

Abstract
Rationale
Lipoprotein apheresis (LA) reduces low-density lipoprotein (LDL) levels in patients with severe familial hypercholesterolemia (FH). We have recently reported that >30% of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) is bound to LDL, thus we predicted that LA would also reduce plasma PCSK9 levels by removing LDL.
Objective
Pre- and post-apheresis plasma from 6 patients with familial hypercholesterolemia on 3 consecutive treatment cycles was used to determine changes in PCSK9 levels.
Methods and Results
LA drastically reduced plasma LDL (by 77±4%). Concomitantly, PCSK9 levels fell by 52±5%, strongly correlating with the LDL drop (P=0.0322; r2=0.26), but not with decreases in triglyceride (49±13%) or high-density lipoprotein levels (18±2%). Levels of albumin, creatinine, and CK-MB did not show significant changes after LA. Similar to LDL, PCSK9 levels returned to pretreatment values between cycles (2-week intervals). Fractionation of pre- and post-apheresis plasma showed that 81±11% of LDL-bound PCSK9 and 48±14% of apolipoprotein B–free PCSK9 were removed. Separation of whole plasma, purified LDL, or the apolipoprotein B–free fraction through a scaled-down, experimental dextran sulfate cellulose beads column produced similar results.
Conclusions
Our results show, for the first time, that modulation of LDL levels by LA directly affects plasma PCSK9 levels, and suggest that PCSK9 reduction is an additional benefit of LA. Because the loss of PCSK9 could contribute to the LDL-lowering effect of LA, then (1) anti-PCSK9 therapies may reduce frequency of LA in patients currently approved for therapy, and (2) LA and anti-PCSK9 therapies may be used synergistically to reduce treatment burden.
Keywords: cholesterol, familial hypercholesterolemia, lipoprotein apheresis, LDL, PCSK9
Familial hypercholesterolemia (FH) is an inherited disease characterized by extremely high levels of low-density lipoprotein cholesterol (LDL-C) leading to premature coronary artery disease (CAD).1 Patients with CAD and LDL >200 mg/ dL, despite maximal therapy or when therapy is not tolerated, qualify for lipoprotein apheresis (LA) as an interventional LDL-lowering maneuver. LA selectively removes apolipoprotein (apo) B–containing lipoproteins from plasma, thereby lowering LDL by 70% to 80%.2 The procedure also significantly reduces lipoprotein(a), triglycerides, and high-density lipoprotein levels. Depending on the LA method, several other CAD biomarkers are partially removed from plasma by the procedure.3–5
Proprotein convertase subtilisin/kexin 9 (PCSK9) is a secreted regulator of cell surface LDL receptor (LDLR) levels, whose function results in elevation of plasma LDL levels.6 Inhibition of PCSK9 is an active and promising area of therapeutic development for hypercholesterolemia. A major target of these drug development efforts is aimed at reducing levels of PCSK9 by using monoclonal antibodies.7–9 We and others have found that PCSK9 is associated with LDL in plasma.10–13 As much as 25% of plasma PCSK9 in transgenic mice, and 40% in human subjects, is in the LDL fraction.12,13 Various studies have reported a significant correlation between PCSK9 and LDL-C,14–16 likely because of PCSK9’s effect on cellular LDLR and plasma LDL clearance. It remains to be established whether primary changes in LDL levels affect plasma PCSK9 levels,17 as can be expected given that significant amounts of PCSK9 are carried by this lipoprotein.
PCSK9 circulates in plasma in different molecular forms: (1) a 62 kDa band, representing the full-length circulating protein minus the prodomain; (2) a 55 kDa fragment, product of cleavage of the 62 kDa protein by the protease furin18,19; and (3) higher-molecular-weight forms, likely PCSK9 homo- or heteromultimers.10,12 The physiological role of the smaller and larger molecular weight PCSK9 forms is not clear.
