

Abstract
To identify potential antivirals against BTV, we have developed, optimized and validated three assays presented here. The CPE-based assay was the first assay developed to evaluate whether a compound showed any antiviral efficacy and have been used to screen large compound library. Meanwhile, cytotoxicity of antivirals could also be evaluated using the CPE-based assay. The dose-response assay was designed to determine the range of efficacy for the selected antiviral, i.e. 50% inhibitory concentration (IC50) or effective concentration (EC50), as well as its range of cytotoxicity (CC50). The ToA assay was employed for the initial MoA study to determine the underlying mechanism of the novel antivirals during BTV viral lifecycle or the possible effect on host cellular machinery. These assays are vital for the evaluation of antiviral efficacy in cell culture system, and have been used for our recent researches leading to the identification of a number of novel antivirals against BTV.
Keywords: Immunology, Issue 80, Drug Discovery, Drug Evaluation, Preclinical, Evaluation Studies as Topic, Drug Evaluation, Feasibility Studies, Biological Assay, Technology, Pharmaceutical, High-Throughput Screening Assays, Animal Diseases, Investigative Techniques, Antiviral, Efficacy, Bluetongue Virus, Cytopathic effect, Dose response, Time-of-Addition, Mechanism-of-Action
Introduction
BTV is a prototype double-stranded RNA virus in the genus Orbivirus, family Reoviridae. BTV is one of the most important diseases of domestic livestock, including sheep, goat, cattle and other domestic animals, with $3 billion/year loss worldwide1,2. The exotic BTV serotype is an important animal pathogen listed in the "USDA High Consequence Livestock Pathogens." Recently, the re-emerging of BTV has caused a major outbreak of disease in cattle and sheep in several countries across Northern Europe3,4. As a result of its economic significance and as a model system, BTV has been the subject of extensive molecular, genetic and structural studies, and several vaccines have been developed. However, due to the lack of proper assays for antiviral drug discovery, there are no antivirals available against BTV.
In a recent high throughput screening (HTS) campaign using BTV as the model system, we developed, optimized and validated a CPE-based assay to identify potential broad-spectrum antivirals against arboviruses5. CPE-based assay is a well-recognized assay that has been used in antiviral drug discovery against a number of viruses that induced rapid and observable CPE/apoptosis5-7. In our system, post BTV infection, CPE is evident in vertebrate cells, including HeLa, BSR, and HEK 293T8. BTV-induced CPE could be monitored and quantified using various cell viability detection methods, including the CellTiter Glo cell viability reagent kit (CTG kit)9. This kit determines the number of viable cells in culture based on quantitation of cellular ATP presented, which signals the presence of metabolically active living cells. Under optimized conditions, the CPE-based assay presented here showed its feasibility with the "mix and measure" one step protocol, and flexibility with stable luminescent signals. Meanwhile, toxic compounds reducing cell viability will be excluded in this CPE-based assay. The CPE-based assay showed its robustness and reliability for antiviral drug discovery against BTV, and has been used to screen the NIH Molecular Libraries Small Molecule Repository (MLSMR), which leads to the identification of six novel cluster of potential antiviral lead compound(s)5.
When a potential antiviral compound has been identified using the CPE-based assay, it will need to be subjected to the ten-concentration dose-response assay to determine the range of antiviral efficacy and cytotoxicity2. The antiviral efficacy, represented as the 50% inhibitory concentration (IC50) or the 50% effective concentration (EC50), is the concentration of a drug which inhibits virus-induced CPE halfway between the baseline and maximum. The cytotoxicity of the antivirals, i.e. the 50% cytotoxicity concentration (CC50), is the concentration of a drug inducing 50% of cytotoxicity between the baseline and maximum. The selective index (SI), denoted as 50% SI (SI50) is calculated from CC50/IC50 which determines the specificity of the antiviral against virus-induced CPE. The IC50 (or EC50), CC50 and SI50 values are critical measures to determine whether an antiviral compound is potent and selective for further drug development.
