

Abstract
Mast cell leukemia (MCL) is a life-threatening disease associated with high mortality and drug-resistance. Only few patients survive more than 12 months. We report on a 55-year-old female patient with acute MCL diagnosed in May 2012. The disease was characterized by a rapid increase in white blood cells and mast cells (MC) in the peripheral blood, and a rapid increase of serum tryptase levels. The KIT D816H mutation was detected in the blood and bone marrow (BM). Induction chemotherapy with high-dose ARA-C and fludarabine (FLAG) was administered. Unexpectedly, the patient entered a hematologic remission with almost complete disappearance of neoplastic MC and a decrease of serum tryptase levels to normal range after 2 cycles of FLAG. Consecutively, the patient was prepared for allogeneic stem cell transplantation. However, shortly after the third cycle of FLAG, tryptase levels increased again, immature MC appeared in the blood, and the patient died from cerebral bleeding. Together, this case shows that intensive chemotherapy regimens, like FLAG, may induce remission in acute MCL. However, treatment responses are short-lived and the overall outcome remains dismal in these patients. We propose to separate this acute type of MCL from more subacute or chronic variants of MCL.
Keywords: Mast cells, Mast cell leukemia, KIT, Chemotherapy, FLAG
1. Introduction
Advanced systemic mastocytosis (SM) is a life-threatening condition characterized by uncontrolled growth and expansion of neoplastic mast cells (MC) in various organ systems [1–4]. In most patients, MC are resistant against various targeted drugs and conventional cytostatic drugs. Mast cell leukemia (MCL) is a rare form of advanced SM, defined by a leukemic spread of immature MC and a short survival [1–4].
So far, only a few publications have reported on successful treatment of patients with MCL [4,5]. Responses to polychemotherapy or targeted drugs are usually incomplete and short-lived. Therefore, hematopoietic stem cell transplantation (SCT) is usually recommended for eligible patients. However, only a few patients are transplantable because of multiorgan damage and the poor response to poly-chemotherapy. Although several different treatment strategies have been proposed, it remains unclear what type of poly-chemotherapy is most effective for debulking in MCL, and how many cycles are required to achieve partial or complete remission prior to SCT.
We here report on a patient with acute MCL in whom polychemotherapy, consisting of fludarabine and high-dose cytosine arabinoside (ARA-C), was administered. Unexpectedly, the patient entered a good partial remission, but unfortunately, remission was only short-lived and was followed by a treatment-resistant relapse.
2. Case report and methods
2.1. Case report
A 55-year old female patient was referred in May 2012 because of rapidly progressing leukocytosis, anemia and thrombocytopenia. The case history did not reveal mutagenic events, relevant co-morbidities or a pre-phase of mastocytosis. Physical examination disclosed mild peripheral edema and a small hematoma on her right leg. No skin lesions and no palpable splenomegaly or lymphadenopathy was found. The peripheral blood count showed 53,300 leukocytes per microliter blood, 9.1 g/dL hemoglobin, and 74,000 platelets. A differential count revealed 17% neutrophils, 15% lymphocytes, 1% monocytes, 4% basophils, 17% metamyelocytes, 5% myelocytes, 1% promyelocytes, and 40% highly atypical immature MC. The serum tryptase level was 904 ng/mL. Moreover, an elevated alkaline phosphatase (aP, 237 U/L) and a markedly elevated lactate dehydrogenase (LDH, 2150 U/L) were found. Bone marrow (BM) investigations confirmed the diagnosis MCL. A sonographic examination revealed a slightly enlarged liver with abnormal density suggesting diffuse infiltration and mild splenomegaly (13 cm diameter). Leukocytes were found to rapidly increase over time, with a doubling-time of less than 10 days.
2.2. Treatment
Before and during chemotherapy, prophylactic histamine receptor blockers and prednisolone as well as prophylactic antibiotics were administered. From May 22, 2012, polychemotherapy (FLAG) was given. The patient received 30 mg/m2 fludarabine i.v. on days 1–5 and 2 g/m2 ARA-C i.v. on days 1–5. From day 6, she also received 30 million units G-CSF s.c. daily until granulocyte recovery. The patient was hospitalized until hematopoietic recovery and discharged between her FLAG cycles. Between May and August 2012, she received 3 cycles of FLAG.