To test the direct effect of modulating LDL levels by LA on PCSK9 plasma levels, we analyzed plasma from patients with FH before and after removal of apoB-containing lipoproteins via LA and also measured PCSK9 in the apheresis column eluate. We show that LA reduces PCSK9 levels by >50%, via column trapping of both LDL-bound and apoB-free PCSK9.
Methods
An expanded Methods section is available in the Online Data Supplement.
Patients
Six subjects with severe FH (according to Simone Broome criteria)20 and history of CAD undergoing LA every 2 weeks at Vanderbilt University Medical Center (VUMC) were studied. Control subjects were not taking lipid-lowering agents. The study protocol was approved by VUMC’s institutional review board (IRB 101615). All participants gave informed consent.
Experimental Dextran Sulfate Column
One milliliter of dextran sulfate cellulose beads (from an unused Liposorber column) was packed into vertical columns and 6 mL of plasma were loaded on the column in each experiment.
Recombinant PCSK9 With Glutathione S-Transferase Affinity Tag
Using the Mammalian Transfection System purchased from Promega, HEK293T cells were transfected with pcDNA-PCSK9-GST and purified from culture media.
Plasma Lipoprotein Separation
Lipoproteins from pre- and post-apheresis plasma were isolated by ultracentrifugation using natural gradient media (Optiprep) and OptiSeal tubes spun at 90 000 rpm for 2.5 hours in a TLN100 rotor.
Results
LA Reduces Plasma PCSK9 Levels
Because PCSK9 binds to LDL,10–13 we predicted that plasma PCSK9 would also be removed by LA. We studied 6 patients undergoing routine LA with dextrose sulfate columns on 3 consecutive treatment cycles and found a significant reduction (52±5%) in plasma PCSK9 after treatment (Figure 1A). The reduction in PCSK9 was higher than predicted by LDL reduction alone (≈80% of LDL removed with ≈40% of PCSK9 associated with LDL should result in a 32% reduction in total PCSK9). No differences were seen in plasma levels of control parameters, such as albumin, creatinine, and CK-MB. Mean changes in plasma lipids and proteins for each patient are summarized in Table 1. PCSK9 levels returned to pretreatment values between each cycle (2-week intervals) in all 6 patients (Figure 1B). As shown in Figure 1C, there was a significant correlation between LDL and PCSK9 decreases (P=0.0322; r2=0.26). No correlation was found between PCSK9 reduction and triglyceride or high-density lipoprotein changes after treatment (Online Figure IA and IB).
Figure 1. Lipoprotein apheresis (LA) reduces plasma proprotein convertase subtilisin/kexin 9 (PCSK9) levels.
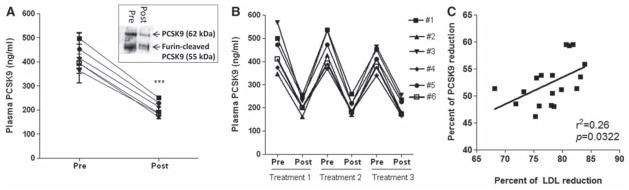
A, Pre- and postapheresis plasma PCSK9 levels in 6 patients with familial hypercholesterolemia (FH). Insert: Representative PCSK9 immunoblot from pre- and post-apheresis plasma. B, Pre- and post-apheresis plasma PCSK9 levels during 3 consecutive treatments in 6 FH patients. C, Correlation between low-density lipoprotein (LDL) reduction and PCSK9 reduction after LA. ***P<0.001.
Table 1.