When an antiviral showed no overt toxicity in vitro, yet prevented virus induced CPE and the productive viral life-cycle, it is important to characterize its MoA2. We initiated such characterization by carrying out ToA assay to determine the possible step(s) of viral life-cycle that is affected by the antiviral. Generally, antiviral compound were added to cells at different times pre- or post-virus infection. If the antivirals were added to the infected cells post to its target step during the course of infection, it would result in lower activity when compared to the one which was added prior to the step. Thus, ToA study is critical for determining the antiviral efficacy of a compound, and its potential target, either on the viral life-cycle or the host machinery involved in the viral life-cycle.
For all three assays, cell viability was determined using the CTG kit following manufacturer's instruction5. This detection system outputs adequate luminescence signals that could be analyzed using various in-house software. Each assay was validated and performed at least in triplicate with eight replicas. For all the obtained data, three parameters, including mean value (AVE), standard deviation (STDEV), and co-efficient variation (CV) were analyzed to determine the robustness of the assay. Once the robustness of the assay has been determined, the data will be further analyzed and plotted using various biostatics and graphic tools2.
Protocol
1. Cells, Virus and the Antiviral Compounds
Maintain BSR cells, a derivative of baby hamster kidney (BHK) cells10, in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal calf serum (FCS), 100U/ml penicillin and 100 μg/ml streptomycin.
For all three assays, plate cells in DMEM with 1% FCS, 100U/ml penicillin and 100 μg/ml streptomycin, as optimized previously5. This medium is referred as assay medium for all three assays.
Incubate all cells in the incubator at 37 °C, with 5% CO2 and 80-95% humidity.
Plaque-purify and propagate the type 10 BTV (BTV-10) as described previously8. Dilute BTV-10 in assay medium for each designated assays.
Dissolve all testing compounds in DMSO to form a stock with concentration at 10 mM. Store the stocks at -20 °C.
Dilute compounds to desired concentrations using assay medium for the designated assays2.
2. CPE-based Assay Using CTG Kit
Seed BSR cells into a 384-well microplate (black; format by 16 x 24) via MicroFlo select dispenser. The seeding density is 5,000 cells/well, and the seeding volume is 20 μl for antiviral efficacy analysis.
Incubate cells for 2-3 hr till cells get adherent to the plate thoroughly.
Add antiviral compound with a final concentration of 10 μM to each well. Mix it completely.
Dilute BTV to desired titer and add 5 μl BTV with denoted MOI to each well. For the control well, add 5 μl of assay media. Incubate the infected cells for 72 hr at 37 °C, with 5% CO2 and 80-95% humidity.
At 72 h.p.i., thaw and equilibrate CTG buffer and the lyophilized CTG substrate to room temperature prior to use. Reconstitute the homogeneous CTG reagent solution by mixing the lyophilized enzyme/substrate and the buffer reagent according to the manufacturer's instructions.
Equilibrate the assay plates to room temperature for 15 min.
Add an equal volume (25 μl) of CTG reagents to each well by a dispenser. After incubating for 15 min at room temperature in darkness, measure luminescence signals using a multi-mode reader with an integration time of 0.1 sec.
3. Dose-response Assay
Seed BSR cells into a 384-well microplate (black; format by 16 x 24) via a dispenser, with a seeding density of 5,000 cells/well in a seeding volume of 20 μl for antiviral efficacy assay and 25 μl for cytotoxicity assay, respectively.
Incubate cells for 2-3 hr at 37 °C, with 5% CO2 and 80-95% humidity till cells are well attached to the plate.
Process assay in a quarter area of the 384-well plate for each compound (equivalent to a 96-well plate), as shown in Table 1. Allocate eight replicates for each concentration in a single assay. Assign the first column as the positive control without adding compound and virus, and the last column (12th) as negative control by adding virus only without compounds.
