ, Satchi-Fainaro R. Biomaterials. 2011;32:3862. doi: 10.1016/j.biomaterials.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van S, Das SK, Wang XH, Feng ZL, Jin Y, Hou Z, Chen F, Pham A, Jiang N, Howell SB, Yu L. Int. J. Nanomedicine. 2010;5:825. doi: 10.2147/IJN.S13482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beer TM, Ryan C, Alumkal J, Ryan CW, Sun J, Eilers KM. Anti-Cancer Drugs. 2010;21:433. doi: 10.1097/CAD.0b013e3283355211. [DOI] [PubMed] [Google Scholar]
- 46.Sabbatini P, Sill MW, O'Malley D, Adler L, Secord AA. Gynecol. Oncol. 2008;111:455. doi: 10.1016/j.ygyno.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 47.Zhang SY, Zou J, Elsabahy M, Karwa A, Li A, Moore DA, Dorshow RB, Wooley KL. Chem. Sci. ASAP; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen XJ, Parelkar SS, Henchey E, Schneider S, Emrick T. Bioconjugate Chem. 2012;23:1753. doi: 10.1021/bc200667s. [DOI] [PubMed] [Google Scholar]
- 49.Bae Y, Fukushima S, Harada A, Kataoka K. Angew. Chem. Int. Ed. 2003;42:4640. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]
- 50.She WC, Luo K, Zhang CY, Wang G, Geng YY, Li L, He B, Gu ZW. Biomaterials. 2013;34:1613. doi: 10.1016/j.biomaterials.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Tannock IF, Rotin D. Cancer Res. 1989;49:4373. [PubMed] [Google Scholar]
- 52.Engin K, Leeper DB, Cater JR, Thistlethwaite AJ, Tupchong L, Mcfarlane JD. Int. J. Hyperther. 1995;11:211. doi: 10.3109/02656739509022457. [DOI] [PubMed] [Google Scholar]
- 53.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong SQ. Biomaterials. 2009;30:5757. doi: 10.1016/j.biomaterials.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 54.Su J, Chen F, Cryns VL, Messersmith PB. J. Am. Chem. Soc. 2011;133:11850. doi: 10.1021/ja203077x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee ES, Gao ZG, Bae YH. J. Control. Release. 2008;132:164. doi: 10.1016/j.jconrel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan L, Chen WL, Li J, Hu JH, Yan JJ, Yang D. J. Polym. Sci. Pol. Chem. 2012;50:4579. [Google Scholar]
- 57.Zhu SJ, Hong MH, Tang GT, Qian LL, Lin JY, Jiang YY, Pei YY. Biomaterials. 2010;31:1360. doi: 10.1016/j.biomaterials.2009.10.044. [DOI] [PubMed] [Google Scholar]
- 58.Zhu SJ, Hong MH, Zhang LH, Tang GT, Jiang YY, Pei YY. Pharmaceut. Res. 2010;27:2030. doi: 10.1007/s11095-010-0217-4. [DOI] [PubMed] [Google Scholar]
- 59.Zhang SY, Li A, Zou J, Lin LY, Wooley KL. Acs Macro Lett. 2012;1:328. doi: 10.1021/mz200226m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang SY, Zou J, Zhang FW, Elsabahy M, Felder SE, Zhu JH, Pochan DJ, Wooley KL. J. Am. Chem. Soc. 2012;134:18467. doi: 10.1021/ja309037m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clement B, Grignard B, Koole L, Jerome C, Lecomte P. Macromolecules. 2012;45:4476. [Google Scholar]
- 62.Dan K, Pan R, Ghosh S. Langmuir. 2011;27:612. doi: 10.1021/la104445h. [DOI] [PubMed] [Google Scholar]
- 63.Oishi M, Sasaki S, Nagasaki Y, Kataoka K. Biomacromolecules. 2003;4:1426. doi: 10.1021/bm034164u. [DOI] [PubMed] [Google Scholar]
- 64.Oishi M, Nagasaki Y, Itaka K, Nishiyama N, Kataoka K. J. Am. Chem. Soc. 2005;127:1624. doi: 10.1021/ja044941d. [DOI] [PubMed] [Google Scholar]
- 65.Du JZ, Du XJ, Mao CQ, Wang J. J. Am. Chem. Soc. 2011;133:17560. doi: 10.1021/ja207150n. [DOI] [PubMed] [Google Scholar]
- 66.Xiong MH, Bao Y, Yang XZ, Wang YC, Sun BL, Wang J. J. Am. Chem. Soc. 2012;134:4355. doi: 10.1021/ja211279u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.