Pre- and Post-apheresis Plasma Lipids and Protein Levels of 6 Subjects With FH
| TC, mg/dL
|
TG, mg/dL
|
HDL, mg/dL
|
LDL, mg/dL
|
Lp(a), mg/dL
|
Albumin, g/dL
|
Creatinine, mg/dL
|
CK-MB, ng/mL
|
PCSK9, ng/mL
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | |
| Patient #1 | 342±35 | 117±7 | 121±65 | 47±13 | 38±3 | 30±2 | 279±27 | 78±5 | N/A | N/A | 4.1±0.3 | 3.8±0.2 | 0.61±0.05 | 0.66±0.03 | 1.04±0.38 | 1.08±0.19 | 496±38 | 251±21 |
| Patient #2 | 315±7 | 96±3 | 217±31 | 110±17 | 45±2 | 35±0 | 226±12 | 39±3 | 2.9±1.8 | 0.1±0.1 | 4.0±0.1 | 3.6±0.1 | 0.9±0.1 | 0.8±0.1 | 1.5±0.1 | 1.3±0.1 | 394±42 | 171±10 |
| Patient #3 | 308±43 | 116±5 | 229±96 | 102±14 | 55±14 | 45±12 | 207±38 | 50±49 | 5.3±1.2 | 0.2±0.1 | 3.9±0.1 | 3.9±0.1 | 0.7±0.2 | 0.6±0.1 | 0.8±0.2 | 0.7±0.2 | 362±18 | 184±17 |
| Patient #4 | 265±21 | 80±8 | 83±12 | 53±21 | 36±2 | 29±2 | 213±16 | 40±5 | 2.9±0.3 | 0.1±0.1 | 3.6±0.0 | 3.4±0.1 | 0.9±0.1 | 0.8±0.1 | 1.1±0.2 | 1.1±0.1 | 522±54 | 224±19 |
| Patient #5 | 337±27 | 127±13 | 132±24 | 92±14 | 54±4 | 42±5 | 257±29 | 64±17 | 17±4 | 6±4 | N/A | N/A | N/A | N/A | N/A | N/A | 452±35 | 230±13 |
| Patient #6 | 244±0 | 65±8 | 177±102 | 91±46 | 36±6 | 28±4 | 173±14 | 19±2 | 6.1±4.4 | 0.1±0.1 | 3.9±0.2 | 3.7±0.3 | 1.1±0 | 1.0±0 | 0.9±0.2 | 1.1±0.1 | 395±17 | 186±13 |
| % Reduction | 65±4 | 49±13 | 18±2 | 77±4 | 89±15 | 6±3 | 8±7 | 8±12 | 52±5 | |||||||||
| P value (pairwise) | 1.50E-06 | 0.004 | 0.03715 | 8.20E-06 | 0.0096 | 0.08 | 0.19 | 0.8 | 3.40E-05 | |||||||||
Values are mean±SD or percent±coefficient of variation (n=3 treatment cycles). P values were calculated using pairwise analysis with Prism 4 analytic software. FH indicates familial hypercholesterolemia; HDL, high-density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein(a) N/A, not analyzed; PCSK9, proprotein convertase subtilisin/kexin 9; TC, total cholesterol; and TG, triglycerides.
LA Removes Both LDL-Bound and apoB-Free PCSK9
As PCSK9 reduction was greater than predicted, we set out to analyze the degree of PCSK9 reduction in different plasma fractions and the quantitative recovery of PCSK9 from the column. We show that 81±11% and 48±14% of PCSK9 was removed from the LDL-bound and apoB-free fraction of the plasma, respectively (Figure 2A). The combined reduction in both fractions explains >80% of PCSK9 removal from the circulation. Additionally, in the column eluate we were able to recover 82±11% of the total PCSK9 removed from the circulation (not shown). Similar to previous reports in mice,10–12 western blot analysis of PCSK9 in plasma from apheresis patients showed no PCSK9 bound to the VLDL (Figure 2B). Purity of each fraction is shown in Online Figure II. PCSK9 bound to LDL was exclusively in the 62 kDa monomeric active form (Figure 2B), whereas the apoB-free fraction showed different molecular forms of PCSK9, mainly the smaller 55 kDa band product of furin cleavage and also low levels of higher molecular weight bands likely due to homo- or hetero-polymerization (Figure 2B).
Figure 2. Lipoprotein apheresis (LA) removes both low-density lipoprotein (LDL)-bound and apoB-free proprotein convertase subtilisin/kexin 9 (PCSK9).
