

Abstract
Hypertriglyceridemia (HTG) is a well established but underestimated cause of acute (AP) and recurrent AP (RAP). The clinical presentation of HTG-induced pancreatitis (HTG pancreatitis) is similar to other causes. Pancreatitis secondary to HTG is typically seen in the presence of one or more secondary factors (uncontrolled diabetes, alcoholism, medications, pregnancy) in a patient with an underlying common genetic abnormality of lipoprotein metabolism (Familial combined hyperlipidemia or Familial HTG). Less commonly, a patient with rare genetic abnormality (Familial chylomicronemic syndrome) with or without an additional secondary factor is encountered. The risk of AP in patients with serum triglycerides >1000 mg/dl and >2000 mg/dl is ∼5% and 10-20% respectively. It is not clear whether HTG pancreatitis is more severe than when it is due to other causes. Clinical management of HTG pancreatitis is similar to that of other causes. Insulin infusion in diabetic patients with HTG can rapidly reduce triglyceride levels. Use of apheresis is still experimental and better designed studies are needed to clarify its role in management of HTG pancreatitis. Diet, lifestyle changes, control of secondary factors are key to the treatment and medications are useful adjuncts to long term management of triglyceride levels. Control of triglyceride levels to well below 500 mg/dl can effectively prevent recurrences of pancreatitis.
Keywords: Hyperlipidemia, pancreatitis, hypertriglyceridemia
Introduction
Hypertriglyceridemia (HTG) is a well established cause of acute (AP) and recurrent acute pancreatitis (RAP). A recognition of HTG as a cause or contributing factor for AP is often delayed or completely missed. Patients with HTG-induced pancreatitis (HTG pancreatitis) often have recurrent attacks which may require repeated hospitalizations. Optimum control of serum triglyceride (TG) levels can prevent recurrences of pancreatitis.
In a review on this topic about ten years ago1, we discussed issues that clinicians face in the diagnosis and management of patients with HTG pancreatitis. Since then, new data has emerged on distribution of HTG and other pancreatitis risk factors in the general population, and prevalence of pancreatitis in patients with severe HTG. Furthermore, clinical and experimental studies have evaluated the role of HTG in the severity of AP and the mechanisms by which free fatty acids (FFA) may cause pancreatic injury. In this update, we incorporate recent data in discussing clinical questions relevant to the practicing physicians, define disease risk and review potential mechanisms for HTG pancreatitis.
Question 1: In what clinical scenarios should I suspect HTG pancreatitis?
HTG can be difficult to establish as the etiology of AP since mild to moderate elevations in serum TG are seen in up to one third of all cases of AP, regardless of etiology2,3. Mild elevation in TG in the setting of AP appears to be an epiphenomenon without an established association with pathogenesis or severity. Conversely, markedly elevated levels of TG are an uncommon but well-established cause of AP, seen in 2-4% cases4,5.
In a patient presenting with AP, HTG as the potential etiology should be suspected in the following scenarios with decreasing frequency: poorly controlled diabetes (established or new diagnosis) in the absence of other risk factors like gallstones, significant alcohol consumption or medications; an alcoholic patient who has very high TG levels; the use of medications known to cause HTG; and, during the third trimester of pregnancy. The common theme in these scenarios is that presence of secondary factor(s) in a patient with an underlying common genetic abnormality of lipoprotein metabolism (Familial combined hyperlipidemia [FCHL] or Familial HTG) can result in elevations of serum TG to levels that can cause pancreatitis. Less commonly, a patient with a genetic abnormality (Familial chylomicronemic syndrome) will present with HTG pancreatitis spontaneously or in the presence of a secondary factor. Detection of lactascent or lipemic serum, physical examination findings such as eruptive xanthomas or lipemia retinalis, or features of metabolic syndrome (e.g. abdominal obesity, premature coronary artery disease, nonalcoholic fatty liver disease, etc.) should raise suspicion for underlying HTG.
Question 2: How should we define HTG and how prevalent is it in the general population?
In its recent clinical practice guidelines6, the Endocrine Society has proposed the following criteria to classify serum TG levels: normal (<150 mg/dl) [1 mmol = 88.5736 mg/dl]; mild HTG (150-199 mg/dl), moderate HTG (200-999 mg/dl); severe HTG (1000-1999 mg/dl); and, very severe HTG (≥2000 mg/dl). These definitions differ from the NCEP ATP III criteria7 to recognize the risk of pancreatitis associated with severe and very severe HTG.
The NHANES study has longitudinally evaluated distributions of serum lipids in a representative sample of US adults. Using NHANES data from 2001-2006, Christian et al8 found serum TG levels to be 150-200 mg/dl, 200-500 mg/dl and 500-2000 mg/dl in 14.2%, 16.3% and 1.7% US adults respectively. Unfortunately, the latter group (500-2000 mg/dl) was not subdivided to determine the prevalence of TG >1000 mg/dl. Very severe HTG (i.e. >2000 mg/dl) was seen in only 3/5680 (0.0005%) individuals and they were excluded from further analyses. Subjects with TG over 500 mg/dl were more likely to be men, of middle age and have a higher prevalence of diabetes, chronic renal disease and concurrent abnormalities in other serum lipids (high non-HDL and low HDL).
Question 3: What is the risk of pancreatitis with HTG?
The absolute risk of pancreatitis based on serum TG has not been clearly defined. A recent population based Swedish study of 33,260 adults evaluated the risk of AP based on baseline serum TG levels after controlling for demographic factors and lifestyle habits during a 25 year follow up9. TG elevation – any, >200 mg/dl, >500 mg/dl and >1000 mg/dl was noted in about 25%, 10%, 4% and 0.05% individuals, respectively. Although no threshold could be determined, a significant trend for an increase in the risk of AP was observed from the lowest to the highest quantile, with the highest quantile showing statistical significance. AP risk was limited to patients with a clinical history suggestive of non-obstructive, non-alcoholic AP (i.e. idiopathic AP). A limitation of this study is that TG estimation at one time point was correlated with outcome that occurred several years later.
Three recent retrospective studies used prevalence of pancreatitis as a surrogate for lifetime risk in patients with severe/very severe HTG10-12. Among 95 patients with TG levels of >1771 mg/dl (mean TG 3376 mg/dl) evaluated at a tertiary care outpatient lipid clinic over a 12 year period12, Sandhu et al noted a history of pancreatitis in 15.8%. The lowest TG during episodes of pancreatitis was 1815 mg/dl. In an additional cohort of 91 patients who had TG levels between 886-1771 mg/dl, a history of pancreatitis was present in only 3.3%, and in each of these patients, TG at the time of pancreatitis was >1771 mg/dl. Linares et al11studied 129 consecutive patients referred for an inpatient endocrinology consultation for severe HTG (TG level >1000mg/ml) over a 6 year period. The vast majority of these patients had Frederickson type IV hyperlipidemia (92%), while the remainder had type V hyperlipidemia (8%). A history of AP was present in 20.2% patients after exclusion of other etiologies. The mean maximum TG level was significantly higher in patients with AP when compared with patients without AP (4470 vs. 2450 mg/ml, p<0.0001). Furthermore, over 85% of patients who had AP had maximal TG levels >3000 mg/ml and 65% had levels >5000 mg/ml. When compared with patients without AP, those with AP were significantly younger at the time of admission (43.1 vs. 48 years) and at diagnosis of HTG (32.4 vs. 40.2 years). In another study of 300 in- and out-patients with at least one TG level of >1000 mg/dl, Bessembinders et al10 found a history of pancreatitis in 8% and documented pancreatitis in 4% patients. The prevalence of pancreatitis increased with TG levels: history of pancreatitis and documented pancreatitis was noted in 8% and 4% in the third quartile (1415 - 2411 mg/dl) and in 19 and 11% in the highest quartile (>2411 mg/dl). Over two-third of patients with pancreatitis were in the highest TG quartile and about one-fourth were in the third quartile. These data tend to support the commonly held notion that the risk of pancreatitis is increased only with severe HTG (>1000 mg/dl).