Dilute compounds to 50 μM in assay medium and add to the 2nd column at 20 μl/well for antiviral efficacy assay. Mix the compound five times using 8-channel semi-automatic pipette to a concentration of 25 μM.
Using 8-channel semi-automatic pipette, transfer 20 μl mixture in 2nd column to the next column (3rd), mix well to form a concentration of 12.5 μM. Repeat this process by transferring 20 μl mixture from 3rd column to the next column (4th) to form another two-fold dilution with a concentration of 6.25 μM. Repeat this two-fold serial dilution till the 11th column. Aspirate and discard 20 μl of the mixture in 11th column after adding and mixing the compound.
Add BTV-10, based on MOI of 0.01, to each well from column 2 to column 11 with a volume of 5 μl/well. After adding the virus, the final compound concentration in each column should be: 20 μM in column 2, 10 μM in column 3, and continued with a two-fold dilution down to column 11 with a final concentration of 0.04 μM.
Add 5 μl of medium to column 1 as cell only control (positive) and 5 μl/well of BTV to column 12 as BTV infection only control (negative).
Dilute compounds to an initial concentration of 200 μM for cytotoxicity assay. Similarly, add 25 μl/well of compound to column 2 and mixed 5x with 8-channel semi-automatic pipette to a concentration of 100 μM. Do not add BTV.
Carry out the two-fold series dilution by aspirating 25 μl from column 2 to the neighboring column 3, and continued till the last column (12th). At column 12, after mixing, aspirate and discard 25 μl of mixture. The final concentration at column 2 should be 100 μM and the 12th column should be at 0.2 μM. The column 1 is the cell only control.
For both antiviral efficacy and cytotoxicity assays, incubate the plates at 37 °C, with 5% CO2 and 80-95% humidity for 72 hr post treatment. Measure cell viability using the CTG kit as described above (protocol steps 2.5-2.7).
4. Time-of-Addition (ToA) Assay
Seed BSR cells from column 1-24 in a 384-well microplate (black; format by 16 x 24) via a dispenser at 5,000 cells/well and the seeding volume is 15 μl/well.
For each compound, utilize all twenty-four columns with eight replicas for each time-point in a half of the 384-well plate (Table 2). Assign column 1 as cells only control by adding 10 μl/well assay medium. Mark column 24 as BTV infection only control (negative control) by adding 5 μl/well assay medium and 5 μl/well virus at MOI of 0.01 to a final volume of 25 μl/well.
Select the even-numbered columns from column 2-22 as antiviral efficacy evaluation column at different hours post infection (h.p.i.). In these columns, infect cells with 5 μl/well BTV at MOI of 0.01. At different h.p.i., also add 5 μl/well diluted compound to each well to form a final volume of 25 μl/well. For the denoted -2 and -1 h.p.i. add compound to BSR cells prior to BTV infection. For 0 h.p.i., add the compound and BTV to the culture simultaneously.
In parallel, designate the odd-numbered columns from column 3-23 as compound only controls, of which compound were added at different time-points as designated (-2, -1, 0, 1, 2, 4, 8, 16, 24, 48 h.p.i.). In these columns, add 5 μl/well assay medium and 5 μl/well diluted compound to form a final volume of 25 μl/well.
After treatment, incubate cells at 37 °C, 5% CO2 with 80-95% humidity.
Determine cell viability at 72 h.p.i. using the CellTiter-Glo kit as described previously (protocol steps 2.5-2.7).
5. Data Analysis
Process all data first using proper in-house software based on the luminescent signals obtained via the multi-mode reader. Determine the mean value (Average), standard deviation (STDEV) for each treatment as well as the coefficient variations (CV), which requires a value no greater than 10%.
Transfer the processed data from the above software to a biostatic and graphic software tool. Carry out the non-linear regression analysis to determine the values of EC50 and CC50.