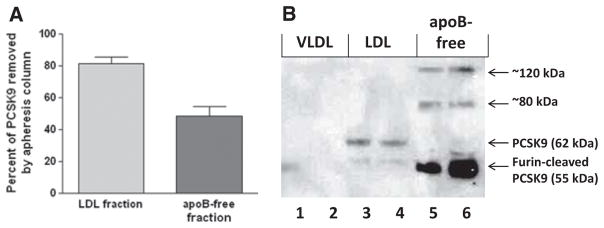
A, Percent of PCSK9 removed from LDL and apoB-free fractions of plasma after LA (n=6). B, PCSK9 immunoblot of plasma fractions isolated by ultracentrifugation from 2 different patients with LA. Lanes 1,2 indicate VLDL; lanes 3,4, LDL; and lanes 5,6, apoB-free (concentrated 2-fold for better visualization of high molecular bands).
Quantification Studies Using a Scaled-Down LA Column
To study PCSK9 removal from each fraction directly, we used a commercial grade, scaled-down dextrose sulfate column with cellulose beads (Liposorber). Using plasma, 61±4% of cholesterol (mainly LDL) and 51±7% of PCSK9 were removed. Using the purified LDL fraction, 68±16% of cholesterol and 61±5% of PCSK9 were removed. Using the apoB-free fraction, 17±9% of cholesterol (mainly high-density lipoprotein) and 38±6% of PCSK9 were removed (Figure 3A and 3B). To test whether purified PCSK9 directly binds the column, we used GST-tag PCSK9 (62 kDa) produced in transfected HEK293T cells (Figure 3B, inset). Figure 3B shows that the scaled-down dextran sulfate column did not remove the purified GST-tag PCSK9 (62 kDa) from the saline solution (pH=7.4). To determine whether the ability to remove both LDL and PCSK9 from plasma is unique to dextran sulfate, we also used polyethylene glycol 8000 (20%) precipitation, a known method to precipitate apoB particles and other proteins from plasma.21 Polyethylene glycol precipitation almost completely removed plasma LDL (97±1.6%) and PCSK9 (95±0.2%; not shown).
Figure 3. Cholesterol and proprotein convertase subtilisin/kexin 9 (PCSK9) adsorption to a scaled-down dextran sulfate column.
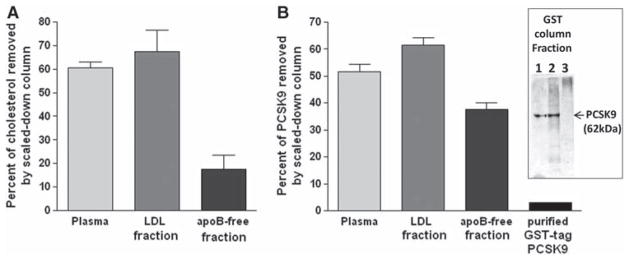
A, Percent of cholesterol removed from total plasma and its low-density lipoprotein (LDL) and apoB-free fractions by a scaled-down dextran sulfate column (n=3). B, Percent of PCSK9 removed from plasma, its fractions, and puri3ed GST-tagged PCSK9 by the scaled-down dextran sulfate column (n=3). Insert: PCSK9 immunoblot of GST-tagged eluted fraction from glutathione agarose column of media from HEK293T cells transfected with pcDNA-PCSK9-GST. The presence of a single band con3rms the purity of the preparation.
Discussion
We were the first to describe the association of PCSK9 with LDL in murine plasma10; we and others have recently reported that more than one third of circulating PCSK9 is associated with plasma LDL in humans.12,13 Here, we tested the effect of direct LDL removal via apheresis on plasma PCSK9 levels. Using pre- and post-apheresis plasma from patients with severe hypercholesterolemia and CAD, we show that PCSK9 is removed from the circulation during the LA through both LDL-dependent and LDL-independent mechanisms.