While more studies are needed to better define the risk of pancreatitis with TG elevations of less than 1000 mg/dl, the following conclusions can be drawn from available data – when compared with the lifetime risk of pancreatitis in the general population (about 0.5-1%), the risk in patients with severe HTG (TG levels >1000 mg/dl) is similar to or slightly higher (∼5%) than among heavy drinkers13. In individuals with very severe HTG (TG >2000 ,g/dl), the risk is much greater (10-20%).
Question 4: What are the risk factors for HTG pancreatitis?
Serum TG level is a reflection of dietary intake, endogenous production and clearance of TG containing lipids and lipoproteins. A detailed review of TG metabolism is beyond the scope of this review. To summarize6,14, TG is mostly carried in the blood as VLDL, and VLDL concentrations are about 10 times higher than chylomicrons. FFA generated by the hydrolysis of dietary TG by luminal lipases enter the enterocytes where most are incorporated into chylomicrons and released into the circulation for utilization by peripheral tissues for energy. In peripheral tissues, chylomicrons are broken down by lipoprotein lipase (LPL) bound to capillary endothelium to release FFA. Unused chylomicrons (chylomicron-remnants) and FFA (not used by tissues or generated from other sources) released into the circulation are taken up by the liver and converted to VLDL. LPL in peripheral tissues also generate FFA from TG present in VLDL. VLDL metabolism is regulated by several apolipoproteins, for e.g. apoC-II is a cofactor for LPL, apoC-III blocks the uptake of lipoproteins by liver receptors and impairs LPL activity, and, apoE is a ligand for hepatic uptake of TG rich remnants.
HTG can occur from a primary (genetic) abnormality of lipid metabolism or presence of secondary factor(s) (Tables 1 and 2), due to an overproduction of TG (VLDL) (e.g. insulin resistance, visceral obesity, alcohol use, pregnancy, oral estrogens); reduced clearance of TG due to defective LPL activity (e.g. LPL deficiency, defects in apoC-II, defective association of LPL with vascular wall [antibodies to heparin]); reduced clearance of VLDL and chylomicron remnants by the liver (e.g. mutations in apoC-III, apoE, apoA-V, angiopoietin-like protein 4); or from a combination of these6,15.
Table 1. Causes of Hypertriglyceridemia associated with Pancreatitis.
| Genetic factors | Familial Combined Hyperlipidemia (FCHL) |
| Familial Hypertriglyceridemia (FHTG) | |
| Familial Dysbetalipoproteinemia | |
| Familial Chylomicronemia Syndrome | |
|
| |
| Secondary factors | Untreated/poorly controlled diabetes mellitus |
| Alcohol abuse | |
| Pregnancy | |
| Medications (oral estrogens, tamoxifen, propofol, valproic acid, isotretinoin, clomiphene, beta-blockers, protease inhibitors, mirtazapine) | |
Table 2. Genetic (primary) causes of hypertriglyceridemia associated with pancreatitis.
| Name | Molecular defect | Lipoprotein elevated | Lipoprotein phenotype | Lipid profile | Clinical findings | Genetic transmission | Estimated Incidence |
|---|---|---|---|---|---|---|---|
| Familial Combined Hyperlipidemia (FCHL) | Unknown | VLDL and LDL | Type IIb, sometimes IIa or IV | Elevated LDL and/or TG | Premature atherosclerosis | AD | 1/200 |
| Familial Hypertriglyceridemia (FHTG) | Unknown | VLDL, occasionally chylomicrons | Type IV, occasionally V | Elevated TG | Usually none | AD | 1/500 |
| Familial Dysbetalipo-proteinemia | Abnormal ApoE (i.e. apoE 2/2) | Chylomicron and VLDL remnants | Type III | Elevated total cholesterol, TG | Palmer and tuberoeruptive xanthomas, premature atherosclerosis | AR or AC | 1/5000 |
| Familial Chylomicronemia Syndrome | LPL deficiency, apo CII deficiency | Chylomicrons | Type I | Elevated TG | Eruptive xanthomas, hepatosplenomegaly | AR | 1/1,000,000 |
Reproduced in part (with permission) from ref. 15.
VLDL: very low density lipoprotein; LDL: low density lipoprotein; AD: autosomal dominant; AR: autosomal recessive; AC: autosomal codominant; apo: apolipoprotein; LPL: lipoprotein lipase
FCHL patients may have HTG without elevation in LDL - this can be differentiated from FHTG by concomitant elevation of apoB and non-HDL cholesterol
Patients with Familial Chylomicronemia Syndrome can have severe/very severe HTG in the absence of secondary factors. In other genetic causes of HTG, fasting serum TG levels are usually between 250-1000 mg/dl and require the presence of one or more secondary factor to result in severe/very severe HTG and increased risk of pancreatitis. In fact, chylomicronemia is more commonly due to combination of genetic causes of HTG and presence of a secondary factor than with Familial Chylomicronemia Syndrome.
Diabetes is the most common secondary factor physicians would encounter in patients with HTG pancreatitis. TG levels are higher in patients with poorly controlled diabetes and in the setting of diabetic ketoacidosis16, thereby increasing the risk of pancreatitis. The prevalence of diabetes in HTG pancreatitis is much higher than the general population and patients with HTG but no pancreatitis. In the NHANES study, the prevalence of diabetes in US adults with normal TG level was 5.2% and in those with TG levels between 500-2000 mg/dl was 14.6%8. In contrast, the prevalence of diabetes was 42.3% and 72% in two series of patients with HTG pancreatitis4,11. The mechanism of HTG in diabetes is linked to insulin resistance which leads to excess FFA return to the liver, increased VLDL production and decreased apoB synthesis; and to hyperinsulinemia which promotes de novo TG synthesis17. It is to be remembered that diabetes can independently increase the risk of AP by 1.5-2.5 folds after controlling for demographic and other risk factors18,19, including HTG20.
Ingestion of alcohol can increase TG levels, even in individuals without underlying HTG. While the effect of moderate alcohol consumption on TG level does not appear to be clinically significant, chronic heavy alcohol consumption can result in significant HTG21. Alcohol increases TG levels from an increase in synthesis of large VLDL particles in the liver, increase in splanchnic extraction of TG from VLDL remnants and chylomicrons, decreased LPL activity, and decreased lipogenesis and glucose oxidation in adipose tissue21.