2.3. Laboratory investigations, staging and follow-up
All examinations were performed within the frame of routine diagnostics-, routine staging-, and routine follow up investigations regarded as standard in mast cell proliferative neoplasms [6]. The patient provided written informed consent before being examined and before BM or blood was obtained and analyzed. In the follow up, serial determinations of all laboratory parameters, including blood counts, differential counts, and the serum tryptase level, were performed.
2.4. Examination of the bone marrow (BM)
BM aspirate smears were stained with Wright–Giemsa and examined for the presence and percentage of MC, blast cells, and signs of dysplasia. Multicolor flow cytometry was performed according to generally accepted recommendations and published techniques [7] using fluorochrome-conjugated monoclonal antibodies (mAb) against CD2, CD25, CD30, CD34, CD45, CD52, and CD117. A specification of mAb is shown in Table 1A. Isolated BM cells were also examined by conventional karyotyping and fluorescence in situ hybridization (FISH). Histological and immunophenotypic analyses of the BM were performed at diagnosis and after 2 cycles of FLAG. Formalin-fixed and paraffin-embedded BM sections (trephine) were stained with mAb against CD117 (KIT), tryptase, chymase, CD2, CD25, CD30 and CD34 (Table 1B). In addition, BM sections were double-stained with an antibody against the proliferation-associated antigen Ki-67 (blue color) and an antibody against KIT (brown color).
Table 1.
(A) Specification of antibodies (Ab) used for immunohistochemistry.
| CD/Ag | Clone | Source | Isotype | Dilution | Retrieval | Provider |
|---|---|---|---|---|---|---|
| Tryptase | G3 | Mouse | IgG1 | 1:50 | MW | Chemicon |
| Chymase | B7 | Mouse | IgG1 | 1:100 | Proteinase | Chemicon |
| CD117/KIT | Polyclonal | Rabbit | IgG1 | 1:200 | MW | Dako |
| CD2 | 6F10.3 | Mouse | IgG1 | 1:50 | MW | Novocastra |
| CD25/IL-2RA | Tu-69 | Mouse | IgG1 | 1:50 | MW | Novocastra |
| CD30/Ki-1 | Ber-H2 | Mouse | IgG1 | 1:20 | MW | Dako |
| CD34/HPCA-1 | QBEND10 | Mouse | IgG1 | 1:10 | MW | Novocastra |
| (B) Specification of antibodies (Ab) used for flow cytometry. | |||||
| CD/Ag | Clone | Source | Isotype | Conjugate | Provider |
| CD2/LFA-2 | RPA-2.10 | Mouse | IgG1 | PE | BD Biosciences |
| CD25/IL-2RA | 2A3 | Mouse | IgG1 | PE | BD Biosciences |
| CD30/Ki-1 | BerH8 | Mouse | IgG1 | PE | BD Biosciences |
| CD34/HPCA-1 | 581 | Mouse | IgG1 | PE | BD Biosciences |
| CD45/LCA | 2D1 | Mouse | IgG1 | PerCP | BD Biosciences |
| CD52/Campath-1 | HI186 | Mouse | IgG1 | PE | BD Biosciences |
| CD117/SCR/KIT | 104D2 | Mouse | IgG1 | PE | BD Biosciences |
CD, cluster of differentiation; IL-2RA, interleukin-2R-alpha; HPCA-1, human progenitor cell antigen-1; MW, microwave; LCA, leukocyte common antigen; SCFR, stem cell factor receptor; PE, phycoerythrin; PerCP, peridinin chlorophyll protein. Provider: BD Biosciences (San Jose, CA, USA), Chemicon (Temecula, CA), Dako (Glostrup, Denmark), Novocastra (Newcastle upon Tyne, UK).
2.5. Molecular studies
BM and blood cells were examined for the presence of codon 816 mutations according to published methods [8]. In addition, cells were subjected to multiplex PCR and conventional PCR to screen for leukemia-related translocations and other gene defects, including BCR/ABL, FIP1L1/PDGFRA, JAK2 V617F, PML/RARA, AML1/ETO, NPM1 mutations, and FLT3 mutations.