Representative Results
1. Antiviral efficacy of compound
The cell-based CPE assay was developed, optimized and validated in vitro using the luminescent-based CTG kit to identify novel antivirals against BTV as described previously2,5. The ten-dose response assay was employed to reflect the antiviral efficacy and cytotoxicity of an identified lead compound by measuring the number of metabolically viable cells in culture based on quantitation of cellular ATP presented in the living cells5,11. In our previous report, a number of potential antiviral compounds were evaluated, including compounds from each cluster identified via HTS against BTV5, and their derivatives via de novo synthesis2. Based on their EC50, CC50 and SI50, several promising lead compounds were identified with potent antiviral efficacy, low toxicity and high selectivity. For example, compound052 (C052) was determined to have an EC50 of 0.27±0.12 μM (Figure 1)2 and a CC50 of 82.69 μM, both showing typical regressive curves under the non-linear regression analyses2. The SI50 of C052 was determined at 306 based on its EC50 and CC50 values. The nanomolar scale antiviral efficacy, low toxicity, and consequently high SI50 indicated that C052 might be a potent and selective antiviral against BTV.
2. Potential MoA for the antiviral compound
The ToA assay was aimed to determine the possible stage(s) of viral life-cycle targeted by compounds. When adding C052 at 1 or 2 hr prior to BTV infection, i.e. -1 and -2 h.p.i., the antiviral efficacies remained at the nanomolar scale (Figure 2)2, indicating that C052 might act beyond the early stage of viral life-cycle, such as virus entry. Furthermore, the antiviral efficacy remained unchanged until C052 was added to infected cells as later as 24 h.p.i.. When added at 32 h.p.i., the percentage of viable cells decreased in C052 treatment cells, indicating that C052 was less protective at this stage of viral life-cycle. When added at 48 h.p.i., there was no protection to BSR cells from BTV-induced CPE. Since the first cycle of BTV viral replication usually completed in infected cells within 24 h.p.i., our results suggested that C052 might act at the late stages of BTV viral life-cycle, such as virus replication, packaging, maturation and egress. Meanwhile, it is also possible that C052 may act on host cellular machineries that were involved during late viral life-cycle2.
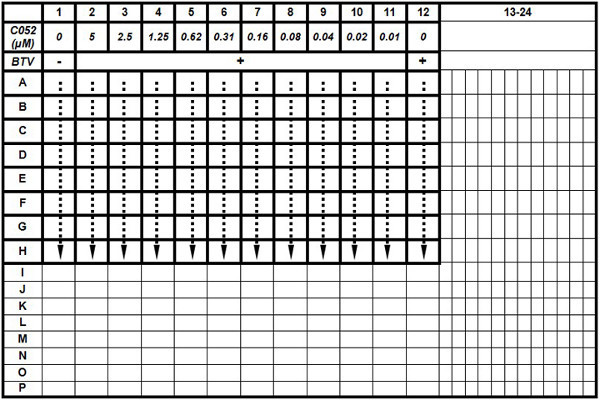 Table 1. The plate layout for dose-response assay. The antiviral efficacy of C052 was evaluated in a 96-well scale within the 384-well plate. Each treatment, including BTV infection plus different C052 concentrations, was performed with eight replicas. At 72 h.p.i., cell viability was determined using the CTG kit.
Table 1. The plate layout for dose-response assay. The antiviral efficacy of C052 was evaluated in a 96-well scale within the 384-well plate. Each treatment, including BTV infection plus different C052 concentrations, was performed with eight replicas. At 72 h.p.i., cell viability was determined using the CTG kit.