Estrogens increase TG levels due to stimulation of VLDL production in the liver6. The effect of estrogens can be seen during replacement or contraception22 and during pregnancy23. Since transdermal estrogens bypasses the liver, it does not result in an increase in TG levels24. Medications commonly associated with HTG and pancreatitis are listed in Table 125-32. HTG pancreatitis has also been reported in rare disorders such as glycogen storage disease33 and certain lipodystrophies34.
Presence of more than one secondary factor further increases the risk of HTG. Bessembinders et al10reviewed records of 300 patients seen either as an out- or in-patient at their institution and had at least one documented serum TG measurement of >1000 mg/dl. The prevalence of diabetes, overweight (BMI >25 kg/m2 or obesity diagnosis) or alcoholism (>21 drinks/week men; >14 drinks/week women) in this cohort was 49%, 80% and 28% respectively. Mean TG levels were significantly higher in patients with alcohol abuse but did not differ based on the presence of diabetes or overweight. The prevalence of alcoholism and pancreatitis increased with the levels of TG – 41% patients in the highest quartile had alcohol abuse when compared with 14% in the lowest quartile; over two-third of patients with pancreatitis were in the highest TG quartile (>2411 mg/dl) and about one-fourth were in the third quartile (1415 - 2411 mg/dl). Alcohol, overweight and diabetes were seen in some combination in close to 90% of all patients. The combination of any two risk factors resulted in higher TG levels than when present alone; and patients with all three risk factors had the highest TG levels.
From a conceptual standpoint, it is important to understand the relationship between primary and secondary factors in increasing the risk of HTG pancreatitis (Figure 1). With the exception of patients with Familial chylomicronemic syndrome who can have spontaneous elevations of TG levels of >2000 mg/dl, TG levels in other genetic causes of TG usually range between 250-1000 mg/dl. As a prerequisite, these conditions require the presence of secondary risk factor(s) to result in severe or very severe HTG and thus increase the risk of pancreatitis. Similarly, presence of secondary factor alone in the absence of a genetic abnormality may not be sufficient to cause severe or very severe HTG. Therefore, individuals with a TG level >2000mg/ml commonly have both a primary genetic defect and a secondary risk factor. Since FCHL and FHTG are seen frequently in the general population (1:200 and 1:500 respectively), the most common scenarios for encountering a patient with HTG pancreatitis is someone with either FCHL or FHTG who also has one or more of the following secondary factors: uncontrolled diabetes, alcoholism, pregnancy/oral estrogen use or another medication that can cause HTG.
Figure 1. Relationship between primary (genetic) and secondary factors in increasing the risk for severe/very severe HTG and pancreatitis.
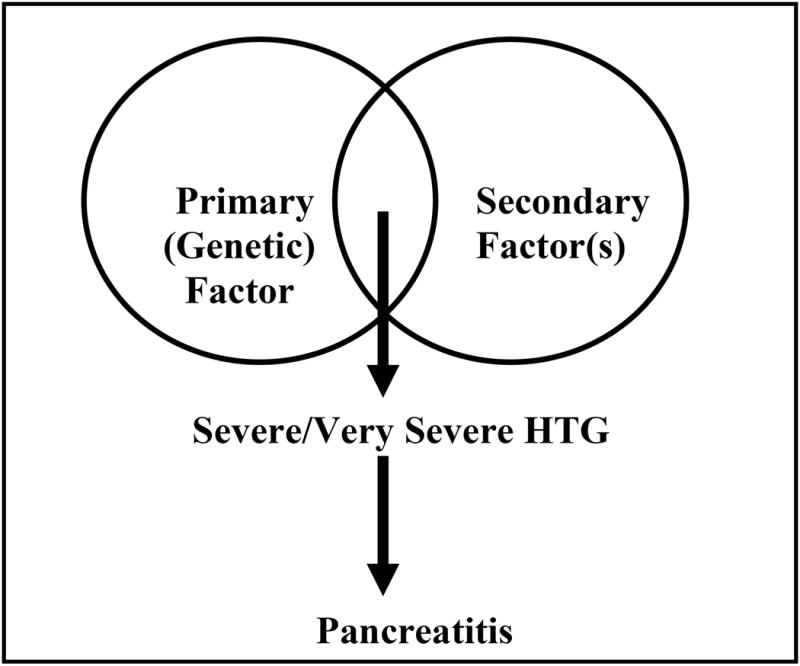
Footnote for Figure 1: Severe/Very severe HTG usually occurs due to presence of secondary factor(s) in a subject with an underlying genetic abnormality of lipid metabolism. Pancreatitis occurs in a subset of patients with severe/very severe HTG.
Question 5: How should I make a diagnosis of HTG as the cause of pancreatitis?
Elevation of serum TG levels to ≥1000 mg/dl in a patient with AP provides strong support for HTG as the cause. TG level of ≥500 mg/dl should raise a high degree of suspicion, especially if no other obvious etiology of AP is apparent or when estimation of TG has been delayed. Ideally, serum TG level needs to be sent within 24 hours of presentation (as close to the onset of pain or presentation as possible), as the influx of TG-rich chylomicrons into the blood stream diminishes quickly during the fasting state. Therefore, delayed estimation of TG levels may lead to falsely low levels. Serum TG levels are further decreased by administration of hypocaloric intravenous fluids which cut off VLDL output from the liver. Dominguez-Munoz et al found that TG levels decreased markedly to near normal levels after 72 hours of fasting in the majority of patients with HTG-induced AP who had levels >1750mg/ml2. The majority of this patient cohort had TG levels that remained slightly above upper limit of normal when checked 2 weeks after initial presentation.
Furthermore, levels of serum pancreatic enzymes can be falsely low when TG levels are >500 mg/ml, likely from interference with the assay to detect amylase and lipase activity which although corrected with serial dilution is not be related to lipid themselves35,36. Fortson et al noted elevations in serum amylase and lipase to >2 times the upper limit of normal in only 54% and 67% cases respectively4, while in another study by Saligram et al pancreatic enzyme elevations of ≥3 times the upper limit of normal were noted in 58% patients5.
The diagnosis of HTG as the etiology of AP or RAP may be delayed or missed completely if TG level is not checked shortly after admission. A subset of patients who are termed idiopathic likely have HTG as the etiology. A high index of suspicion in appropriate clinical setting and obtaining serum TG levels as close to the onset of pain during initial or recurrent attack(s) of AP is therefore critical to identify patients with HTG induced AP. Fasting serum TG levels during follow up after the patient has started oral diet can help to detect HTG.
Question 6: Is the clinical presentation of HTG pancreatitis similar to other causes?
A diagnosis of AP is established when two out of the following three findings are present - epigastric abdominal pain, elevated serum enzyme levels to three or more times the upper limit of normal, or radiologic imaging consistent with AP37,38. The clinical presentation of AP due to HTG is similar to that of other etiologies. As highlighted above, presence of risk factors and physical examination findings should prompt suspicion of HTG.
Question 7: What are the potential mechanisms for HTG to cause pancreatitis?
The exact mechanism by which HTG causes AP has not been fully elucidated. Three approaches have been used to study the basic pathophysiology of HTG-associated pancreatitis. These include studies on hyperlipoproteinemic mink that spontaneously develop pancreatitis39-41, effect of HTG on the severity of caerulein induced pancreatitis in rodents42-44 and in-vitro studies to determine the mechanisms of how these may occur42,44,45.