FH is mainly caused by mutations in the LDLR and less commonly by mutations in apoB or gain-of-function mutations in PCSK9.22 Certain LDLR mutations (eg, receptor-negative) may also result in increased plasma PCSK9 levels12,23 and lack of responsiveness to anti-PCSK9 therapy.24 LA is used to treat hypercholesterolemia in patients with FH by acutely lowering levels of apoB-containing particles2 and other cardiovascular risk factors.3–5
Our goal was to investigate the direct effect of LA on plasma PCSK9. Here, we show that LA with dextran sulfate cellulose beads decreases plasma PCSK9 levels by more than 50%, and that >80% of PCSK9 lost from plasma is recovered from the column eluate. The decreases in plasma PCSK9 levels caused by LA are similar to those attained in bi-weekly injections of low-dose anti-PCSK9 antibody therapy, which resulted in an average LDL drop of 24%.25 This means that the PCSK9-lowering effect of LA should explain a portion of the maintenance of low LDL levels after each apheresis session and that LDL levels would more rapidly return to baseline without the drop in PCSK9. After LA, plasma LDL levels progressively increase and return to baseline after 10 to 14 days.26,27 Similarly, in our study, PCSK9 levels returned to pretreatment values between treatment cycles. The reduction in PCSK9 during apheresis significantly correlated (r2=0.26) with LDL reduction. These results imply that the correlation between PCSK9 and LDL-C or apoB levels14,15,16 reported in the literature is not only due to the known effect of PCSK9 on LDLR, but also due to the physical association between PCSK9 and LDL.
We show that LA results in removal of both LDL-bound and apoB-free PCSK9, and that LDL-bound and apoB-free PCSK9 are different with nonoverlapping molecular forms. We show for the first time that the 62 kDa band, which is the intact and functional form of PCSK9, is only present on the LDL fraction of these patients. This observation was previously described by us and others only in transgenic mice expressing human PCSK9.10–12 This suggests either that only intact PCSK9 has affinity for LDL or that the association with the LDL particle protects PCSK9 from cleavage and multi-merization. We also show that both the furin-cleaved 55 kDa form and the low levels of higher-molecular-weight forms (likely homo- or hetero-multimers)28 of PCSK9 are found exclusively in the apoB-free fraction of the plasma. These observations imply that: (1) If intact PCSK9 is the functional protein, then current ELISA methods that test for total plasma PCSK9 levels should be replaced with more specific methods to quantify the 62 kDa form. (2) Because the 62 kDa form is exclusively carried by the LDL particle, then the catabolic destiny of PCSK9 and its effect on LDLR are both driven by the canonical ligand for this receptor. (3) Immune complexes between therapeutic antibodies and target PCSK9 should form on the LDL particle and may be removed via LDL-dependent mechanisms.
Using a Web-based tool (http://isoelectric.ovh.org/),29 we predicted that PCSK9 is negatively charged at plasma pH (full length protein pI=6.6; furin-cleaved fragment pI=7.02) and, as such, not likely to bind the dextran sulfate apheresis column directly, which is also negatively charged. Using purified GST-tagged PCSK9, we show that the intact PCSK9 protein is not removed by the dextran sulfate column, which suggests that the significant loss of plasma PCSK9 we see during LA is mediated by interacting partner protein(s).
Anti-PCSK9 antibodies are seen by most as likely to achieve Food and Drug Administration approval for cholesterol control in patients with less severe hypercholesterolemia than that required for LA qualification.30 More recently, anti-PCSK9 antibodies have been shown to cause significant LDL lowering even in homozygous FH subjects, a group of patients that is currently treated mainly by LA.24 Because the reduction in PCSK9 could contribute to the long-term LDL control caused by LA therapy, anti-PCSK9 therapies may reduce the frequency of LA in patients currently approved for therapy, and LA and anti-PCSK9 therapies may be used synergistically to reduce treatment burden.
