

SUMMARY
Axonal degeneration arises as a consequence of neuronal injury and is a common hallmark of a number of neurodegenerative diseases. However, the genetic causes and the cellular mechanisms that trigger this process are still largely unknown. Based on forward genetic screening in C. elegans, we have identified the α-tubulin acetyltransferase gene mec-17 as causing spontaneous, adult-onset, and progressive axonal degeneration. Loss of MEC-17 leads to microtubule instability, a reduction in mitochondrial number, and disrupted axonal transport, with altered distribution of both mitochondria and synaptic components. Furthermore, mec-17-mediated axonal degeneration occurs independently from its acetyltransferase domain; is enhanced by mutation of coel-1, a tubulin-associated molecule; and correlates with the animal’s body length. This study therefore identifies a critical role for the conserved microtubule-associated protein MEC-17 in preserving axon integrity and preventing axonal degeneration.
INTRODUCTION
Maintenance of axonal structure is critical for neuronal function. Under normal conditions, the axon is maintained in a healthy state through a constant supply of materials via its attachment to the cell body. This connection is lost after nerve injury and becomes compromised in a number of neurodegenerative diseases, leading to degeneration of the axon (Coleman, 2005; Raff et al., 2002). Specific cellular mechanisms that have been linked to axonal degeneration include deficits in axonal transport, mitochondrial dysfunction, increase in intra-axonal calcium, and calcium-dependent cytoskeletal breakdown (Coleman, 2005; Hilliard, 2009; Wang et al., 2012). A major component of the cytoskeleton is formed by the microtubules, cylindrical structures assembled from chains of α- and β-tubulin heterodimers termed protofilaments (Desai and Mitchison, 1997). These highly dynamic structures are essential for many cellular functions, including intracellular transport and organelle positioning, as well as cell division and polarity (Westermann and Weber, 2003). Microtubules are subjected to a wide range of posttranslational modifications, including acetylation (Janke and Bulinski, 2011), the major site of which occurs on lysine 40 of α-tubulin inside the microtubule lumen (Nogales et al., 1998). Although it has largely been considered as a passive marker of stable microtubules (Hammond et al., 2008), several studies have postulated that acetylation of α-tubulin is required for efficient axonal transport, allowing greater binding and movement of motor proteins (Cai et al., 2009; d’Ydewalle et al., 2011; Dompierre et al., 2007; Hammond et al., 2010; Konishi and Setou, 2009; Reed et al., 2006). Loss or reduction in the levels of acetylated α-tubulin has also been associated with a number of pathological conditions, including familial dysautonomia and Alzheimer’s, Huntington’s, and Charcot-Marie-Tooth diseases (d’Ydewalle et al., 2011; Dompierre et al., 2007; Gardiner et al., 2007; Hempen and Brion, 1996).
Despite acetylation of K40 first being described in 1985 (L’Hernault and Rosenbaum, 1985), the identity of the α-tubulin acetyltransferase enzyme and the precise functional consequence of the modification remained unresolved until recently, when MEC-17/αTAT1 was identified as the acetylating enzyme conserved across all ciliated organisms (Akella et al., 2010; Shida et al., 2010). Significant insights into its function have now emerged, with two recent studies in C. elegans demonstrating that MEC-17 is critical for microtubule organization, stabilizing their number and length, and defining protofilament number (Cueva et al., 2012; Topalidou et al., 2012). Furthermore, Cueva et al. (2012) propose that K40 acetylation promotes the formation of stabilizing salt bridges between protofilaments, thereby creating structural supports within the microtubule lumen. In spite of the apparent importance of MEC-17, only a few morphological alterations have been linked to its loss. These include an increase in microtubule dynamics in Tetrahymena (Akella et al., 2010), a progressive loss of mechanosensory neuron function and minor neurite outgrowth defects in C. elegans (Topalidou et al., 2012; Zhang et al., 2002), and behavior consistent with neuromuscular defects in zebrafish (Akella et al., 2010). In C. elegans, MEC-17 is expressed solely in the six mechanosensory neurons (Zhang et al., 2002) and functions redundantly with its more broadly expressed paralog, ATAT-2 (Akella et al., 2010; Shida et al., 2010).
Here, we show that MEC-17 is critical for maintaining axonal structure, with mutations causing spontaneous, adult-onset, and progressive axonal degeneration in C. elegans. This phenotype is independent of the role of MEC-17 in acetylating α-tubulin, as an enzymatically inactive version retains the ability to protect axons from degeneration. Loss of MEC-17 disrupts axonal transport, causing a decrease in the number and spatial arrangement of axonal mitochondria and an altered distribution of presynaptic loci. We find that MEC-17 functions in synergy with the microtubule-associated protein COEL-1 to stabilize microtubules and protect from axon fragility. Furthermore, our results establish that axonal degeneration can be strongly enhanced by elongation of the axonal shaft resulting from increased body length.
RESULTS
Isolation of the ky850 Strain with Axonal Degeneration
To identify factors required for the maintenance of axonal structure, we performed forward genetic screens using a C. elegans strain expressing GFP in the six mechanosensory neurons (PLML/R, PVM, ALML/R, and AVM; Figure 1A). This wild-type strain, carrying the transgene zdIs5(Pmec-4::GFP), effectively displays no axonal degeneration (Figure S1A). We visually screened for animals displaying spontaneous axonal degeneration and identified the recessive ky850 mutation as presenting GFP interruptions (axonal breaks) in the PLM, ALM, and AVM axons (Figure 1B). Degeneration of the separated distal fragments occurred in a stereotypical Wallerian-like fashion, with thinning, beading, and fragmentation occurring over the 24–96 hr following the initial breaks, but did not lead to a “die-back” phenotype. The defect appeared selectively in adult animals (adult-onset), and the penetrance increased progressively with age, reaching a maximum of 45% in PLM (Figure S1B). ky850 animals displayed a deficit in their response to gentle mechanical stimuli (light-touch assay) applied to either their head or tail, indicating that both the anterior and posterior mechanosensory circuits (mediated by ALMs/AVM and PLMs, respectively) were dysfunctional (Figure S1C). In addition to axonal degeneration, we observed axonal outgrowth defects in ky850 animals that appeared during development and worsened with age (Figures S1D and S1E).
Figure 1. Identification and Mapping of the ky850 Mutation.

(A) Image and schematic of a wild-type zdIs5(Pmec-4::GFP) larval-stage four (L4) animal, illustrating the six mechanosensory neurons (PLML/R, PVM, ALML/R, and AVM). Anterior is left and ventral is down in this and all subsequent images.
(B) Image and schematic of a typical intact PLM neuron from a 3-day-old adult wild-type animal (B1) and that of a typical axon break (bracketed region) in a 3-day-old adult ky850 mutant (B2).