3. Results
3.1. Histologic findings and cytology of neoplastic cells
The BM histology showed a hypercellular marrow with an excess of atypical immature MC expressing tryptase and KIT (Fig. 1). As determined by tryptase-staining, normal hematopoietic cells were almost completely replaced by the MC infiltrate (90% infiltration of the BM). The hypercellular BM smear was also found to contain an excess of MC. In fact, about 70–80% of all nucleated BM cells were classified as MC or immature atypical MC precursors by Wright–Giemsa staining (Fig. 1A). Most of these MC were immature and many of them contained bi- or poly-lobed nuclei (atypical MC type II) (Fig. 1A) [9]. A few spindle-shaped MC were also detected (atypical MC type I). In addition, numerous metachromatically granulated blast cells were seen (Fig. 1A). As expected, the blood smear also contained an excess of MC (40% of all nucleated cells) as well as numerous metachromatic blasts. No cytological or histological signs for an accompanying hematopoietic non-MC-disease (AHNMD) was found. The diagnosis MCL based on WHO criteria [10–12] was established.
Fig. 1.
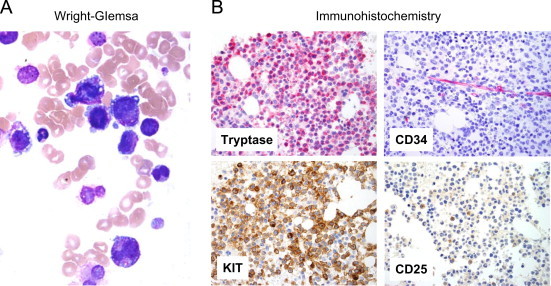
Morphology and phenotype of neoplastic mast cells. Bone marrow (BM) smears (A) and BM sections (B) were examined at the time of diagnosis. BM smears were stained by Wright-Giemsa staining. Original magnification 60×. Note the presence of metachromatic blasts and promastocytes with bi-lobed nuclei. BM sections were stained with antibodies against tryptase, CD34, KIT and CD25. Indirect immunohistochemistry. Original magnification 40×.
3.2. Cytogenetic and molecular results
Conventional karyotyping and FISH revealed a translocation involving chromosomes 7 and 10. The reported karyotype was: 46XX,del(7)(q22),der(10) t(7;10)(q22;q26). Molecular analyses revealed a rare KIT point mutation, namely KIT D816H in BM and blood cells. We also screened for additional mutations in several leukemia-related genes by PCR. However, no additional lesions were detected.
3.3. Phenotype of neoplastic MC
As assessed by immunohistochemistry, neoplastic cells were found to stain positive for tryptase and KIT (Fig. 1B). Some of the neoplastic MC were also found to react with an antibody against CD25, although the staining reaction was weak (Fig. 1B). Neoplastic MC did not stain positive for CD2, CD30 (Ki-1 antigen), CD34 or chymase (Table 2). These cells also stained negative for chloracetate esterase (CAE) and myeloperoxidase (MPO). However, a substantial number of MC (about 20%) were found to react with an antibody against Ki-67. As assessed by multicolor flow cytometry, MC were found to express CD45 and KIT, and low amounts of CD25, CD30 and CD52. By contrast, MC did not stain positive for CD2 (Table 3).
Table 2.
Immunophenotype of neoplastic mast cells (MC) in BM sections.
| Expression detected in |
||||
|---|---|---|---|---|
| The patient's BM MC | MC in patients with |
|||
| Antigen/marker | CD | ISMa | ASMa | |
| LFA-2 | CD2 | +/− | − | − |
| IL-2RA | CD25 | −/+ | + | + |
| Ki-1 | CD30 | −/+ | −/+ | + |
| HPCA-1 | CD34 | − | − | − |
| KIT | CD117 | + | + | + |
| CAE | n.c. | + | +/− | +/− |
| MPO | n.c. | − | − | − |
| Chymase | n.c. | − | +/− | − |
| Tryptase | n.c. | + | + | + |
| Ki-67 | n.c. | +/−b | − | − |
Data were obtained by indirect immunohistochemistry using antibodies directed against various leukocyte (differentiation) antigens.