Limitation of the Study
This study is based on data from only 6 patients undergoing LA. Therefore, there was limited statistical power to study associations between PCSK9 loss and patient characteristics. PCSK9 was measured only in pre- and post-apheresis plasma, and recovery rate of PCSK9 was not studied. In addition, all patients were treated with the most common LA method in the United States (Liposorber, dextran sulfate cellulose beads). It remains to be determined whether other LA interventions such as heparin-induced extracorporeal LDL precipitation or direct adsorption of lipids have similar effects on plasma PCSK9.
Supplementary Material
Novelty and Significance.
What Is Known?
Low-density lipoprotein (LDL) cholesterol lowering is the most common treatment to reduce the risk of atherosclerotic cardiovascular disease, and statins are the drug of choice for many to achieve the desired level.
PCSK9 circulates in plasma and is responsible for post-transcriptional reduction of hepatic surface LDL receptor.
Anti-PCSK9 antibodies are proving to be safe and effective in reducing LDL cholesterol levels and are moving toward Food and Drug Administration approval.
LDL-apheresis (LA) is currently used in patients with familial hyper-cholesterolemia when LDL >200 mg/dL (>300 if no cardiovascular disease).
An association of plasma PCSK9 with the LDL particle has been reported.
What New Information Does This Article Contribute?
We show for the first time that LDLA reduces plasma PCSK9 levels, by >50%.
The loss of PCSK9 is in excess of that predicted by the extent of association between PCSK9 and LDL.
Reduction in plasma PCSK9 levels correlated to reduction in LDL cholesterol levels.
In plasma, the active PCSK9 (62 kDa) is only present on the LDL.
A significant portion of PCSK9 not associated with LDL is also removed by LA.
PCSK9 does not directly bind to the apheresis column.
The loss of LDL-bound PCSK9 is because of the removal of LDL, whereas the loss of LDL-free PCSK9 is likely because of the presence of interacting partner proteins.
Both the existence and the clinical significance of LDL-bound PCSK9 have been a matter of debate. In this study, the physical removal of LDL via LA enabled us to prove definitively that a significant portion of PCSK9 is part of the LDL particle. More importantly, we show that all of the intact and likely most active form of PCSK9 is exclusively associated with LDL. Our study suggests that PCSK9 removal could contribute to LDL lowering of LA. This finding should direct studies on the synergistic effect of LA and anti-PCSK9 therapies and foster investigations on the fate of PCSK9-containing LDL after antibody treatment.
Acknowledgments
We thank Dr Patricia Yancey for insightful discussion and advice, and Kimberly Campbell for performing the lipid analyses.
Sources of Funding
This study was supported by National Institutes of Health (NIH–NHLBI) grants R01-HL106845 to S.F. and R01-HL111945 to M.F.L. We acknowledge support from the Atherosclerosis Core of Vanderbilt’s Mouse Metabolic Phenotyping Center (NIH grant DK59637).
Nonstandard Abbreviations and Acronyms
- apo
apolipoprotein
- FH
familial hypercholesterolemia
- LA
lipoprotein apheresis
- LDL-C
low-density lipoprotein cholesterol
- PCSK9
proprotein convertase subtilisin/kexin 9
Footnotes
Brief UltraRapid Communications are designed to be a format for manuscripts that are of outstanding interest to the readership, report definitive observations, but have a relatively narrow scope. Less comprehensive than Regular Articles but still scientifically rigorous, BURCs present seminal findings that have the potential to open up new avenues of research. A decision on BURCs is rendered within 7 days of submission.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.113.302655/-/DC1.
Disclosures
None.