(C) Axonal breaks quantified in mec-17(ok2109) animals of progressive ages. Similar results were obtained for mec-17(u265) animals (data not shown). DOA, day-old adult.
(D) Penetrance of axonal breaks in ky850, QH4387 (3x outcrossed ky850), and mec-17(ok2109) animals, carrying the Pmec-4::mec-17 rescue transgene versus nontransgenic siblings.
(E) Penetrance of axonal degeneration in ky850 (black bar) animals and the different mec-17 alleles (gray bars). F1 cross progeny (white bars) were analyzed for phenotypic complementation. The scale bars represent 25 μm; error bars represent SE of proportion; n ≥ 100 animals; ***p < 0.001.
The ky850 Mutation Is an Allele of mec-17
Several lines of evidence revealed that ky850 is an allele of the α-tubulin acetyltransferase gene mec-17. First, direct mapping from whole-genome sequencing data (Zuryn et al., 2010) revealed a strong peak on chromosome IV at the precise location of mec-17 (Figure S1F), and we identified a C-T transition at nucleotide position 79 of the gene, resulting in the introduction of a stop codon in the encoded protein, truncating MEC-17 from 262 amino acids to 26 (Figure S1G). Second, cell-autonomous expression of wild-type MEC-17 in the mechano-sensory neurons (using a Pmec-4::mec-17 transgene) provided strong rescue of the degenerative phenotype (Figure 1D). Third, two other alleles of mec-17 (ok2109 and u265; Figure S1G) also presented with progressive, adult-onset axonal breaks in the mechanosensory neurons (Figure 1C). However, although the axon breaks were morphologically identical, the penetrance of degeneration in these strains was lower than in animals with the ky850 mutation (21% compared to 45% in 5-day-old adults). This discrepancy is likely due to a background effect of additional mutations in the ky850 strain, as outcrossing ky850 reduced the penetrance of axonal degeneration to levels similar to those in mec-17(ok2109) animals (Figures 1D and 1E). Importantly, cell-autonomous expression of wild-type MEC-17 in either this outcrossed ky850 strain (QH4387) or in the mec-17(ok2109) strain strongly rescued the degeneration observed in the PLM axon (Figure 1D). As previously described (Topalidou et al., 2012), the two other mec-17 alleles displayed outgrowth defects in PLM and ALM, which were similar to those of ky850 mutants, but again to a lower penetrance (Figure S1E). Finally, as we found all three alleles of mec-17 (ky850, ok2109, and u265) to be recessive (no axonal degeneration in heterozygous animals; n > 100), we conducted complementation analyses of ky850 or the outcrossed ky850 strain (QH4387) with ok2109 and u265, which revealed noncomplementation (Figure 1E), thereby confirming that the three mutations are alleles of the same gene.
Loss of mec-17 Leads to Disruption of Mitochondria and Axonal Transport
To characterize the intra-axonal mechanisms disrupted by loss of MEC-17 function, we first analyzed mitochondria using a fluorescently tagged version of the translocase of outer mitochondrial membrane 20 protein (Kanaji et al., 2000; Figure 2A). The average number of mitochondria in mec-17 (ok2109) animals was reduced compared to wild-type at both the L4 and adult stages (Figures 2A–2C). Furthermore, mec-17(ok2109) animals displayed a striking disruption in the localization of their mitochondria. Wild-type animals presented a relatively even distribution of mitochondria in the PLM axon in the L4 stage and a slightly skewed distribution toward the cell body in adulthood (Figure 2D). In contrast, mec-17(ok2109) animals had a skewed distribution of mitochondria at the L4 stage, with a reduced number of mitochondria in the distal segment. This defect was severely enhanced in adult animals, with the distal segment becoming largely devoid of mitochondria (Figures 2B and 2D). Interestingly, it was in these distal regions with reduced mitochondrial number that we observed the majority of the axonal breaks. In addition, we found that mec-17(ok2109) animals had a large increase in the number of mitochondria localized in the posterior PLM neurite (Figure 2E), corresponding to the additional outgrowth defects observed in mec-17 mutants. We also observed similar mitochondrial defects in ALM neurites (Figures S2A–S2C). Taken together, these results uncover a critical role of MEC-17 in regulating the number and localization of mitochondria in the mechanosensory neurons.
Figure 2. mec-17 Mutants Display a Reduction in Axonal Mitochondria and a Clustering toward the Cell Body.

(A and B) Image and schematic of mitochondria in PLM in both wild-type (A) and mec-17(ok2109)
(B) 3-day-old adults. Mitochondria (arrowheads) were visualized with the vdEx484[Pmec-4::tomm-20::mRFP] transgene. (B1 and B2) Magnified view of the corresponding regions in the mec-17 mutant animal to highlight faint mitochondria.
(C) Average number of mitochondria in PLM in wild-type and mec-17(ok2109) animals.
(D) Quantification of mitochondria in proximal (closest third to the cell body), central (middle third), and distal (final third) regions of the PLM axon in L4 and 3-day-old adults.
(E) Average number of mitochondria in the PLM posterior neurite in wild-type and mec-17(ok2109) animals.
The scale bars represent 25 μm; the error bars represent SEM; *p < 0.05; **p < 0.01; ***p < 0.001; n ≥ 26 animals.
A possible explanation for the mitochondrial defects is a disruption in axonal transport. We analyzed a fluorescently tagged version of UNC-104/kinesin-3 (Kumar et al., 2010), one of the main motor molecules responsible for transport of synaptic vesicles to presynaptic loci (Hall and Hedgecock, 1991). Wild-type animals exhibited a consistent distribution of fluorescence in PLM, with a smooth increase in expression along the axon in a proximal-to-distal fashion, and pooling at the distal end and at presynaptic sites (Figure 3A). By comparison, the vast majority of mec-17(ok2109) mutant animals displayed a bipolar distribution, with strong expression also observed in the posterior neurite (Figures 3B and 3C), and approximately 7% of animals completely lacked expression of UNC-104 at the presynaptic loci of PLM (Figure 3C). Furthermore, more than 25% of animals lacked accumulation at the distal end of the axon (Figure 3D) and instead displayed accumulation of bright puncta along more proximal sections of the axon (Figure 3E), indicative of disrupted axonal transport. To correlate these findings with localization of presynaptic components, we studied the distribution of the synaptic vesicle-associated small guanosine triphosphatase, RAB-3 (Nonet et al., 1997). Expression of RAB-3 in wild-type animals was strongly localized to the presynaptic terminals and only weakly within the cell body and proximal anterior axon (Figure 4A). On the contrary, 24% of mec-17(ok2109) mutant animals completely lacked expression of RAB-3 at the presynaptic sites of PLM (Figure 4C). Similar to the distribution of UNC-104, we also observed bipolar distribution of RAB-3 in mec-17(ok2109) animals, with clusters visible in the ends of the posterior neurite (Figures 4B and 4C). Similar deficits in the localization of UNC-104 and RAB-3 were observed in ALM neurons of mec-17 mutants (Figures S2D and S2E).