MC, mast cells; BM, bone marrow; ISM, indolent SM; ASM, aggressive SM; IL-2RA, interleukin-2 receptor alpha chain; HPCA-1, human progenitor cell antigen-1; CAE, chloroacetate esterase; MPO, myeloperoxidase; n.c., not yet clustered.
Data refer to the published literature and own unpublished observations.
As assessed by KIT/Ki-67 double-staining experiments, about 20% of all mast cells were found to stain positive for Ki-67 in this patient.
Table 3.
Cell surface phenotype of neoplastic mast cells assessed by flow cytometry.
| CD | Antigen | Expression of cell surface antigens on KIT+MC at |
|
|---|---|---|---|
| Diagnosis | Relapse | ||
| CD2 | LFA-2 | + | − |
| CD13 | Aminopeptidase N | ++ | ++ |
| CD15 | Lewis X antigen | − | − |
| CD25 | IL-2RA | + | +/− |
| CD26 | DPPVI | − | − |
| CD30 | Ki-1 antigen | − | − |
| CD34 | HPCA-1 | − | − |
| CD43 | Leukosialin | ++ | ++ |
| CD51 | VNRA | − | − |
| CD52 | Campath-1 | + | − |
| CD56 | NCAM | − | − |
| CD61 | VNRB | − | − |
| CD90 | Thy-1 | − | − |
| CD117 | KIT | + | + |
| CD123 | IL-3RA | + | +/− |
| CD133 | AC133 | − | − |
| CD183 | CXCR4 | − | − |
Expression of cell surface antigens on bone marrow mast cells (MC) was analyzed by multicolor flow cytometry using fluorochrome-conjugated antibodies.
3.4. Clinical course and response to chemotherapy
Because of rapid progression, we decided to start treatment with FLAG. After the first cycle, leukocytes (WBC), circulating MC, and serum tryptase levels decreased substantially (Fig. 2). After 3 cycles of FLAG, the serum tryptase level even decreased to normal range (<20 ng/mL) and neoplastic MC in the peripheral blood disappeared. In the BM, neoplastic cells also decreased in number, but residual MC were still detectable (Fig. 3). Overall, the response was judged as an incomplete (almost complete) hematologic remission. At that time, the blood count was normal without substantial cytopenia, and all disease-related symptoms had disappeared. Therefore, allogeneic hematopoietic SCT was planned. However, unfortunately, the patient relapsed with drug-resistant MCL shortly after the third cycle of FLAG and died from cerebral bleeding after a survival time of 5 months.
Fig. 2.
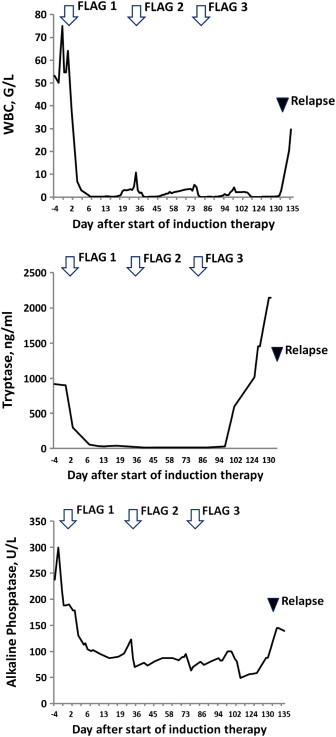
Leukocyte counts, and tryptase levels before and during therapy. Before and during therapy, the white blood cell count (WBC) (A), serum tryptase levels (B) and alkaline phosphatase levels (C) were determined. Serum tryptase was measured by a commercial immunoassay (normal range: 0–15 ng/mL). Chemotherapy treatment (FLAG—arrows) and the time of relapse (time of BM investigation revealing relapse) are also indicated.
Fig. 3.