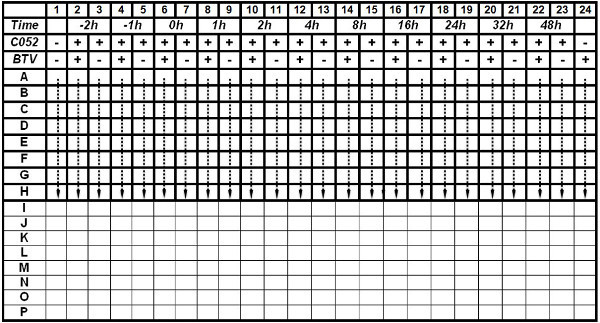 Table. 2 The plate layout for the Time-of-Addition (ToA) assay. The ToA assay of C052 was evaluated in the 384-well plate as indicated in the layout. Each treatment, including BTV infection plus time of adding C052, was carried out with eight replicas. The -2 and -1 h.p.i. indicate that C052 was added to the cells before BTV infection. At 0 h.p.i., BTV and C052 were added simultaneously. At 72 h.p.i., cell viability was determined using the CTG kit. Click here to view table.
Table. 2 The plate layout for the Time-of-Addition (ToA) assay. The ToA assay of C052 was evaluated in the 384-well plate as indicated in the layout. Each treatment, including BTV infection plus time of adding C052, was carried out with eight replicas. The -2 and -1 h.p.i. indicate that C052 was added to the cells before BTV infection. At 0 h.p.i., BTV and C052 were added simultaneously. At 72 h.p.i., cell viability was determined using the CTG kit. Click here to view table.
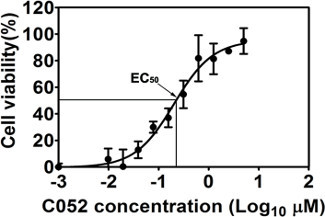 Figure 1. The antiviral efficacy of C052. Cells were infected with BTV at MOI of 0.01 in the presence of ten different concentrations of C052, as indicated in the figure . Cell viability was determined at 72 h.p.i, using the CTG kit. Each data point represents means and SD from five replicates. This figure has been modified from Gu et al. 20122.
Figure 1. The antiviral efficacy of C052. Cells were infected with BTV at MOI of 0.01 in the presence of ten different concentrations of C052, as indicated in the figure . Cell viability was determined at 72 h.p.i, using the CTG kit. Each data point represents means and SD from five replicates. This figure has been modified from Gu et al. 20122.
 Figure 2. The time-of-addition assay for C052. C052 at 2.5 mM and 0.27 mM, respectively, was added to BTV infected cells at different h.p.i. as indicated, and the protection of C052 against BTV induced CPE, or cell viability, was measured using the CTG kit at 72 h.p.i. Each data points represented the average values and SD from of eight independent replicates. This figure has been modified from Gu et al. 20122.
Figure 2. The time-of-addition assay for C052. C052 at 2.5 mM and 0.27 mM, respectively, was added to BTV infected cells at different h.p.i. as indicated, and the protection of C052 against BTV induced CPE, or cell viability, was measured using the CTG kit at 72 h.p.i. Each data points represented the average values and SD from of eight independent replicates. This figure has been modified from Gu et al. 20122.
Discussion
For the initial identification of antiviral hits, one of the key steps for antiviral drug discovery and development is to develop robust assays, which includes selecting a quantifiable marker, developing a simple protocol, obtaining sufficient signals and less than 10% CV. Most biochemical or cell-based screens are designed to provide a chemical starting point based upon the most robust, simple and inexpensive assay, due to the required reproducibility in the screening process and the potentially large number of molecules to be screened. The CPE-based assay was designated to meet these requirements to identify effective hits from a large compound library. CPE refers to the adverse effect on the cultured cells associated with the multiplication of viruses. Various assays are available to use the CPE indicator through measuring a variety of different markers indicating the number of dead cells (cytotoxicity), the number of live cells (viability), and the mechanism of cell death (apoptosis). Commercial reagents for CPE-based assay are available with a simple assay protocol. For example, the CTG kit includes the single "mix and measure" step and has been widely used to quantify virus induced CPE. However, CPE-based assay is limited to viruses that could induce rapid CPE. For viruses that do not induce rapid CPE, various assays have been developed. For example, the replicon-based assay using the replicon-harboring cell line allows screening for inhibitors of viral replication, including translation, polyprotein processing, and minus- and plus-strand RNA synthesis12-14. The antisense RNA strategies and virtual screening of small-molecule libraries also could be used to identify possible antivirals15,16. Nevertheless, it is critical that the efficacy of an antiviral drug be validated in the in vitro cell-based assay which allows the evaluation of antiviral efficacy against the entire viral life-cycle. CPE-based assay has been successfully adapted into high throughput format and is one of the most reliable and robust assay for the screening of large compound libraries, including recently accomplished HTS screenings against influenza virus6,17, severe acute respiratory syndrome coronavirus (SARS-CoV)18, arenaviruses19, and Reovirus (Bluetongue virus)5.