Figure 3. mec-17 Animals Display Aberrant Axonal Transport.

(A and B) Image and schematic of UNC-104/ kinesin-3 in PLM of wild-type (A) and mec-17(ok2109) (B) 3-day-old adult animals used to monitor axonal transport. UNC-104 was visualized using the jsIs1111[Pmec-4::unc-104::GFP] transgene. Insets show a magnified view of the distal axon tips. Arrowheads point to the distal tip of the PLM axon. The scale bars represent 25 μm.
(C) Quantification of UNC-104::GFP localization in PLM in wild-type and mec-17(ok2109) animals. Anterior neurite defined as per image shown in (A), with anterior localization and accumulation at presynaptic sites. Gray bars indicate additional localization in the PLM posterior neurite. White bars designate localization in both the anterior and posterior neurites with no presynaptic accumulation.
(D) Comparison of the number of animals displaying UNC-104::GFP pooling at the distal end of the PLM axon.
(E) Accumulation of UNC-104::GFP measured in different regions along the PLM axon in wild-type and mec-17(ok2109) animals at the L4 stage and in 3-day-old adults. This analysis excludes pooling at the distal tip.
The error bars represent SE of proportion; *p < 0.05; ***p < 0.001; n ≥ 51 animals.
Figure 4. mec-17 Animals Display Aberrant Localization of Presynaptic Components.
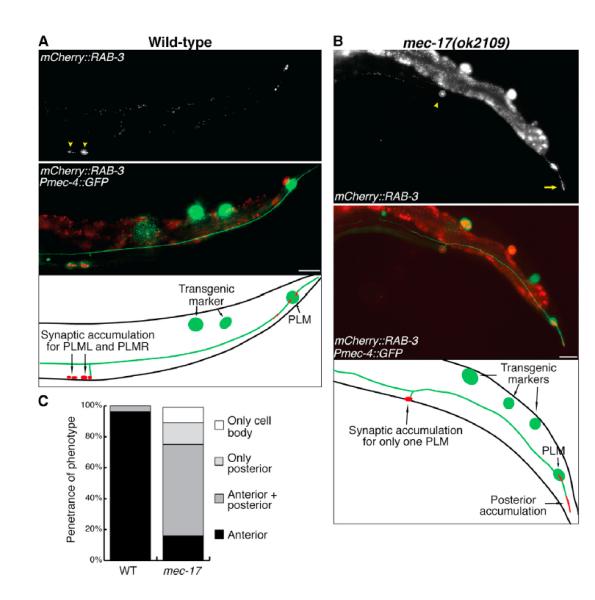
(A and B) Image and schematic of PLM presynaptic loci in wild-type (A) and mec-17(ok2109)
(B) 3-day-old adult animals visualized using the vdEx262[Pmec-4::mCherry::rab-3] transgene. Arrowheads point to presynaptic sites; arrow points to accumulation in the posterior neurite. The scale bars represent 25 μm.
(C) Localization of mCherry::RAB-3 analyzed in wild-type and mec-17(ok2109) animals. Anterior refers to localization in the anterior neurite (as in A); posterior refers to localization in the posterior neurite (as in B). n ≥ 52 animals.
Collectively, these findings suggest that, in the absence of MEC-17, axonal transport is defective, and as a result, the localization of presynaptic components and the number and distribution of mitochondria are disrupted, culminating in degeneration of the axon.
MEC-17-Mediated Acetylation of MEC-12 Is Not Required for Axonal Maintenance
MEC-17 has been demonstrated to function redundantly with its paralog, ATAT-2, in the acetylation of MEC-12/α-tubulin (Akella et al., 2010; Shida et al., 2010). To determine if ATAT-2 is also required in the maintenance of axonal structure, we analyzed atat-2(ok2415) animals, which have a deletion eliminating almost 60% of the atat-2 transcript. Single mutants displayed no degenerative phenotype in the mechanosensory neurons, and double mutants with mec-17 were not different from mec-17 single mutants (Figure S3A), demonstrating that ATAT-2 does not function in axonal maintenance in these neurons. Next, to determine if the acetyltransferase domain of MEC-17 is needed for axonal maintenance, we generated a mutant version (MEC-17 [D144N]) lacking the ability to acetylate MEC-12/α-tubulin (Shida et al., 2010; Topalidou et al., 2012) and introduced it selectively in the mechanosensory neurons of mec-17 mutant animals. As shown in Figure 5A, this version of MEC-17 could still rescue axonal breakage. Moreover, this acetyltransferase-dead version of MEC-17 could also completely rescue the mitochondrial distribution defect observed in mec-17(ok2019) mutant animals (Figures 5B and S3B), demonstrating that the acetyltransferase domain is not required for MEC-17 to prevent axonal degeneration. To determine whether MEC-12/α-tubulin functions in the mec-17-mediated degenerative phenotype, we studied animals carrying mutations in the mec-12 gene. Two different alleles of mec-12 (either u63 or u241; Figure S3C) generated a highly penetrant, largely adult-onset, and severe degenerative phenotype in all six mechanosensory neurons (Figures 5C and S3E– S3G). Axonal degeneration of PLM in the double mutants with mec-17 showed an allele-dependent effect (increased degeneration with u63 and decreased with the dominant allele u241) confirming the functional interaction between MEC-17 and MEC-12. Interestingly, loss of function of the other microtubule subunit, MEC-7/β-tubulin, also caused severe axonal degeneration in the mechanosensory neurons (Figures 5D, S3D, and S3H) that was exacerbated by mutation in mec-17, again indicating functional synergy. Furthermore, we found similar defects in the distribution of mitochondria in mec-12 and mec-7 mutant animals as those in mec-17 mutants (Figures S3I and S3J). To further investigate the role of the MEC-17 acetyltransferase domain in axonal degeneration, we analyzed a mutant version of MEC-12 that could not be acetylated at the K40 site (K40R; Akella et al., 2010). As shown in Figure 5E, MEC-12(K40R) could rescue the degeneration observed in mec-12(e1607) animals to similar levels as wild-type (WT) MEC-12. Finally, we found that MEC-12(K40R) animals display mitochondria along the PLM axon as per WT animals (Figures 5B and S3B). Together, these data conclusively demonstrate that acetylation of MEC-12 is redundant for MEC-17’s role in maintaining axonal structure and reveal a critical role for each microtubule subunit, MEC-12/α-tubulin and MEC-7/β-tubulin, in maintaining axonal integrity after development.
Figure 5. MEC-17 Acetyltransferase Activity Is Not Required for Axonal Maintenance.

(A) Expression of MEC-17 lacking acetyltransferase activity from the Pmec-4::mec-17(D144N) transgene rescues the axonal degeneration phenotype in ky850, QH4387 (3x outcrossed ky850), and mec-17(ok2109) animals, compared to their nontransgenic siblings.