Severe hyperlipdemia in mink with a recessive inheritance pattern was noted on a farm in Norway39. These animals have grossly lipemic plasma and elevated plasma TG (60-170 mmol/liter, with normal values 0.7-0.9 mmol/liter), and the TG trend towards normal on a low-fat diet. The animals have detectable lipoprotein lipase (LPL) amounts but no activity. The defect was identified as a Pro214Leu point mutation in the LPL gene similar to those found in human hyperlipoproteinemia type I46. These mink are often found dead with evidence of pancreatitis and lipogranulomas in the mesentery on necropsy39. Histologically, the pancreas shows hemorrhage and necrosis. This begins as intralobular necrosis progressing to total lobular destruction, with lipid-filled macrophages in necrotic areas41. The earliest pathology shows vacuolation of exocrine cells resulting mainly from swollen mitochondria40.
Hypertrilgyceridemic mice with LPL deficiency42,44 and rats with HTG (>1000mg/dl) induced by Triton WR133943 have worse pancreatic injury than their normal counterparts. This is evident as higher serum amylase levels and worse histological damage to the pancreas42,43 in hypertriglyceridemic animals.
Studies to determine how HTG may result in the above outcomes include those focusing on the lipolytic generation of FFA from the TG present in chylomicrons42,44. The exocrine pancreas is rich in lipases47 which are secreted into the extracellular environment when acinar cells harvested from rodents are stimulated by hormones such as cholecystokinin48. Co-incubation of acinar cells with chylomicrons and cholecystokinin results in an increase in FFA in the medium with a corresponding decrease in TG42,44. Pharmacologic inhibition of these lipases using the lipase inhibitor orlistat in vitro prevents the injury to acinar cells induced by this co-incubation42,44. Orlistat also prevents the lipolytic generation of free fatty acids induced by these cells45. Strong evidence, including electron microscopy showing mitochondrial damage in hypertriglyceridemic mink exocrine pancreas40, and recent findings demonstrating a decrease in acinar ATP levels, necrosis via inhibition of mitochondrial complexes I and V by fatty acids45 support the role of fatty acid induced mitochondrial toxicity in the adverse outcomes. The roles of protein kinase C in hypertriglyceridemic pancreatitis remains to be explored43.
Question 8: What is the relationship between HTG and the clinical severity of AP?
Clinical studies assessing the impact of HTG on the severity of AP have had conflicting results. Balachandra et al prospectively enrolled 43 consecutive patients with AP of all etiologies to assess the relationship of admission TG level and severity3. TG levels were elevated in 33% of patients (mean 265 mg/ml). No difference detected in severity, based on APACHE II scores or complications in patients with and without HTG. This small study suggests that mild elevations in serum TG level in AP do not appear to hold prognostic value.
Anderson et al prospectively evaluated 230 admissions with AP to a regional hospital in South Africa49. Any and severe HTG was seen in 24% and 6% admissions; more frequently in the Indian race when compared with other ethnicities. Patients with HTG showed a trend towards increased severity. Although the mortality in patients with and without HTG was similar, all deaths in the dyslipidemia group were in patients with HTG.
In the study by Fortson et al4, the prevalence of pancreatic changes (64%), peripancreatic fluid (54%), necrosis (10%), pseudocysts (23%) and pleural effusion (20%) on CT scan in 70 patients with HTG pancreatitis was generally similar to historical data on these complications in AP from other causes.
Finally, Deng et al studied 176 patients with severe AP, of which 45 had HTG (≥500 mg/dl)50. Patients with HTG were younger and more likely to have alcohol abuse, overeating and high fat diet. On univariate analyses, patients with HTG tended to have more severe disease as reflected by a higher prevalence of hypoalbuminemia, hypocalcemia, APACHE-II scores, renal failure, shock, infection and overall mortality.
Based on available data from clinical studies, it is safe to assume that mild elevations in TG levels do not affect the severity of AP. The role of TG elevation of >500 mg/dl on the clinical course of AP is unclear. Future studies using adequate sample size should compare outcomes in patients with HTG (>500 and >1,000 mg/dl) with other major causes of AP.
Question 9: What is the natural history of HTG pancreatitis?
The natural history in patients with HTG pancreatitis depends on how well serum TG levels and concomitant secondary risk factors are controlled. Among 70 patients with HTG pancreatitis, Fortson et al noted a history of prior attacks in 44%4. In a series of 17 patients with HTG pancreatitis, over a mean follow up period of 42 months, recurrences were seen only in 1 patient who was non compliant to diet and medications51. In another series of 35 patients with Type V hyperlipoproteniemia, when compared to patients with no pain (n=16), those with episodes of pain only (n=8) and pain and pancreatitis (n=11) were more likely to be younger and had higher mean TG levels (5865 vs. 2573 mg/dl). The number and severity of pain episodes (HTG abdominal crisis) and frequency of attacks of pancreatitis decreased considerably during a follow up of 1-11 years with treatment (diet, medications or jejunoilieal bypass) and correlated well with TG levels of ≥2000 mg/dl.
While the association of HTG and recurrent episodes of pancreatitis is well known, whether HTG can cause CP has not been well studied. CP has been reported in patients with type I and V hyperlipidemia52-55. HTG in susceptible subjects with alcohol abuse is likely a contributing factor for recurrent attacks and an eventual transition to CP.
Question 10: Is there a genetic predisposition to HTG pancreatitis?
Presence of pancreatitis in a small fraction of patients with HTG has led to a search of genetic susceptibility factors. Chang et al evaluated the frequency of mutations in cationic trypsinogen (PRSS1), serine protease inhibitor Kazal type 1 (SPINK1), cystic fibrosis transmembrane conductance regulator (CFTR), and tumor necrosis factor superfamily member 2 (TNF2) genes in 128 Taiwanese patients - 80 with HTG and 46 with HTG pancreatitis56. The prevalence of polymorphisms in CFTR (M470V) and TNF (863A) genes was significantly higher when compared with patients with HTG alone.
More studies are needed to better understand the role of genetic factors that increase the risk of pancreatitis in patients with severe/very severe HTG.
Question 11: How do I manage patients with HTG induced AP?
Initial treatment of AP includes pancreas rest by limiting oral intake, aggressive intravenous hydration, and analgesia. Other etiologies of AP should be quickly ascertained with a thorough history and physical examination, basic laboratory testing, imaging as indicated, and medication review. As mentioned previously, serum TG level should be checked shortly after admission given rapid decline in levels with fasting. If there is evidence of severe AP and need for prolonged fasting, pancreatic rest via post-ligament of Treitz nasojejunal enteral feeding tube57 (preferred) or total parenteral nutrition (TPN) should be entertained58. For obvious reasons, if TPN is chosen, lipids should be avoided. Once HTG is the established cause of AP, multiple mostly experimental therapies including insulin, heparin, and apheresis have been studied in the acute setting (Table 3).