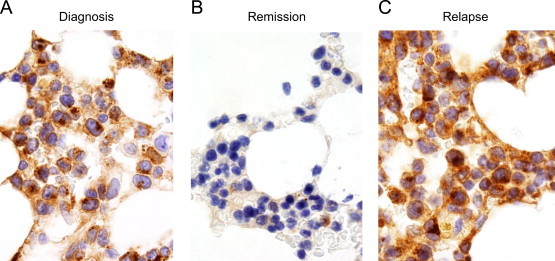
Bone marrow (BM) examination after therapy and at the time of relapse. Before chemotherapy (FLAG) (A), after 2 cycles of FLAG (B) and at the time of relapse after 3 cycles of FLAG (C), BM biopsy sections were stained with an antibody against KIT. After successful treatment, only a few residual KIT+ mast cells were detected (B). At that time, tryptase levels had returned to normal range (<20 ng/mL).
4. Discussion
In patients with MCL, the clinical course is usually characterized by invasive expansion of MC in various internal organs and consecutive multi-organ failure [2–5]. Overall, the prognosis in MCL remains poor. However, MCL itself is a heterogeneous disorder, and the clinical course is unpredictable. In some of these patients, the proliferation-rate of MC is rather low, and organ damage (C-Findings) develops only after several weeks or months. In other cases, however, MC are rapidly proliferating cells that increase in the BM and also in the peripheral blood. We here describe a patient with MCL in whom an excessive proliferation of MC with extremely rapid expansion in the peripheral blood was seen. Because of rapid clinical deterioration, we decided to start chemotherapy shortly after admission. Unexpectedly, the patient responded well to FLAG and entered an almost complete hematologic remission with disappearance of MC in the blood and decrease of serum tryptase levels to normal range. However, although the patient received another cycle of FLAG and was prepared for SCT, she relapsed with resistant MCL and died from cerebral bleeding after an observation time of 5 months.
In most patients with MCL, multi-organ involvement with organ damage is seen [2–5,10]. In most cases, the BM is the primary site of disease evolution [13]. However, only a smaller subset of patients with MCL present with circulating MC, and only very few cases present with rapidly increasing numbers of MC in the peripheral blood. In our patient, leukocyte counts increased rapidly within a very short time, and most of the expanding leukocytes were immature MC and metachromatic blasts, suggesting a high proliferation rate of MC. Indeed, we found that a substantial proportion of neoplastic MC (20%) in our patient stained positive for Ki-67, a proliferation-associated antigen that is usually not expressed in neoplastic MC in SM. The rapid proliferation of MC was also supported by the very high serum tryptase level that had increased rapidly shortly before and at the time of her relapse.
In advanced SM, MC exhibit a characteristic phenotype, including CD25 and CD30 [1–3,14]. In our patient, neoplastic MC stained positive for CD117 (KIT) and tryptase, but most MC did not react with antibodies against CD25 and CD30. This was a somehow unexpected result. Notably, CD30 has been considered to serve a marker of aggressive MC neoplasms [14]. However, apparently, CD30 is not expressed in MC in all cases of MCL. Otherwise, the phenotype of MC was found to correspond well with an aggressive type of SM. In fact, MC did not express chymase and CD2. The LFA-2 antigen (CD2) is of special interest, as it has been described already that CD2 levels are lower in advanced SM (compared to indolent SM=ISM) and may even decrease in MC with the progression of the disease from ISM to ASM or MCL.
In most patients with SM, a transforming mutation in codon 816 of KIT is detectable. In the vast majority of ISM and ASM patients, KIT D816V is found [8,15–18]. However, in MCL, other mutations in KIT, including rare codon 816 mutations are detectable. In our patient, the KIT mutation D816H was detectable in neoplastic cells. This KIT mutant may trigger differentiation of MC in the same way as KIT D816V. However, this mutation is unable to explain the rapid malignant expansion of the clone. Rather, additional mutations and lesions are considered to be responsible for malignant proliferation of MC in MCL [1,2,5]. In our patient, we were able to detect additional lesions by karyotyping. In particular, we were able to detect a t(7;10)(q22;q26) translocation in neoplastic cells. Whether this lesion was involved in malignant progression remains unknown. Alternatively, additional mutations and lesions contributed to malignant expansion of MC in this case.