(B) Expression of the Pmec-4::mec-17(D144N) transgene rescues the mitochondrial distribution defect found in 3-day-old adult mec-17(ok2109) animals, restoring their localization to the WT pattern. MEC-12(K40R) (lacking acetylation residue) animals display a normal distribution of mitochondria.
(C) Quantification of axonal degeneration (L4 stage) in animals carrying single and double mutations in MEC-17 and MEC-12/α-tubulin.
(D) Comparison of axonal degeneration (L4 stage) in mec-17 and mec-7 (encoding β-tubulin) single and double mutants.
(E) Rescue of PLM axonal degeneration in mec-12(e1607) animals with WT MEC-12 or with the MEC-12(K40R) mutated version.
The error bars represent SE of proportion (A and C–E) and SE (B); *p < 0.05; **p < 0.01; ***p < 0.001; n ≥ 100 animals (A and C–E) and ≥24 animals (B).
Mutation of mec-17 Leads to Microtubule Instability
Although the acetylation activity of MEC-17 was not required for its function in axonal maintenance, our results with MEC-12 and MEC-7 suggested that loss of mec-17 might negatively affect microtubule stability. To directly visualize microtubule dynamics in PLM, we analyzed a GFP-tagged version of end-binding protein 2 (EBP-2), a marker of plus-end-growing microtubules (Srayko et al., 2005). As the majority of EBP-2::GFP “comets” (growing microtubules) have previously been demonstrated to occur close to the cell body under resting conditions (Ghosh-Roy et al., 2012), we used spinning-disk confocal microscopy and kymographs to analyze the initial 25 μm of the PLM axon (Figures 6A and 6B; Movie S1). Remarkably, as shown in Figure 6C and Movie S2, animals carrying the mec-17(ok2109) mutation displayed a 2-fold increase in the number of growing microtubules compared to WT animals. Moreover, whereas the vast majority of microtubules grew in the anterograde direction in WT animals, mec-17(ok2109) mutants presented a significantly higher proportion of EBP-2::GFP comets moving in the retrograde direction (Figure 6D). Whereas no changes in microtubule track length, growth duration, or catastrophe frequency were observed, the velocity of microtubule growth was significantly reduced in adult mec-17(ok2109) animals (Figures S4A–S4D). These data indicate that loss of MEC-17 function leads to a destabilization of the microtubule network, causing a greater frequency of microtubule growth and disrupting microtubule polarity within the axon.
Figure 6. Loss of mec-17 Causes Microtubule Instability and Axon Fragility.

(A) A representative image of PLM in a L4 WT animal carrying the Pmec-4::ebp-2::GFP transgene that highlights the 25 μm region of interest (ROI) analyzed in the kymograph shown below the image. Schematic adjacent to kymograph illustrates how comets were traced and measured for growth length and duration. White arrows point to comets growing in the anterograde direction.
(B) Representative image of EBP-2::GFP expressed in PLM of a mutant mec-17(ok2109) L4 animal, with an increased number of microtubule comets, as evident in the kymograph below the image. White arrows point to comets growing in the anterograde direction; red arrows to those in the retrograde direction. For both (A) and (B), the scale bar in the images represents 10 μm; scale bars in the kymographs represent 10 μm for the x axis, and 10 s for the y axis.
(C) Mutation of mec-17(ok2109) causes a significant increase in the number of EBP-2::GFP comets compared to WT in L4 and 3-day-old adult animals. n = 15 animals for WT, and n ≥ 11 for mec-17(ok2109).
(D) EBP-2::GFP comets grow significantly more in the retrograde direction in both L4 and 3-day-old adult mec-17(ok2109) mutants compared to WT. n ≥ 25 comets for WT and n ≥ 69 for mec-17(ok2109).
(E) mec-17(ok2109) animals were grown on control agar containing 1% DMSO or agar containing either 1 μM paclitaxel (microtubule stabilizer) or 0.1 M colchicine (microtubule destabilizer) and scored as 2-day-old adults.
(F) Penetrance of axonal degeneration in 3-day-old adult coel-1(gk1236), mec-17(ok2109), or coel-1; mec-17 double mutants.
(G) Animals carrying the ky850 or mec-17(ok2109) mutations were paralyzed by microinjection of unc-54 dsRNA and the penetrance of breaks compared to that in nontreated animals.
The error bars represent SE (C and D) and SE of proportion (E–G); *p < 0.05; **p < 0.01; ***p < 0.001; n ≥ 100 animals for (E–G).
We extended our analyses of microtubule stability by treating animals with microtubule-stabilizing (paclitaxel) or -destabilizing (colchicine) compounds (Fojo, 2008). Treatment of mec-17 mutants with paclitaxel led to a more than 6-fold decrease in the penetrance of PLM axonal degeneration (Figures 6E, S4E, S4G, and S4H). Next, using a concentration that had no effect on wild-type animals, we treated mec-17 mutants with colchicine and found a more than 5-fold increase in axonal degeneration (Figures 6E and S4F–S4H). Interestingly, the mitochondrial distribution defects observed in mec-17 mutants (Figure 2D) were rescued with paclitaxel treatment and exacerbated by colchicine (Figures S4I and S4J), supporting a correlation between mitochondrial mislocalization and axonal degeneration.
We further tested the effect of mec-17 mutation on axonal stability by investigating the genetic interaction between mec-17 and coel-1. COEL-1 is homologous to the tubulin-folding cofactor E-like molecule, which is involved in tubulin stability (Bartolini et al., 2005). Mutation of coel-1 by itself was not sufficient to induce a degenerative phenotype in the mechano-sensory neurons and did not present an altered distribution of mitochondria (Figures 6F and S5A–S5C). However, when combined with mutation of mec-17, we observed a 2-fold enhancement in the penetrance of axonal breaks in both PLM and AVM (Figure 6F), demonstrating a genetic interaction between the two genes.
Finally, to determine if MEC-17-induced axon degeneration was due to an acquired axonal fragility, we investigated the effect of physical tension from the animal’s movement on axonal structure. We analyzed mec-17 mutant animals paralyzed by RNAi against unc-54, the gene encoding the major C. elegans myosin heavy chain (MacLeod et al., 1977). Paralysis of mec-17 mutants induced a strong suppression of axonal degeneration (Figure 6G), indicating that the mechanical stress applied to the axons by simple body movement was sufficient to cause spontaneous breaks. These results support a model in which MEC-17 acts to stabilize the microtubules so as to prevent the axon becoming vulnerable to physical strain.