Table 3. Treatment of Hypertriglyceridemic Pancreatitis.
| Setting | Treatment | Mechanism | Pro | Con | |
|---|---|---|---|---|---|
| Acute/Recurrent Acute Pancreatitis | Acute Phase | NPO, Analgesia Intravenous fluid hydration | Pancreas rest Maintain blood flow to pancreas | Standard of care, transition to oral feeding when ready | |
| Severe Acute Pancreatitis (prolonged recovery expected) | Post-ligament of Trietz enteral feeding (as early as feasible) | Pancreas rest | Decreases ileus and risk of infection | Invasive (NGJ/NJ tube) | |
| Select cases | Insulin | Increased peripheral activity of LPL; reverses hepatic effects of insulin resistance | Readily available, especially useful in known diabetics with hyperglycemia | Hypoglycemia | |
| Experimental (clinical benefit not proven) | Apheresis* | Quickly remove TG from serum | Normalizes TG levels usually with 1-3 sessions | Invasive , expensive line infection/deep vein thrombosis, bleeding (requires anti-coagulation) | |
| Long term management/Prevention | Lifestyle Modifications (weight loss, low-fat diet, alcohol abstinence, control of diabetes, stop offending medication) | Remove / control co-factors responsible for HTG | Integral to HTG management and preventing recurrent pancreatitis | None | |
| TG-lowering medications (fibrates, nicotinic acid, omega-3 fatty acids/fish oil) | Reduce hepatic TG production, enhance TG clearance | Can reduce TG levels by up to 60% | Myopathy , rhabdomyolysis (fibrates) Flushing, GI intolerance, hepatotoxicity (niacin) Diarrhea (fish oil) | ||
| Consultation with Dietetian and Endocrinology | Education, optimize medication regimen | Enhance patient understanding and adherence | |||
NPO – nil per os; TG – Triglycerides; HTG – hypertriglyceridemia; LPL – lipoprotein lipase; NGJ – nasogastro-jejunal; NJ – nasojejunal
Role in prevention not established.
If facility for NGJ/NJ is not available, consider transfer to a center with experience. Avoid TPN if possible unless feeding intolerance, ileus or complications.
Apheresis has been repeatedly shown in cases reports and series to be effective in quickly removing TG from the serum of patients with HTG pancreatitis of different causes59-64. The premise of using apheresis is that it removes available TG in VLDL and chylomircons from serum and prevents generation of FFA which cause local and systemic effects44,45,65,66. In most patients, TG levels decrease by ∼65% and 85% after one or two sessions respectively63. However, a clear benefit of apheresis in reducing the severity and improving outcomes in AP has not been conclusively shown.
Chen et al retrospectively analyzed clinical outcomes in HTG pancreatitis patients before (n=34) and after (n=60) the availability of apheresis at their institution. These groups had statistically similar background and clinical characteristics; ultimately 20 patients in the latter group opted for apheresis with a median time of initiation of 3 days after symptom onset. There was no significant difference in mortality and local or systemic complications between the two time periods or among patients with severe AP (defined by Ranson's score >3) who did (n=10) or did not (n=19) get plasmapheresis60. The limitations of this study are its retrospective design, experience from a single center and small sample size.
Apheresis is expensive and is not without risk. It requires central intravenous access and transient anti-coagulation with associated complications of line-associated bacteremia, deep venous thrombosis, and bleeding. Potential candidates for apheresis may be patients with predicted severe or severe AP who continue to have TG levels above 1000 mg/dl after the first 24-48 hours. However, given the lack of proven efficacy, a firm recommendation for apheresis cannot be made at this time and the treatment should be highly individualized. In fact, the recent American Society for Apheresis (ASFA) guidelines placed apheresis in patients with HTG pancreatitis as 2C grade (weak recommendation) and that other alternatives may be equally reasonable based on data from observational studies or cases series (category III evidence)67. Apheresis as a therapeutic modality needs stronger evidence, ideally in the form of a randomized clinical trial before consideration as a standard treatment strategy.
Infusion of insulin and heparin has been utilized to treat HTG in the acute setting68,69 and in patients with HTG pancreatitis70-72. Insulin leads to an increase in peripheral LPL activity and helps to reverse hepatic effects of insulin resistance. Insulin infusion is especially helpful in patients with poorly controlled diabetes who have hyperglycemia in addition to severe HTG69. Enthusiasm for heparin has waned, as it has been shown that although it results in an increases in serum LPL activity, this effect is transient and is quickly followed by reduced activity of LPL, which may ultimately lead to a rise in TG73,74. Therefore, insulin infusion should be considered in select patients while the use of heparin is controversial and should be avoided.
Question 12: How do I prevent future episodes of pancreatitis?
A multi-faceted approach is required in patients with HTG pancreatitis to prevent recurrences. Lifestyle modifications directed at weight loss, limiting intake of fat and simple carbohydrates, abstinence from alcohol, control of secondary risk factors such as diabetes, and discontinuation of offending medication is integral to the management. In conjunction, lipid lowering medications are often required. Patients should seek formal consultation with a dietitian to discuss food choices and an endocrinologist to help control TG levels and diabetes. Although TG levels close to normal may be preferable, levels <500 mg/dl represents a safe therapeutic target for prevention of recurrences. In a large study evaluating claims data in 41,210 patients with serum TG >500 mg/dl, the risk of pancreatitis, other clinical events (cardiovascular, diabetes, renal) and overall cost were evaluated during follow up period (average 2.26 years) based on whether TG levels were decreased to <500 mg/dl (79%) or not (21%). Patients in whom TG levels continued to be over 500 mg/dl were about 2 times more likely to have pancreatitis (HR 1.79, 95% CI 1.47-2.18), as well as other clinical events and overall cost of care 75.
Fibrates are the first line medications for treating HTG6. HMG-CoA reductase inhibitors, i.e. statins have a weak TG lowering effect, and should not be used as monotherapy for HTG. Statins can have a synergistic lipid-lowering effect in combination with fibrates and should be considered in patients in whom severe HTG is not controlled on fibrates alone76. Fibrates are generally well-tolerated but caution should be exercised when used with statins given the increased, albeit small risk of myopathy or rhabdomyolysis77. This risk is mainly attributed to older fibrates such as gemfibrozil and appears to be much less with newer fibrate derivatives such as fenofibric acid78. Nicotinic acid, or niacin, is considered a second-line agent. However, its efficacy is often limited by side effects such as facial flushing, gastrointestinal intolerance, and hepatotoxicity. Omega-3 fatty acids can be used in combination with other agents such as fibrates but not as monotherapy79.
In a recent small study of 14 patients with LPL deficiency with severe HTG and history of pancreatitis, promising results were seen in the reduction of fasting HTG and rates of pancreatitis with alipogene tiparvovec (AAV1-LPLS477X) gene therapy80. Nonpharmacologic approaches have been considered in patients refractory to standard medical treatment. Stefanutti et al reported a small retrospective case series of 17 medically-refractory patients with severe HTG (mean maximum TG level 1929 mg/dl) of whom 12 had a history of AP treated with serial apheresis (as many as 46 sessions in one patient). Although the length of follow-up was not explicitly reported, TG levels <1000 mg/dl were maintained without development of recurrent AP81. Another case series utilized apheresis for the prevention of recurrent HTG pancreatitis in 6 patients. Apheresis was used up to 8 sessions per month until goal TG level of <150 mg/dl was reached. The authors reported a significant reduction in TG as well as a reduction in the frequency of recurrent AP82. Since control of TG levels is needed over the long term (which can be few decades) to prevent pancreatitis episodes, the role of maintenance apheresis needs further study.