As mentioned above, the clinical course in MCL is unpredictable. In a few patients, a more indolent course may be found during the first few months. By contrast, in our patient, rapid clinical deterioration based on an extremely rapid (explosive) expansion of MC in the blood and BM was found and required immediate treatment with chemotherapy. We propose that such cases of acute MCL should be discriminated distinctively from less aggressive or even more chronic forms of MCL.
Because of drug resistance, the overall prognosis of MCL remains dismal and the survival is short in most cases [1–4,19,20]. Therefore, patients with MCL are candidates for chemotherapy and SCT. In our patient, we were surprised to see that the patient´s leukemia responded well to induction chemotherapy. We selected the FLAG protocol because this chemotherapy regimen is well known to work well in a subset of patients with chemotherapy-refractory acute leukemias. In our patient, we obtained an almost complete remission after two cycles of FLAG. At that time we prepared the patient for SCT. However, unfortunately, the patient relapsed after her third cycle of FLAG and died from cerebral bleeding 5 months after diagnosis.
Together, our case shows that FLAG may induce remission even in acute MCL. Despite the dismal outcome in our patient, we recommend this protocol as induction treatment in patients with MCL, prior to SCT.
Contributions
P.V. designed the study, wrote the paper and approved the final version of the manuscript. G.E., K.B., H.H., and I.S. isolated mast cells and contributed flow cytomtery staining experiments. S.C.R. performed immunohistochemistry and molecular studies. G.H. performed molecular studies and KIT mutation analyses. R.T. and I.S. performed cytomorphologic investigations. L.M. performed histopathology, chromosome analyses and FISH. W.R.S. contributed the patient, analyzed clinical variables and performed treatment. C.M. provided molecular data and logistic support. H.P.H. contributed immunohistochemical evaluations as well as logistic support.
Disclosures
P.V. and H.P.H. are Novartis-Consultants in a global trial examining the effects of PKC412 (midostaurin) in advanced mastocytosis. P.V. received a Research grant from Novartis and a Research grant from The Mastocytosis Society.
Acknowledgments
This study was supported by the Austrian National Science Fund (FWF), Grant # SFB-F4704.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Valent P., Akin C., Sperr W.R., Horny H.P., Arock M., Lechner K. Diagnosis and treatment of systemic mastocytosis: state of the art. Br J Haematol. 2003;122(5):695–717. doi: 10.1046/j.1365-2141.2003.04575.x. [DOI] [PubMed] [Google Scholar]
- 2.Valent P., Akin C., Sperr W.R., Escribano L., Arock M., Horny H.P. Aggressive systemic mastocytosis and related mast cell disorders: current treatment options and proposed response criteria. Leuk Res. 2003;27(7):635–641. doi: 10.1016/s0145-2126(02)00168-6. [DOI] [PubMed] [Google Scholar]
- 3.Georgin-Lavialle S., Lhermitte L., Dubreuil P., Chandesris M.O., Hermine O., Damaj G. Mast cell leukemia. Blood. 2013;121(8):1285–1295. doi: 10.1182/blood-2012-07-442400. [DOI] [PubMed] [Google Scholar]
- 4.Travis W.D., Li C.Y., Hoagland H.C., Travis L.B., Banks P.M. Mast cell leukemia: report of a case and review of the literature. Mayo Clin Proc. 1986;61(12):957–966. doi: 10.1016/s0025-6196(12)62636-6. [DOI] [PubMed] [Google Scholar]
- 5.Gotlib J., Berubé C., Growney J.D., Chen C.C., George T.I., Williams C. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood. 2005;106(8):2865–2870. doi: 10.1182/blood-2005-04-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valent P., Akin C., Escribano L., Födinger M., Hartmann K., Brockow K. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435–453. doi: 10.1111/j.1365-2362.2007.01807.x. [DOI] [PubMed] [Google Scholar]