Body Length Correlates with Axonal Degeneration
In outcrossing the ky850 mutant animals, we serendipitously identified a mutant animal with an elongated body (LONg phenotype), which displayed a significant increase in the number of axonal breaks. We mapped and cloned this spontaneous mutation and found it to be an allele (vd037) of the gene lon-3 (encoding for a cuticle collagen; Nyström et al., 2002). A second known lon-3 allele (e2175) was also able to significantly enhance the axonal degeneration phenotype of mec-17 mutants (Figure 7). To test if the enhancement of axonal degeneration was caused by the specific disruption of lon-3, or more generically by the longer body shape, we analyzed other mutant strains displaying similar elongated morphology (Figures 7 and S6A). We studied another two conserved genes from the transforming growth factor β (TGF-β)-like-signaling pathway, lon-1 (a pathogenesis-related protein; Morita et al., 2002) and lon-2 (a glypican; Gumienny et al., 2007), as well as the TGF-β-signaling independent gene, lon-8 (Soete et al., 2007). Mutations in these genes caused a mild axonal degenerative phenotype (Figure S6B). However, when combined with the mec-17(ok2109) mutation, all four genes caused a remarkable increase in axonal degeneration (Figure 7).
Figure 7. Axon Degeneration Is Modulated by Body Morphology in mec-17 Mutants.

Schematics show percentage increase or decrease in body length of 3-day-old adults caused by mutations in different genes. Bar graph represents the increase or decrease in the penetrance of degeneration in mec-17(ok2109) mutants when combined with body-elongating mutations (lon-1(e185), lon-2(e678), lon-3(e2175), lon-8(hu187)) or shortening mutations (dbl-1(nk3), sma-3(e491), sma-4(e729), sma-6(e1482)).
The error bars represent SE of proportion; n ≥ 160 animals.
We postulated that this enhancement in degeneration could be due to the increased demands placed upon the cell body in order to maintain a significantly longer axon. To test this hypothesis, we analyzed shorter animals to determine if axonal degeneration was suppressed. As previously described, mutation of the genes sma-3 (a receptor-regulated Smad), sma-4 (a Smad protein), sma-6 (a TGF-β-like type I receptor), and dbl-1 (a bone-morphogenic-protein-like TGF-β-signaling molecule) results in reduced body size (Krishna et al., 1999; Savage et al., 1996; Savage-Dunn et al., 2000; Suzuki et al., 1999; Figures 7 and S6A). We found no degenerative phenotype in single mutants (n > 100 animals analyzed), and all four mutations significantly suppressed the degenerative phenotype in mec-17(ok2109) mutants (Figure 7). To ensure that these modulating effects were not due to underlying differences in the developmental status of the long versus short animals, we performed lifespan experiments. We found no correlation between body morphology and lifespan (Figures S6C and S6D) and extended these findings by analyzing degeneration in animals mutant for daf-2, a receptor tyrosine kinase molecule that when mutated causes a doubling in the lifespan of C. elegans (Kenyon et al., 1993). The penetrance of axonal degeneration in mec-17; daf-2 double mutants was not significantly different from that in mec-17 single-mutant animals (Figure S6E). These results reveal an underlying role of the animal’s body morphology in the capacity of the neuron to preserve axonal structure over time.
DISCUSSION
The axon frequently extends many times the length of the cell body. As such, maintenance of the axonal structure over an organism’s lifetime imposes considerable strain and requires effective transport of organelles, specific molecules, and trophic factors along the axonal shaft. In particular, cumulative evidence supports a critical role for mitochondrial transport and function in axonal maintenance (Court and Coleman, 2012). Furthermore, maintenance of axonal structure is dependent on an intact microtubule network that is not only vital for providing structural support but also allows the movement of motor proteins, which rely on the microtubules for axonal trafficking (Falnikar and Baas, 2009). Our results identify the crucial contribution of MEC-17 in preserving axonal structure and suggest a model in which MEC-17 stabilizes the microtubules to allow efficient binding and movement of motor proteins, thus permitting correct transport of mitochondria and other essential cargo throughout the axon.
The disrupted localization of UNC-104/kinesin-3, combined with mislocalization of RAB-3 and mitochondria, suggest a role for MEC-17 in maintaining efficient axonal transport. Mitochondria are critical for axonal health, carrying out essential functions in generating adenosine triphosphate, buffering intracellular Ca2+ and reactive oxygen species, and synthesizing various cellular components (Court and Coleman, 2012; Martin, 2012). Efficient, long-distance transport of mitochondria relies upon kinesin and dynein motor proteins, as well as the microtubule tracks that guide them (Hollenbeck, 1996). MEC-17 is critical for microtubule architecture (Cueva et al., 2012; Topalidou et al., 2012), and here we show that, in the absence of mec-17, microtubules are destabilized, axonal transport disrupted, and axon segments furthest from the cell body become largely devoid of mitochondria. The vast majority of axon breaks we observed occurred within these mitochondria-barren regions, suggesting that MEC-17 is required to keep a consistent distribution and number of mitochondria for proper neuronal maintenance. However, it is equally plausible that loss of axonal stability in the absence of MEC-17 prevents the movement of mitochondria out of the distal axon regions, leading to their degradation and thus a reduction in their numbers.
Our data support a role for MEC-17 in stabilizing the microtubules. We find a strong enhancement of microtubule dynamics in animals mutant for mec-17, suggesting that loss of MEC-17 causes instability of the microtubule network, leading to excessive microtubule turnover and more microtubules entering the growth phase in compensation. Furthermore, axonal degeneration and mitochondrial-distribution defects induced by the absence of mec-17 could be strongly suppressed with a microtubule-stabilizing drug and significantly enhanced by mild destabilization. In addition, we found qualitatively similar, albeit more severe, axonal degeneration phenotypes in animals lacking MEC-12/α-tubulin and MEC-7/β-tubulin, and a genetic interaction of mec-17 with mutations in either of these tubulins. Our genetic analysis defines a synergic role of MEC-17 with the tubulin-folding cofactor-E-like molecule COEL-1, a molecule related to a murine model of motor neuron disease (progressive motor neuronopathy) characterized by progressive axonal degeneration (Bommel et al., 2002).
Previous studies have shown that acetylation of α-tubulin does not affect microtubule stability and that the acetyltransferase domain of MEC-17 is not required for correct neurite outgrowth or neuronal function (Perdiz et al., 2011; Cueva et al., 2012; Topalidou et al., 2012). However, the enzymatic activity of MEC-17 is required for regulating the organization and number of microtubules and the protofilaments within them. Two recent publications have made the surprising discovery that the microtubules of fibroblasts with knockdown of mec-17/aTAT1 expression, or derived from MEC-17/aTAT1-null mice, show resistance to the depolymerizing agent nocodazole (Kalebic et al., 2013a, b). These results add to the growing intrigue of the MEC-17 molecule and may point toward cell-specific effects on microtubule stability, with the molecule promoting stability in the nervous system but acting to destabilize the microtubules network in order to promote cytoskeletal rearrangements and cell motility in other tissues. Our data showing that MEC-17 functions independently from its acetyltransferase domain in preventing axonal degeneration support the notion of enzymatic-dependent and -independent functions, with MEC-17 binding within the microtubule lumen and likely acting as a structural support to modulate microtubule organization (Cueva et al., 2012; Topalidou et al., 2012). Whereas at present we cannot exclude the existence of other domains capable of still-unidentified enzymatic activities, our study reveals a critical role for MEC-17 in modulating microtubule stability and protecting from axonal degeneration.