References
- 1.Liyanage KE, Burnett JR, Hooper AJ, van Bockxmeer FM. Familial hypercholesterolemia: epidemiology, Neolithic origins and modern geographic distribution. Crit Rev Clin Lab Sci. 2011;48:1–18. doi: 10.3109/10408363.2011.565585. [DOI] [PubMed] [Google Scholar]
- 2.Stein EA. Drug and alternative therapies for hyperlipidemia. Atherosclerosis. 1994;108 (suppl):S105–S116. doi: 10.1016/0021-9150(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi S, Oka M, Moriya H, Maesato K, Okamoto K, Ohtake T. LDL-apheresis reduces P-Selectin, CRP and fibrinogen – possible important implications for improving atherosclerosis. Ther Apher Dial. 2006;10:219–223. doi: 10.1111/j.1744-9987.2006.00332.x. [DOI] [PubMed] [Google Scholar]
- 4.Dihazi H, Koziolek MJ, Söllner T, Kahler E, Klingel R, Neuhoff R, Strutz F, Mueller GA. Protein adsorption during LDL-apheresis: proteomic analysis. Nephrol Dial Transplant. 2008;23:2925–2935. doi: 10.1093/ndt/gfn127. [DOI] [PubMed] [Google Scholar]
- 5.Stefanutti C, Morozzi C, Petta A. Lipid and low-density-lipoprotein apheresis. Effects on plasma inflammatory profile and on cytokine pattern in patients with severe dyslipidemia. Cytokine. 2011;56:842–849. doi: 10.1016/j.cyto.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 6.Benjannet S, Rhainds D, Essalmani R, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 7.Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, Wu R, Pordy R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]
- 8.Koren MJ, Scott R, Kim JB, Knusel B, Liu T, Lei L, Bolognese M, Wasserman SM. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as mono-therapy in patients with hypercholesterolaemia (mendel): a randomised, double-blind, placebo-controlled, phase 2 study. The Lancet. 2012;380:1995–2006. doi: 10.1016/S0140-6736(12)61771-1. [DOI] [PubMed] [Google Scholar]
- 9.Giugliano RP, Desai NR, Kohli P, Rogers WJ, Somaratne R, Huang F, Liu T, Mohanavelu S, Hoffman EB, McDonald ST, Abrahamsen TE, Wasserman SM, Scott R, Sabatine MS. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (laplacetimi 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet. 2012;380:2007–2017. doi: 10.1016/S0140-6736(12)61770-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan D, Yancey PG, Qiu S, Ding L, Weeber EJ, Linton MF, Fazio S. Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry. 2008;47:1631–1639. doi: 10.1021/bi7016359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun H, Samarghandi A, Zhang N, Yao Z, Xiong M, Teng BB. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol. 2012;32:1585–1595. doi: 10.1161/ATVBAHA.112.250043. [DOI] [PubMed] [Google Scholar]
- 12.Tavori H, Fan D, Blakemore JL, Yancey PG, Ding L, Linton MF, Fazio S. Serum proprotein convertase subtilisin/kexin type 9 and cell surface low-density lipoprotein receptor: evidence for a reciprocal regulation. Circulation. 2013;127:2403–2413. doi: 10.1161/CIRCULATIONAHA.113.001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low-density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (pcsk9) in human plasma and inhibits pcsk9-mediated ldl receptor degradation. J Biol Chem. 2013;288:8279–8288. doi: 10.1074/jbc.M112.421370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert G, Ancellin N, Charlton F, Comas D, Pilot J, Keech A, Patel S, Sullivan DR, Cohn JS, Rye KA, Barter PJ. Plasma PCSK9 concentrations correlate with LDL and total cholesterol in diabetic patients and are decreased by fenofibrate treatment. Clin Chem. 2008;54:1038–1045. doi: 10.1373/clinchem.2007.099747. [DOI] [PubMed] [Google Scholar]