The dose-response assay was designated to further validate the antiviral efficacy of the hits identified via the single dose CPE-based assay, usually after screening a large compound library. These hits, although identified with antiviral activity, need further confirmation to reveal their potentials to become a drug. Antiviral efficacy of a compound could be revealed through the analysis via EC50, CC50 and SI50 value using the dose response analysis. Meanwhile, out off-target or false positives will be identified, and the number of lead compounds could be narrowed down. Via this analysis, the order of compound potencies and toxicities could be ranked to direct future antiviral drug discovery, especially for the structure-activity relationship (SAR) analysis and future medicinal chemistry modification. In fact, compound C052 was a derivative of compound C003, which associated with good drug-like properties both in vitro and in vivo.
To meet the requirements for a drug, it is a must to determine its MoA, i.e. to characterize the interaction of a compound with its target at physiologic concentrations. Various assays have been developed of particular antivirals, i.e. to not only inhibit the target but to have acceptable solubility, permeability, protein binding, selectivity, metabolism and toxicity profiles. Since MoA studies were low-throughput assay needing laborious efforts, only a few selected molecules, with potent EC50, high CC50 and high SI50 value, could be readily analyzed. An understanding the MoA of a compound at this stage can add depth to the interpretation of cellular activity or its absence. The ToA study is designated to narrow down the stage when the antiviral interacts with viral or host targets. Knowing a compound is competitive with a substrate in certain stage viral life-cycle could provide an immediate direction for further MoA analysis. Alternatively, more potent cell based assay with known mechanism, including assay directly screen for HIV-1 integrase inhibitors20, inhibiting virus binding to surface protein21 could be used which provide known MoA before actual screenings22. While hits from these screening may well-related to the designated MoA, there actually MoA may also need to subject to ToA and other mechanism of action studies.
In summary, using the three assays reported here, we have successfully screened and identified several potent antivirals against BTV, including C003 and C0522. In particular, the SI50 (of C052 was up at 306, which suggested that C052 is highly selective against BTV. Via the ToA and other MoA studies presented in the original paper, we proposed that C052 could be a potential antiviral agent interacting with host autophagy machinery, could be developed into an anti-BTV drug.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This project was supported by grant 1R03MH08127-01 and 7R03MH08127-02 from NIH to Q. Li, and by the IMPACT funds from Department of Medicine at UAB to Q. Li. Support from the Molette Fund and Auburn University is appreciated. We also thank the technical assistances from Ms. Pulin Che and Mr. Volodymyr Musiienko during the course of the work.