EXPERIMENTAL PROCEDURES
Strains
Strains were cultured under standard conditions (Brenner, 1974), and all experiments were performed at room temperature (22°C). The wild-type N2 Bristol and CB4856 Hawaiian isolates and the following mutations were used: LGII: sma-6(e1482); LGIII: daf-2(e1370), mec-12(u63), mec-12(u241), mec-12(e1607), sma-4(e729), lon-1(e185), sma-3(e491); LGIV: mec-17(ok2109), mec-17(u265), mec-17(ky850); LGV: dbl-1(nk3), lon-3(vd037), lon-3(e2175), lon-8(hu187); and LGX: coel-1(gk1236), lon-2(e678), atat-2(ok2415), mec-7(ok2152).
Transgenes used were: zdIs5(Pmec-4::GFP), vdEx267/vdEx539/vdEx546 (Pmec-4::mec-17 [10 ng/μl]; Plad-2::mCherry), vdEx484(Pmec-4::tomm-20::mRFP [0.5ng/μl]; Punc-122::GFP [25 ng/μl]), jsIs1111(Pmec-4::unc-104::GFP; a gift from Sandhya Koushika), vdEx262(Pmec-4::mCherry::rab-3 [0.5 ng/μl]; Punc-122::GFP [25 ng/μl]), vdEx411/vdEx543/vdEx548 (Pmec-4::mec-17[D144N] [10 ng/μl]; Podr-1::dsRed [30 ng/μl]), juEx2843(Pmec-4::EBP-2::GFP; a gift from Anindya Ghosh-Roy and Andrew Chisholm); ekSi1(Pmec-12::mec-12::3′ UTR mec-12) II; ekSi3(Pmec-12::mec-12 [K40R]::3′ UTR mec-12) II (both were gifts from Jacek Gaertig).
Isolation of Mutants and Genetic Mapping
Animals of genotype zdIs5; sir-2.1(ok434) were mutagenized using 50 mM ethyl methanesulfonate (Sigma) for 4 hr. The ky850 mutation was isolated from a clonal F2 progeny screen, and the sir-2.1 mutation was found not to influence the phenotype. These animals were crossed to wild-type CB4856 males, and single nucleotide polymorphism (SNP) mapping was performed using 11 previously described SNPs per chromosome (Davis et al., 2005; Wicks et al., 2001), which mapped the mutation to a central region on chromosome IV. The ky850 isolate was backcrossed 103 with the parental zdIs5; sir-2.1 line, before whole-genome sequencing was conducted on each (Australian Genome Research Facility), reaching a mean depth of 16.6–16.9.
The lon-3(vd037) allele consists of a 3,525 bp deletion spanning from the left flank sequence 5′-ttgccagagagagaaagttgacaaaacaat-3′ to the right flank sequence 5′-tcctgttgggcaagtattctccacagtaca-3′ and an insertion of 12 bp (gggcaagtattc). Genotyping was performed using the following primers: outer left 5′-agagcacaatgacactctttcc-3′ ; outer right 5′-agatatccattgtcgttcttccc-3′ ; inner left 5′-aagcaaatgaccgatcgacc-3′ ; and inner right 5′-tcacgtatgttgacaaata tcagg-3′.
Molecular Biology
Standard molecular biology techniques were used. The Pmec-4::mec-17 plasmid was generated by replacing the GFP sequence from the Pmec-4::GFP plasmid with an XmaI/AgeI full-length genomic mec-17 sequence (amplified from N2 genomic DNA using 5′-atgcaagtcgacgccgacctcc-3′ and 5′-tcaccacaatggcttgctcgacag-3′ primers). Pmec-4::tomm-20::mRFP was created by replacing the myo-3 promoter from Pmyo-3::tomm-20::mRFP (Ichishita et al., 2008) with an HindIII/XbaI Pmec-4 sequence (amplified from Pmec-4::GFP using 5′-aagcttcaatacaagctcaa-3′ and 5′-tctataacttgatagcgata-3′ primers). The Pmec-4::mCherry::rab-3 plasmid was created by replacing GFP in Pmec-4::GFP with mCherry and subsequently inserting a FseI/AscI rab-3 coding sequence downstream from mCherry. The rab-3 sequence was amplified from the Prab3::eGFP::RAB3::rim-3 plasmid (a gift from Michael Nonet, Washington University in St. Louis; using 5′-gctggcggacaacctcaagg-3′ and 5′-ttagcaattgcattgctgttgagc-3′ primers). QuikChange Site Directed Mutagenesis (Agilent Technologies) was used to create the Pmec-4::mec-17(D144N) plasmid, with Pmec-4::mec-17 used as the template and the following primers: sense: 5′-gaaccttatcaattggctcttaataatccatcagtcactcttc-3′ and antisense: 5′-gaagagtgactgatggattattaagagccaattgataaggttc-3′.
To induce paralysis, P0 animals were microinjected with double-stranded RNA (dsRNA) against unc-54, transcribed with a MEGAscript T7 In Vitro Transcription Kit (Life Technologies) using a DNA template amplified from N2 genomic DNA (with T7 tagged primers: 5′-tgagcaacagctcaaggaga-3′ and 5′-acaaaaatagggggtgggag-3′) and F1 progeny were analyzed.
Microscopy
Animals were immobilized using 0.05% tetramisole hydrochloride on 3% agar pads, imaged with a Zeiss Axio Imager Z1 equipped with a Photometrics Cool Snap HQ2 camera, and analyzed using Metamorph software.
Analysis of Microtubule Dynamics
EBP-2::GFP dynamics were analyzed similarly as previously described (Ghosh-Roy et al., 2012). Animals were immobilized without anesthetics using a suspension of 0.1 μm polystyrene beads on 12.5% agarose pads made with M9. Live imaging was performed with a Yokogawa Confocal Scanner Unit CSU-W1 attached to a Zeiss Axio Observer and fitted with a Hamamatsu C11440-22C sCMOS camera (2,048 × 2,048, with 2 × 2 binning) and Slidebook 5.5.4 software. EBP-2::GFP dynamics were captured with 114 ms exposures captured every 230 ms for 200 frames. ImageJ 1.47u was used to generate kymographs from the initial 25 mm of the PLM axon, from which EBP-2::GFP tracks were manually traced to calculate their growth size and duration (as shown in the schematic in Figure 6A). Only tracks that were completely imaged and longer than 0.5 μm were analyzed. Catastrophe frequency represents the ratio of the total number of tracks divided by the duration of all growth events within the kymographs (Stepanova et al., 2010).