Future Directions
Studies in recent years have provided population estimates of HTG and some clarity on the risk of pancreatitis with HTG. Future studies should address the role of HTG in disease severity, mechanism of HTG pancreatitis, reason why a subset of individuals with severe/very severe HTG develop pancreatitis, and, the role of insulin and apheresis in the management of HTG pancreatitis.
Acknowledgments
Supported in part by NIH - RO1DK 077906 (DY), Department of Army (DOA - PR110417) (VPS) and NIH - R01DK 092460 (VPS).
Grant Support: None
Footnotes
Views presented in this article belong solely to the authors.
Conflict of Interest: The authors have no conflicts that are relevant to the manuscript.
References
- 1.Yadav D, Pitchumoni CS. Issues in hyperlipidemic pancreatitis. J Clin Gastroenterol. 2003;36:54–62. doi: 10.1097/00004836-200301000-00016. [DOI] [PubMed] [Google Scholar]
- 2.Dominguez-Munoz JE, Malfertheiner P, Ditschuneit HH, et al. Hyperlipidemia in acute pancreatitis. Relationship with etiology, onset, and severity of the disease. Int J Pancreatol. 1991;10:261–7. [PubMed] [Google Scholar]
- 3.Balachandra S, Virlos IT, King NK, et al. Hyperlipidaemia and outcome in acute pancreatitis. Int J Clin Pract. 2006;60:156–9. doi: 10.1111/j.1742-1241.2005.00645.x. [DOI] [PubMed] [Google Scholar]
- 4.Fortson MR, Freedman SN, Webster PD., 3rd Clinical assessment of hyperlipidemic pancreatitis. Am J Gastroenterol. 1995;90:2134–9. [PubMed] [Google Scholar]
- 5.Saligram S, Lo D, Saul M, et al. Analyses of hospital administrative data that use diagnosis codes overestimate the cases of acute pancreatitis. Clin Gastroenterol Hepatol. 2012;10:805–11 e1. doi: 10.1016/j.cgh.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2969–89. doi: 10.1210/jc.2011-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–421. [PubMed] [Google Scholar]
- 8.Christian JB, Bourgeois N, Snipes R, et al. Prevalence of severe (500 to 2,000 mg/dl) hypertriglyceridemia in United States adults. Am J Cardiol. 107:891–7. doi: 10.1016/j.amjcard.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Lindkvist B, Appelros S, Regner S, et al. A prospective cohort study on risk of acute pancreatitis related to serum triglycerides, cholesterol and fasting glucose. Pancreatology. 2012;12:317–24. doi: 10.1016/j.pan.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Bessembinders K, Wielders J, van de Wiel A. Severe hypertriglyceridemia influenced by alcohol (SHIBA) Alcohol Alcohol. 2011;46:113–6. doi: 10.1093/alcalc/agq088. [DOI] [PubMed] [Google Scholar]
- 11.Lloret Linares C, Pelletier AL, Czernichow S, et al. Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas. 2008;37:13–2. doi: 10.1097/MPA.0b013e31816074a1. [DOI] [PubMed] [Google Scholar]
- 12.Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to a specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157. doi: 10.1186/1476-511X-10-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yadav D, Eigenbrodt ML, Briggs MJ, et al. Pancreatitis: prevalence and risk factors among male veterans in a detoxification program. Pancreas. 2007;34:390–8. doi: 10.1097/mpa.0b013e318040b332. [DOI] [PubMed] [Google Scholar]
- 14.Lambert JE, Parks EJ. Postprandial metabolism of meal triglyceride in humans. Biochim Biophys Acta. 1821:721–6. doi: 10.1016/j.bbalip.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rader DJ. Lipid Disorders. In: Topol E, editor. Textbook of cardiovascular medicine. 3rd. Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 16.Nair S, Yadav D, Pitchumoni CS. Association of diabetic ketoacidosis and acute pancreatitis: observations in 100 consecutive episodes of DKA. Am J Gastroenterol. 2000;95:2795–800. doi: 10.1111/j.1572-0241.2000.03188.x. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian S, Chait A. Hypertriglyceridemia secondary to obesity and diabetes. Biochim Biophys Acta. 2012;1821:819–25. doi: 10.1016/j.bbalip.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Girman CJ, Kou TD, Cai B, et al. Patients with type 2 diabetes mellitus have higher risk for acute pancreatitis compared with those without diabetes. Diabetes Obes Metab. 2010;12:766–71. doi: 10.1111/j.1463-1326.2010.01231.x. [DOI] [PubMed] [Google Scholar]
- 19.Noel RA, Braun DK, Patterson RE, et al. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care. 2009;32:834–8. doi: 10.2337/dc08-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai SW, Muo CH, Liao KF, et al. Risk of acute pancreatitis in type 2 diabetes and risk reduction on anti-diabetic drugs: a population-based cohort study in Taiwan. Am J Gastroenterol. 2011;106:1697–704. doi: 10.1038/ajg.2011.155. [DOI] [PubMed] [Google Scholar]
- 21.Van de Wiel A. The effect of alcohol on postprandial and fasting triglycerides. Int J Vasc Med. 2012;2012:862504. doi: 10.1155/2012/862504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldenberg NM, Wang P, Glueck CJ. An observational study of severe hypertriglyceridemia, hypertriglyceridemic acute pancreatitis, and failure of triglyceride-lowering therapy when estrogens are given to women with and without familial hypertriglyceridemia. Clin Chim Acta. 2003;332:11–9. doi: 10.1016/s0009-8981(03)00129-3. [DOI] [PubMed] [Google Scholar]
- 23.Geng Y, Li W, Sun L, et al. Severe acute pancreatitis during pregnancy: eleven years experience from a surgical intensive care unit. Dig Dis Sci. 2011;56:3672–7. doi: 10.1007/s10620-011-1809-5. [DOI] [PubMed] [Google Scholar]
- 24.Goodman MP. Are all estrogens created equal? A review of oral vs. transdermal therapy. J Womens Health (Larchmt) 2012;21:161–9. doi: 10.1089/jwh.2011.2839. [DOI] [PubMed] [Google Scholar]
- 25.Arbel Y, Weinstein D, Yogev R, et al. Acute pancreatitis following clomiphene citrate treatment: case report and review of the literature. Int J Surg. 2008;6:483–4. doi: 10.1016/j.ijsu.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 26.Chen JL, Spinowitz N, Karwa M. Hypertriglyceridemia, acute pancreatitis, and diabetic ketoacidosis possibly associated with mirtazapine therapy: a case report. Pharmacotherapy. 2003;23:940–4. doi: 10.1592/phco.23.7.940.32725. [DOI] [PubMed] [Google Scholar]