References
- Hemati B, et al. Bluetongue virus targets conventional dendritic cells in skin lymph. J. Virol. 2009;83:8789–8799. doi: 10.1128/JVI.00626-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, et al. Novel Virostatic Agents against Bluetongue Virus. PLoS ONE. 2012;7:e43341. doi: 10.1371/journal.pone.0043341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiswinkel R, et al. The 2006 outbreak of bluetongue in northern Europe--the entomological perspective. Prev. Vet. Med. 2008;87:55–63. doi: 10.1016/j.prevetmed.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Szmaragd C, et al. Mortality and case fatality during the recurrence of BTV-8 in northern Europe in 2007. Vet. Rec. 2007;161:571–572. doi: 10.1136/vr.161.16.571-e. [DOI] [PubMed] [Google Scholar]
- Li Q, Maddox C, Rasmussen L, Hobrath JV, White LE. Assay development and high throughput antiviral drug screening against Bluetongue virus. Antiviral Research. 2009;83:267–273. doi: 10.1016/j.antiviral.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah JW, et al. A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res. 2007;73:50–59. doi: 10.1016/j.antiviral.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Che P, Wang L, Li Q. The development, optimization and validation of an assay for high throughput antiviral drug screening against Dengue virus. Int. J. Clin. Exp. Med. 2009;2:363–373. [PMC free article] [PubMed] [Google Scholar]
- Li Q, Li H, Blitvich BJ, Zhang J. The Aedes albopictus inhibitor of apoptosis 1 gene protects vertebrate cells from bluetongue virus-induced apoptosis. Insect Mol. Biol. 2007;16:93–105. doi: 10.1111/j.1365-2583.2007.00705.x. [DOI] [PubMed] [Google Scholar]
- Petty RD, Sutherland LA, Hunter EM, Cree IA. Comparison of MTT and ATP-based assays for the measurement of viable cell number. J. Biolumin. Chemilumin. 1995;10:29–34. doi: 10.1002/bio.1170100105. [DOI] [PubMed] [Google Scholar]
- Buchholz UJ, Finke S, Conzelmann KK. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 1999;73:251–259. doi: 10.1128/jvi.73.1.251-259.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips T, Jenkinson L, McCrae C, Thong B, Unitt J. Development of a high-throughput human rhinovirus infectivity cell-based assay for identifying antiviral compounds. J. Virol. Methods. 2011;173:182–188. doi: 10.1016/j.jviromet.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Harvey TJ, et al. Tetracycline-inducible packaging cell line for production of flavivirus replicon particles. J. Virol. 2004;78:531–538. doi: 10.1128/JVI.78.1.531-538.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Basagoiti F, et al. High-throughput assays using a luciferase-expressing replicon, virus-like particles, and full-length virus for West Nile virus drug discovery. Antimicrobial Agents and Chemotherapy. 2005;49:4980–4988. doi: 10.1128/AAC.49.12.4980-4988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Basagoiti F, et al. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob. Agents Chemother. 2006;50:1320–1329. doi: 10.1128/AAC.50.4.1320-1329.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan M, et al. In vitro and in vivo protection against the highly pathogenic H5N1 influenza virus by an antisense phosphorothioate oligonucleotide. Antivir. Ther. 2008;13:109–114. [PubMed] [Google Scholar]
- Ray D, Shi PY. Recent advances in flavivirus antiviral drug discovery and vaccine development. Recent Pat. Antiinfect. Drug Discov. 2006;1:45–55. doi: 10.2174/157489106775244055. [DOI] [PubMed] [Google Scholar]
- Severson WE, et al. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2) J. Biomol. Screen. 2008;13:879–887. doi: 10.1177/1087057108323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson WE, et al. Development and validation of a high-throughput screen for inhibitors of SARS CoV and its application in screening of a 100,000-compound library. J. Biomol. Screen. 2007;12:33–40. doi: 10.1177/1087057106296688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolken TC, et al. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antivir. Res. 2006;69:86–97. doi: 10.1016/j.antiviral.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loock M, et al. A novel high-throughput cellular screening assay for the discovery of HIV-1 integrase inhibitors. J. Virol. Methods. 2012;179:396–401. doi: 10.1016/j.jviromet.2011.11.029. [DOI] [PubMed] [Google Scholar]
- Kampmann T, et al. In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses. Antiviral Res. 2009;84:234–241. doi: 10.1016/j.antiviral.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Kirchmair J, et al. Development of anti-viral agents using molecular modeling and virtual screening techniques. Infect. Disord. Drug Targets. 2011;11:64–93. doi: 10.2174/187152611794407782. [DOI] [PubMed] [Google Scholar]