Phenotypic Analyses
Unless otherwise stated, all analyses were performed on 3-day-old adults. Axonal breaks were only classified as breaks when a distal fragment was visible; however, due to an increased rate of distal clearance in mec-12 and mec-7 animals, degeneration was recorded when a distal fragment was present or when the axons were significantly shorter than in wild-type animals. Light-touch assays were performed as previously described (Chalfie and Sulston, 1981). Scoring of mitochondria was performed in animals without axonal degeneration, so as to eliminate bias caused by shortening of axons in mec-17 mutants with breaks. To determine the axonal positioning of mitochondria and accumulation of UNC-104::GFP, the axon was divided into three equal-sized segments using ImageJ 1.47u and labeled as proximal, central, and distal in relation to the cell body. UNC-104::GFP pooling at the distal end was scored when there was a clear accumulation of bright fluorescence, whereas axonal accumulation was recorded when fluorescent puncta were at least twice as bright as adjacent axon segments and when they caused the axon to at least double in width. PLM posterior outgrowth was recorded as a defect when it reached a length significantly longer than wild-type animals (greater than approximately four cell-body lengths and frequently branched), and ALM posterior outgrowth was considered a defect when the neurite was greater than two cell bodies in length. Metamorph software was used to measure body length, with the length of a central line from head to tail being recorded.
Drug Treatment
Animals were grown on nematode growth media (NGM) agar plates containing 0.1 mM colchicine (Sigma-Aldrich) or 1 μM paclitaxel (Sigma-Aldrich) in DMSO as previously described (Chalfie and Thomson, 1982; Kirszenblat et al., 2013). For control plates, animals were grown with 1% DMSO. P0 animals were grown from L4 stage on drug plates and their F1 progeny were scored.
Lifespan Analysis
Lifespan assays were conducted by transferring L4-stage animals to NGM agar plates containing 0.1 mg/ml 5-fluoro-2′-deoxyuridine (Sigma-Aldrich), which causes sterility without affecting adult morphology or lifespan (Mitchell et al., 1979). Animal viability was analyzed daily, with visualization of body movement and pharyngeal pumping providing evidence of life and prodding with a platinum wire pick used to confirm life or death in the absence of these signs.
Statistical Analysis
Statistical analyses were performed using Primer of Biostatistics 3.01 and Microsoft Excel 2011. Error of proportions was used to assess variation across a single population, and two-way comparisons were performed using the Student’s t test.
Supplementary Material
ACKNOWLEDGMENTS
We thank Cori Bargmann in whose lab this project was initiated and who provided reagents; Toshihiko Oka, Sandhya Koushika, Michael Nonet, Martin Chalfie, Anindya Ghosh-Roy, Andrew Chisholm, and Jacek Gaertig for strains and reagents; Steve Zuryn, Luke Hammond, and Paula Mugno Ramirez for technical assistance; and Rowan Tweedale, Paolo Bazzicalupo, Pankaj Sah, Kang Shen, Sean Millard, Rosina Giordano-Santini, Annika Nichols, and members of the Hilliard lab for helpful discussion and comments on the manuscript. Nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) National Center for Research Resources, and the International C. elegans Gene Knockout Consortium. This work was supported by grants from NIH R01NS060129, National Health and Medical Research Council Project Grants 569500 and 631634, and an Australian Research Council Future Fellowship (to M.A.H.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and two movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2013.12.004.
REFERENCES
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J. MEC-17 is an α-tubulin acetyltransferase. Nature. 2010;467:218–222. doi: 10.1038/nature09324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini F, Tian G, Piehl M, Cassimeris L, Lewis SA, Cowan NJ. Identification of a novel tubulin-destabilizing protein related to the chaperone cofactor E. J. Cell Sci. 2005;118:1197–1207. doi: 10.1242/jcs.01719. [DOI] [PubMed] [Google Scholar]
- Bommel H, Xie G, Rossoll W, Wiese S, Jablonka S, Boehm T, Sendtner M. Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease. J. Cell Biol. 2002;159:563–569. doi: 10.1083/jcb.200208001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, McEwen DP, Martens JR, Meyhofer E, Verhey KJ. Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol. 2009;7:e1000216. doi: 10.1371/journal.pbio.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Sulston J. Developmental genetics of the mechano-sensory neurons of Caenorhabditis elegans. Dev. Biol. 1981;82:358–370. doi: 10.1016/0012-1606(81)90459-0. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Thomson JN. Structural and functional diversity in the neuronal microtubules of Caenorhabditis elegans. J. Cell Biol. 1982;93:15–23. doi: 10.1083/jcb.93.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Court FA, Coleman MP. Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 2012;35:364–372. doi: 10.1016/j.tins.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Cueva JG, Hsin J, Huang KC, Goodman MB. Posttranslational acetylation of α-tubulin constrains protofilament number in native micro-tubules. Curr. Biol. 2012;22:1066–1074. doi: 10.1016/j.cub.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011;17:968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics. 2005;6:118. doi: 10.1186/1471-2164-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnikar A, Baas PW. Critical roles for microtubules in axonal development and disease. Results Probl. Cell Differ. 2009;48:47–64. doi: 10.1007/400_2009_2. [DOI] [PubMed] [Google Scholar]
- Fojo T. The Role of Microtubules in Cell Biology, Neurobiology, and Oncology. Humana Press; Totowa, NJ: 2008. [Google Scholar]
- Gardiner J, Barton D, Marc J, Overall R. Potential role of tubulin acetylation and microtubule-based protein trafficking in familial dysautonomia. Traffic. 2007;8:1145–1149. doi: 10.1111/j.1600-0854.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- Ghosh-Roy A, Goncharov A, Jin Y, Chisholm AD. Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev. Cell. 2012;23:716–728. doi: 10.1016/j.devcel.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumienny TL, MacNeil LT, Wang H, de Bono M, Wrana JL, Padgett RW. Glypican LON-2 is a conserved negative regulator of BMP-like signaling in Caenorhabditis elegans. Curr. Biol. 2007;17:159–164. doi: 10.1016/j.cub.2006.11.065. [DOI] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Cai D, Verhey KJ. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 2008;20:71–76. doi: 10.1016/j.ceb.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, Verhey KJ. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell. 2010;21:572–583. doi: 10.1091/mbc.E09-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempen B, Brion JP. Reduction of acetylated α-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1996;55:964–972. doi: 10.1097/00005072-199609000-00003. [DOI] [PubMed] [Google Scholar]