- 7.Escribano L., Diaz-Agustin B., López A., Núñez López R., García-Montero A., Almeida J. Spanish Network on Mastocytosis (REMA). Immunophenotypic analysis of mast cells in mastocytosis: when and how to do it. Proposals of the Spanish Network on Mastocytosis (REMA) Cytom Part B Clin Cytom. 2004;58(1):1–8. doi: 10.1002/cyto.b.10072. [DOI] [PubMed] [Google Scholar]
- 8.Sotlar K., Escribano L., Landt O., Möhrle S., Herrero S., Torrelo A. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol. 2003;162(3):737–746. doi: 10.1016/S0002-9440(10)63870-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sperr W.R., Escribano L., Jordan J.H., Schernthaner G.H., Kundi M., Horny H.P. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk Res. 2001;25(7):529–536. doi: 10.1016/s0145-2126(01)00041-8. [DOI] [PubMed] [Google Scholar]
- 10.Valent P., Horny H.P., Escribano L., Longley B.J., Li C.Y., Schwartz L.B. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25(7):603–625. doi: 10.1016/s0145-2126(01)00038-8. [DOI] [PubMed] [Google Scholar]
- 11.Valent P., Horny H.-P., Li C.Y., Longley J.B., Metcalfe D.D., Parwaresch R.M. Mastocytosis (mast cell disease) In: Jaffe E.S., Harris N.L., Stein H., Vardiman J.W., editors. World Health Organization (WHO) classification of tumours. Pathology & genetics. Tumours of haematopoietic and lymphoid tissues. IARC Press; Lyon, France: 2001. pp. 291–302. [Google Scholar]
- 12.Horny H.P., Akin C., Metcalfe D.D., Akin C., Escribano L., Valent P. Mastocytosis (mast cell disease) In: Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H., Thiele J., Vardiman J.W., editors. World Health Organization (WHO) classification of tumours. Pathology & genetics. Tumours of haematopoietic and lymphoid tissues. IARC Press; Lyon, France: 2008. pp. 54–63. [Google Scholar]
- 13.Horny H.P., Valent P. Diagnosis of mastocytosis: general histopathological aspects, morphological criteria, and immunohistochemical findings. Leuk Res. 2001;25(7):543–551. doi: 10.1016/s0145-2126(01)00021-2. [DOI] [PubMed] [Google Scholar]
- 14.Sotlar K., Cerny-Reiterer S., Petat-Dutter K., Hessel H., Berezowska S., Müllauer L. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24(4):585–595. doi: 10.1038/modpathol.2010.224. [DOI] [PubMed] [Google Scholar]
- 15.Nagata H., Worobec A.S., Oh C.K., Chowdhury B.A., Tannenbaum S., Suzuki Y. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA. 1995;92(23):10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longley B.J., Tyrrell L., Lu S.Z., Ma Y.S., Langley K., Ding T.G. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12(3):312–314. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 17.Fritsche-Polanz R., Jordan J.H., Feix A., Sperr W.R., Sunder-Plassmann G., Valent P. Mutation analysis of C-KIT in patients with myelodysplastic syndromes without mastocytosis and cases of systemic mastocytosis. Br J Haematol. 2001;113(2):357–364. doi: 10.1046/j.1365-2141.2001.02783.x. [DOI] [PubMed] [Google Scholar]
- 18.Féger F., Ribadeau Dumas A., Leriche L., Valent P., Arock M. Kit and c-kit mutations in mastocytosis: a short overview with special reference to novel molecular and diagnostic concepts. Int Arch Allergy Immunol. 2002;127(2):110–114. doi: 10.1159/000048179. [DOI] [PubMed] [Google Scholar]
- 19.Lim K.H., Pardanani A., Butterfield J.H., Li C.Y., Tefferi A. Cytoreductive therapy in 108 adults with systemic mastocytosis: outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am J Hematol. 2009;84(12):790–794. doi: 10.1002/ajh.21561. [DOI] [PubMed] [Google Scholar]
- 20.Pardanani A., Lim K.H., Lasho T.L., Finke C., McClure R.F., Li C.Y. Prognostically relevant breakdown of 123 patients with systemic mastocytosis associated with other myeloid malignancies. Blood. 2009;114(18):3769–3772. doi: 10.1182/blood-2009-05-220145. [DOI] [PubMed] [Google Scholar]