- 27.Devlin JW, Lau AK, Tanios MA. Propofol-associated hypertriglyceridemia and pancreatitis in the intensive care unit: an analysis of frequency and risk factors. Pharmacotherapy. 2005;25:1348–52. doi: 10.1592/phco.2005.25.10.1348. [DOI] [PubMed] [Google Scholar]
- 28.Durrington PN, Cairns SA. Acute-pancreatitis: a complication of beta-blockade. Br Med J (Clin Res Ed) 1982;284:1016. doi: 10.1136/bmj.284.6321.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerstner T, Busing D, Bell N, et al. Valproic acid-induced pancreatitis: 16 new cases and a review of the literature. J Gastroenterol. 2007;42:39–48. doi: 10.1007/s00535-006-1961-4. [DOI] [PubMed] [Google Scholar]
- 30.McCarter TL, Chen YK. Marked hyperlipidemia and pancreatitis associated with isotretinoin therapy. Am J Gastroenterol. 1992;87:1855–8. [PubMed] [Google Scholar]
- 31.Riedel DJ, Gebo KA, Moore RD, et al. A ten-year analysis of the incidence and risk factors for acute pancreatitis requiring hospitalization in an urban HIV clinical cohort. AIDS Patient Care STDS. 2008;22:113–21. doi: 10.1089/apc.2007.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakhri J, Ben Salem C, Harbi H, et al. Severe acute pancreatitis due to tamoxifen-induced hypertriglyceridemia with positive rechallenge. Jop. 2010;11:382–4. [PubMed] [Google Scholar]
- 33.Vivatrat N, Barshop BA, Jones KL. Severe hypertriglyceridemia and recurrent pancreatitis in a girl with type Ia glycogen storage disease and type III hyperlipoproteinemia. Am J Med Genet A. 2009;149A:2557–9. doi: 10.1002/ajmg.a.33046. [DOI] [PubMed] [Google Scholar]
- 34.Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96:3313–25. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesser PB, Warshaw AL. Diagnosis of pancreatitis masked by hyperlipemia. Ann Intern Med. 1975;82:795–8. doi: 10.7326/0003-4819-82-6-795. [DOI] [PubMed] [Google Scholar]
- 36.Warshaw AL, Bellini CA, Lesser PB. Inhibition of serum and urine amylase activity in pancreatitis with hyperlipemia. Ann Surg. 1975;182:72–5. doi: 10.1097/00000658-197507000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banks PA, Freeman ML. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101:2379–400. doi: 10.1111/j.1572-0241.2006.00856.x. [DOI] [PubMed] [Google Scholar]
- 38.Forsmark CE, Baillie J. AGA Institute technical review on acute pancreatitis. Gastroenterology. 2007;132:2022–44. doi: 10.1053/j.gastro.2007.03.065. [DOI] [PubMed] [Google Scholar]
- 39.Christophersen B, Nordstoga K, Shen Y, et al. Lipoprotein lipase deficiency with pancreatitis in mink: biochemical characterization and pathology. J Lipid Res. 1997;38:837–46. [PubMed] [Google Scholar]
- 40.Nordstoga K, Christophersen B, Ytrehus B, et al. Pancreatitis associated with hyperlipoproteinaemia type I in mink (Mustela vison): earliest detectable changes occur in mitochondria of exocrine cells. J Comp Pathol. 2006;134:320–8. doi: 10.1016/j.jcpa.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Nordstoga K, Sorby R, Olivecrona G, et al. Pancreatitis in Hyperlipemic Mink (Mustela Vison) Vet Pathol. 2011 doi: 10.1177/0300985811417248. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Sternfeld L, Yang F, et al. Enhanced susceptibility to pancreatitis in severe hypertriglyceridaemic lipoprotein lipase-deficient mice and agonist-like function of pancreatic lipase in pancreatic cells. Gut. 2009;58:422–30. doi: 10.1136/gut.2007.146258. [DOI] [PubMed] [Google Scholar]
- 43.Wang YJ, Sun JB, Li F, Zhang SW. Hyperlipidemia intensifies cerulein-induced acute pancreatitis associated with activation of protein kinase C in rats. World J Gastroenterol. 2006;12:2908–13. doi: 10.3748/wjg.v12.i18.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang F, Wang Y, Sternfeld L, et al. The role of free fatty acids, pancreatic lipase and Ca+ signalling in injury of isolated acinar cells and pancreatitis model in lipoprotein lipase-deficient mice. Acta Physiol (Oxf) 2009;195:13–28. doi: 10.1111/j.1748-1716.2008.01933.x. [DOI] [PubMed] [Google Scholar]
- 45.Navina S, Acharya C, DeLany JP, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med. 2011;3:107ra10. doi: 10.1126/scitranslmed.3002573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Savonen R, Nordstoga K, Christophersen B, et al. Chylomicron metabolism in an animal model for hyperlipoproteinemia type I. J Lipid Res. 1999;40:1336–46. [PubMed] [Google Scholar]
- 47.Lowe ME. The triglyceride lipases of the pancreas. J Lipid Res. 2002;43:2007–16. doi: 10.1194/jlr.r200012-jlr200. [DOI] [PubMed] [Google Scholar]
- 48.Singh VP, Saluja AK, Bhagat L, et al. Serine protease inhibitor causes F-actin redistribution and inhibition of calcium-mediated secretion in pancreatic acini. Gastroenterology. 2001;120:1818–27. doi: 10.1053/gast.2001.24883. [DOI] [PubMed] [Google Scholar]
- 49.Anderson F, Thomson SR, Clarke DL, et al. Dyslipidaemic pancreatitis clinical assessment and analysis of disease severity and outcomes. Pancreatology. 2009;9:252–7. doi: 10.1159/000212091. [DOI] [PubMed] [Google Scholar]
- 50.Deng LH, Xue P, Xia Q, et al. Effect of admission hypertriglyceridemia on the episodes of severe acute pancreatitis. World J Gastroenterol. 2008;14:4558–61. doi: 10.3748/wjg.14.4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Athyros VG, Giouleme OI, Nikolaidis NL, et al. Long-term follow-up of patients with acute hypertriglyceridemia-induced pancreatitis. J Clin Gastroenterol. 2002;34:472–5. doi: 10.1097/00004836-200204000-00020. [DOI] [PubMed] [Google Scholar]
- 52.Hacken JB, Moccia RM. Calcific pancreatitis in a patient with type 5 hyperlipoproteinemia. Gastrointest Radiol. 1979;4:143–6. doi: 10.1007/BF01887513. [DOI] [PubMed] [Google Scholar]
- 53.Krauss RM, Levy AG. Subclinical chronic pancreatitis in type I hyperlipoproteinemia. Am J Med. 1977;62:144–9. doi: 10.1016/0002-9343(77)90361-8. [DOI] [PubMed] [Google Scholar]
- 54.Hoste P, Lentini F, Rottiers R. Chronic relapsing pancreatitis in twin-brothers with type V hyperlipoproteinemia. Acta Clin Belg. 1978;33:313–22. doi: 10.1080/22953337.1978.11718649. [DOI] [PubMed] [Google Scholar]
- 55.Truninger K, Schmid PA, Hoffmann MM, et al. Recurrent acute and chronic pancreatitis in two brothers with familial chylomicronemia syndrome. Pancreas. 2006;32:215–9. doi: 10.1097/01.mpa.0000202942.93578.dd. [DOI] [PubMed] [Google Scholar]