- Hilliard MA. Axonal degeneration and regeneration: a mechanistic tug-of-war. J. Neurochem. 2009;108:23–32. doi: 10.1111/j.1471-4159.2008.05754.x. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. The pattern and mechanism of mitochondrial transport in axons. Front. Biosci. 1996;1:d91–d102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J. Biochem. 2008;143:449–454. doi: 10.1093/jb/mvm245. [DOI] [PubMed] [Google Scholar]
- Janke C, Bulinski JC. Post-translational regulation of the micro-tubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011;12:773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- Kalebic N, Martinez C, Perlas E, Hublitz P, Bilbao-Cortes D, Fiedorczuk K, Andolfo A, Heppenstall PA. Tubulin acetyltransferase αTAT1 destabilizes microtubules independently of its acetylation activity. Mol. Cell. Biol. 2013a;33:1114–1123. doi: 10.1128/MCB.01044-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalebic N, Sorrentino S, Perlas E, Bolasco G, Martinez C, Heppenstall PA. αTAT1 is the major α-tubulin acetyltransferase in mice. Nat. Commun. 4, 1962. 2013b doi: 10.1038/ncomms2962. [DOI] [PubMed] [Google Scholar]
- Kanaji S, Iwahashi J, Kida Y, Sakaguchi M, Mihara K. Characterization of the signal that directs Tom20 to the mitochondrial outer membrane. J. Cell Biol. 2000;151:277–288. doi: 10.1083/jcb.151.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kirszenblat L, Neumann B, Coakley S, Hilliard MA. A dominant mutation in mec-7/β-tubulin affects axon development and regeneration in Caenorhabditis elegans neurons. Mol. Biol. Cell. 2013;24:285–296. doi: 10.1091/mbc.E12-06-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Setou M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 2009;12:559–567. doi: 10.1038/nn.2314. [DOI] [PubMed] [Google Scholar]
- Krishna S, Maduzia LL, Padgett RW. Specificity of TGFbeta signaling is conferred by distinct type I receptors and their associated SMAD proteins in Caenorhabditis elegans. Development. 1999;126:251–260. doi: 10.1242/dev.126.2.251. [DOI] [PubMed] [Google Scholar]
- Kumar J, Choudhary BC, Metpally R, Zheng Q, Nonet ML, Ramanathan S, Klopfenstein DR, Koushika SP. The Caenorhabditis elegans Kinesin-3 motor UNC-104/KIF1A is degraded upon loss of specific binding to cargo. PLoS Genet. 2010;6:e1001200. doi: 10.1371/journal.pgen.1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Hernault SW, Rosenbaum JL. Chlamydomonas α-tubulin is posttranslationally modified by acetylation on the ε-amino group of a lysine. Biochemistry. 1985;24:473–478. doi: 10.1021/bi00323a034. [DOI] [PubMed] [Google Scholar]
- MacLeod AR, Waterston RH, Fishpool RM, Brenner S. Identification of the structural gene for a myosin heavy-chain in Caenorhabditis elegans. J. Mol. Biol. 1977;114:133–140. doi: 10.1016/0022-2836(77)90287-x. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Biology of mitochondria in neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci. 2012;107:355–415. doi: 10.1016/B978-0-12-385883-2.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DH, Stiles JW, Santelli J, Sanadi DR. Synchronous growth and aging of Caenorhabditis elegans in the presence of fluorodeoxyuridine. J. Gerontol. 1979;34:28–36. doi: 10.1093/geronj/34.1.28. [DOI] [PubMed] [Google Scholar]
- Morita K, Flemming AJ, Sugihara Y, Mochii M, Suzuki Y, Yoshida S, Wood WB, Kohara Y, Leroi AM, Ueno N. A Caenorhabditis elegans TGF-β, DBL-1, controls the expression of LON-1, a PR-related protein, that regulates polyploidization and body length. EMBO J. 2002;21:1063–1073. doi: 10.1093/emboj/21.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH. Structure of the α β tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J. Neurosci. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyström J, Shen ZZ, Aili M, Flemming AJ, Leroi A, Tuck S. Increased or decreased levels of Caenorhabditis elegans lon-3, a gene encoding a collagen, cause reciprocal changes in body length. Genetics. 2002;161:83–97. doi: 10.1093/genetics/161.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiz D, Mackeh R, Poüs C, Baillet A. The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell. Signal. 2011;23:763–771. doi: 10.1016/j.cellsig.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Savage C, Das P, Finelli AL, Townsend SR, Sun CY, Baird SE, Padgett RW. Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor β pathway components. Proc. Natl. Acad. Sci. USA. 1996;93:790–794. doi: 10.1073/pnas.93.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage-Dunn C, Tokarz R, Wang H, Cohen S, Giannikas C, Padgett RW. SMA-3 smad has specific and critical functions in DBL-1/SMA-6 TGFbeta-related signaling. Dev. Biol. 2000;223:70–76. doi: 10.1006/dbio.2000.9713. [DOI] [PubMed] [Google Scholar]
- Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major α-tubulin K40 acetyltransferase alphαTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA. 2010;107:21517–21522. doi: 10.1073/pnas.1013728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soete G, Betist MC, Korswagen HC. Regulation of Caenorhabditis elegans body size and male tail development by the novel gene lon-8. BMC Dev. Biol. 2007;7:20. doi: 10.1186/1471-213X-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srayko M, Kaya A, Stamford J, Hyman AA. Identification and characterization of factors required for microtubule growth and nucleation in the early C. elegans embryo. Dev. Cell. 2005;9:223–236. doi: 10.1016/j.devcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Stepanova T, Smal I, van Haren J, Akinci U, Liu Z, Miedema M, Limpens R, van Ham M, van der Reijden M, Poot R, et al. History-dependent catastrophes regulate axonal microtubule behavior. Curr. Biol. 2010;20:1023–1028. doi: 10.1016/j.cub.2010.04.024. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Yandell MD, Roy PJ, Krishna S, Savage-Dunn C, Ross RM, Padgett RW, Wood WB. A BMP homolog acts as a dose-dependent regulator of body size and male tail patterning in Caenorhabditis elegans. Development. 1999;126:241–250. doi: 10.1242/dev.126.2.241. [DOI] [PubMed] [Google Scholar]
- Topalidou I, Keller C, Kalebic N, Nguyen KC, Somhegyi H, Politi KA, Heppenstall P, Hall DH, Chalfie M. Genetically separable functions of the MEC-17 tubulin acetyltransferase affect microtubule organization. Curr. Biol. 2012;22:1057–1065. doi: 10.1016/j.cub.2012.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003;4:938–947. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 2001;28:160–164. doi: 10.1038/88878. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma C, Delohery T, Nasipak B, Foat BC, Bounoutas A, Bussemaker HJ, Kim SK, Chalfie M. Identification of genes expressed in C. elegans touch receptor neurons. Nature. 2002;418:331–335. doi: 10.1038/nature00891. [DOI] [PubMed] [Google Scholar]
- Zuryn S, Le Gras S, Jamet K, Jarriault S. A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics. 2010;186:427–430. doi: 10.1534/genetics.110.119230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.