- 15.Mayne J, Raymond A, Chaplin A, Cousins M, Kaefer N, Gyamera-Acheampong C, Seidah NG, Mbikay M, Chrétien M, Ooi TC. Plasma PCSK9 levels correlate with cholesterol in men but not in women. Biochem Biophys Res Commun. 2007;361:451–456. doi: 10.1016/j.bbrc.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 16.Chan DC, Lambert G, Barrett PH, Rye KA, Ooi EM, Watts GF. Plasma proprotein convertase subtilisin/kexin type 9: a marker of LDL apolipoprotein B-100 catabolism? Clin Chem. 2009;55:2049–2052. doi: 10.1373/clinchem.2009.128645. [DOI] [PubMed] [Google Scholar]
- 17.Stein EA, Raal FJ. Insights into PCSK9, low-density lipoprotein receptor, and low-density lipoprotein cholesterol metabolism: of mice and man. Circulation. 2013;127:2372–2374. doi: 10.1161/CIRCULATIONAHA.113.003360. [DOI] [PubMed] [Google Scholar]
- 18.Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The pro-protein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem. 2006;281:30561–30572. doi: 10.1074/jbc.M606495200. [DOI] [PubMed] [Google Scholar]
- 19.Lipari MT, Li W, Moran P, Kong-Beltran M, Sai T, Lai J, Lin SJ, Kolumam G, Zavala-Solorio J, Izrael-Tomasevic A, Arnott D, Wang J, Peterson AS, Kirchhofer D. Furin-cleaved proprotein convertase subtilisin/kexin type 9 (PCSK9) is active and modulates low density lipoprotein receptor and serum cholesterol levels. J Biol Chem. 2012;287:43482–43491. doi: 10.1074/jbc.M112.380618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Group SSCobotSBR. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ. 1991;303:893–896. doi: 10.1136/bmj.303.6807.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Briggs CJ, Anderson D, Johnson P, Deegan T. Evaluation of the polyethylene glycol precipitation method for the estimation of high-density lipoprotein cholesterol. Ann Clin Biochem. 1981;18:177–181. doi: 10.1177/000456328101800309. [DOI] [PubMed] [Google Scholar]
- 22.Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4:214–225. doi: 10.1038/ncpcardio0836. [DOI] [PubMed] [Google Scholar]
- 23.Gao F, Ihn HE, Medina MW, Krauss RM. A common polymorphism in the LDL receptor gene has multiple effects on LDL receptor function. Hum Mol Genet. 2013;22:1424–1431. doi: 10.1093/hmg/dds559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the pcsk9 monoclonal antibody, amg 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–2120. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 25.Dias CS, Shaywitz AJ, Wasserman SM, et al. Effects of AMG 145 on low-density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo-controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60:1888–1898. doi: 10.1016/j.jacc.2012.08.986. [DOI] [PubMed] [Google Scholar]
- 26.Kroon AA, van’t Hof MA, Demacker PN, Stalenhoef AF. The rebound of lipoproteins after LDL-apheresis. Kinetics and estimation of mean lipoprotein levels. Atherosclerosis. 2000;152:519–526. doi: 10.1016/s0021-9150(00)00371-3. [DOI] [PubMed] [Google Scholar]
- 27.Gordon BR, Kelsey SF, Bilheimer DW, Brown DC, Dau PC, Gotto AM, Jr, Illingworth DR, Jones PH, Leitman SF, Prihoda JS. Treatment of refractory familial hypercholesterolemia by low-density lipoprotein apheresis using an automated dextran sulfate cellulose adsorption system. The Liposorber Study Group. Am J Cardiol. 1992;70:1010–1016. doi: 10.1016/0002-9149(92)90352-y. [DOI] [PubMed] [Google Scholar]
- 28.Fan D, Qiu S, Overton CD, Yancey PG, Swift LL, Jerome WG, Linton MF, Fazio S. Impaired secretion of apolipoprotein E2 from macrophages. J Biol Chem. 2007;282:13746–13753. doi: 10.1074/jbc.M611754200. [DOI] [PubMed] [Google Scholar]
- 29.Sillero A, Maldonado A. Isoelectric point determination of proteins and other macromolecules: oscillating method. Comput Biol Med. 2006;36:157–166. doi: 10.1016/j.compbiomed.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 30.The New York Times. Jul 10, 2013. Rare mutation ignites race for cholesterol drug. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.