- 56.Chang YT, Chang MC, Su TC, et al. Association of cystic fibrosis transmembrane conductance regulator (CFTR) mutation/variant/haplotype and tumor necrosis factor (TNF) promoter polymorphism in hyperlipidemic pancreatitis. Clin Chem. 2008;54:131–8. doi: 10.1373/clinchem.2007.093492. [DOI] [PubMed] [Google Scholar]
- 57.O'Keefe SJ. A guide to enteral access procedures and enteral nutrition. Nat Rev Gastroenterol Hepatol. 2009;6:207–15. doi: 10.1038/nrgastro.2009.20. [DOI] [PubMed] [Google Scholar]
- 58.Yi F, Ge L, Zhao J, et al. Meta-analysis: total parenteral nutrition versus total enteral nutrition in predicted severe acute pancreatitis. Intern Med. 2012;51:523–30. doi: 10.2169/internalmedicine.51.6685. [DOI] [PubMed] [Google Scholar]
- 59.Al-Humoud H, Alhumoud E, Al-Hilali N. Therapeutic plasma exchange for acute hyperlipidemic pancreatitis: a case series. Ther Apher Dial. 2008;12:202–4. doi: 10.1111/j.1744-9987.2008.00572.x. [DOI] [PubMed] [Google Scholar]
- 60.Chen JH, Yeh JH, Lai HW, et al. Therapeutic plasma exchange in patients with hyperlipidemic pancreatitis. World J Gastroenterol. 2004;10:2272–4. doi: 10.3748/wjg.v10.i15.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gubensek J, Buturovic-Ponikvar J, Marn-Pernat A, et al. Treatment of hyperlipidemic acute pancreatitis with plasma exchange: a single-center experience. Ther Apher Dial. 2009;13:314–7. doi: 10.1111/j.1744-9987.2009.00731.x. [DOI] [PubMed] [Google Scholar]
- 62.Kyriakidis AV, Karydakis P, Neofytou N, et al. Plasmapheresis in the management of acute severe hyperlipidemic pancreatitis: report of 5 cases. Pancreatology. 2005;5:201–4. doi: 10.1159/000085272. [DOI] [PubMed] [Google Scholar]
- 63.Yeh JH, Chen JH, Chiu HC. Plasmapheresis for hyperlipidemic pancreatitis. J Clin Apher. 2003;18:181–5. doi: 10.1002/jca.10063. [DOI] [PubMed] [Google Scholar]
- 64.Kohli RS, Bleibel W, Shetty A, et al. Plasmapheresis in the treatment of hypertriglyceridemic pancreatitis with ARDS. Dig Dis Sci. 2006;51:2287–91. doi: 10.1007/s10620-006-9315-x. [DOI] [PubMed] [Google Scholar]
- 65.Dettelbach MA, Deftos LJ, Stewart AF. Intraperitoneal free fatty acids induce severe hypocalcemia in rats: a model for the hypocalcemia of pancreatitis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1990;5:1249–55. doi: 10.1002/jbmr.5650051210. [DOI] [PubMed] [Google Scholar]
- 66.Domschke S, Malfertheiner P, Uhl W, et al. Free fatty acids in serum of patients with acute necrotizing or edematous pancreatitis. Int J Pancreatol. 1993;13:105–10. doi: 10.1007/BF02786078. [DOI] [PubMed] [Google Scholar]
- 67.Szczepiorkowski ZM, Winters JL, Bandarenko N, et al. Guidelines on the use of therapeutic apheresis in clinical practice--evidence-based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher. 2010;25:83–177. doi: 10.1002/jca.20240. [DOI] [PubMed] [Google Scholar]
- 68.Cole RP. Heparin treatment for severe hypertriglyceridemia in diabetic ketoacidosis. Arch Intern Med. 2009;169:1439–41. doi: 10.1001/archinternmed.2009.221. [DOI] [PubMed] [Google Scholar]
- 69.Henderson SR, Maitland R, Mustafa OG, et al. Severe hypertriglyceridaemia in Type 2 diabetes mellitus: beneficial effect of continuous insulin infusion. Qjm. 2013 doi: 10.1093/qjmed/hcs238. [DOI] [PubMed] [Google Scholar]
- 70.Alagozlu H, Cindoruk M, Karakan T, et al. Heparin and insulin in the treatment of hypertriglyceridemia-induced severe acute pancreatitis. Dig Dis Sci. 2006;51:931–3. doi: 10.1007/s10620-005-9006-z. [DOI] [PubMed] [Google Scholar]
- 71.Monga A, Arora A, Makkar RP, et al. Hypertriglyceridemia-induced acute pancreatitis--treatment with heparin and insulin. Indian J Gastroenterol. 2003;22:102–3. [PubMed] [Google Scholar]
- 72.Twilla JD, Mancell J. Hypertriglyceridemia-induced acute pancreatitis treated with insulin and heparin. Am J Health Syst Pharm. 2012;69:213–6. doi: 10.2146/ajhp110144. [DOI] [PubMed] [Google Scholar]
- 73.Nasstrom B, Olivecrona G, Olivecrona T, et al. Lipoprotein lipase during continuous heparin infusion: tissue stores become partially depleted. J Lab Clin Med. 2001;138:206–13. doi: 10.1067/mlc.2001.117666. [DOI] [PubMed] [Google Scholar]
- 74.Weintraub M, Rassin T, Eisenberg S, et al. Continuous intravenous heparin administration in humans causes a decrease in serum lipolytic activity and accumulation of chylomicrons in circulation. J Lipid Res. 1994;35:229–38. [PubMed] [Google Scholar]
- 75.Christian JB, Arondekar B, Buysman EK, et al. Clinical and economic benefits observed when follow-up triglyceride levels are less than 500 mg/dL in patients with severe hypertriglyceridemia. J Clin Lipidol. 2012;6:450–61. doi: 10.1016/j.jacl.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 76.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) Jama. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 77.Jacobson TA. Myopathy with statin-fibrate combination therapy: clinical considerations. Nat Rev Endocrinol. 2009;5:507–18. doi: 10.1038/nrendo.2009.151. [DOI] [PubMed] [Google Scholar]
- 78.Schima SM, Maciejewski SR, Hilleman DE, et al. Fibrate therapy in the management of dyslipidemias, alone and in combination with statins: role of delayed-release fenofibric acid. Expert Opin Pharmacother. 2010;11:731–8. doi: 10.1517/14656560903575639. [DOI] [PubMed] [Google Scholar]
- 79.Hooper L, Thompson RL, Harrison RA, et al. Risks and benefits of omega 3 fats for mortality, cardiovascular disease, and cancer: systematic review. Bmj. 2006;332:752–60. doi: 10.1136/bmj.38755.366331.2F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaudet D, Methot J, Dery S, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPL(S447X)) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene therapy. 2013;20:361–9. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stefanutti C, Di Giacomo S, Vivenzio A, et al. Therapeutic plasma exchange in patients with severe hypertriglyceridemia: a multicenter study. Artif Organs. 2009;33:1096–102. doi: 10.1111/j.1525-1594.2009.00810.x. [DOI] [PubMed] [Google Scholar]
- 82.Schaap-Fogler M, Schurr D, Schaap T, et al. Long-term plasma exchange for severe refractory hypertriglyceridemia: a decade of experience demonstrates safety and efficacy. J Clin Apher. 2009;24:254–8. doi: 10.1002/jca.20224. [DOI] [PubMed] [Google Scholar]