

Abstract
Aim
To study population pharmacokinetics and pharmacogenetics of ritonavir boosted atazanavir using exactly recorded drug intake time by MEMS.
Methods
The ANRS134–COPHAR 3 trial was conducted in 35 HIV-infected treatment-naïve patients. Atazanavir (300 mg), ritonavir (100 mg), and tenofovir (300 mg) + emtricitabine (200 mg) were supplied in bottles with a MEMS cap to be taken once daily during 6 months. Six concentrations measurements were collected at week 4, then trough levels bimonthly. A model integrating atazanavir and ritonavir pharmacokinetics and pharmacogenetics was developed using non-linear mixed effects.
Results
Use of exact dosing history data halved unexplained variability in atazanavir clearance. Ritonavir-atazanavir interaction model suggested that optimal boosting effect is achievable by lower ritonavir exposures. Patients with at least one copy of the CYP3A5*1 allele exhibited 42% increased oral CL.
Conclusion
We provide evidence that variability in atazanavir pharmacokinetics is defined by adherence, CYP3A5 genotype and ritonavir exposure.
Keywords: Adenine; analogs & derivatives; therapeutic use; Adult; Aged; Alleles; Cytochrome P-450 CYP3A; genetics; Deoxycytidine; analogs & derivatives; therapeutic use; Drug Interactions; Drug Therapy, Combination; Female; Genotype; HIV Infections; drug therapy; HIV Protease Inhibitors; pharmacokinetics; pharmacology; therapeutic use; Humans; Male; Medication Adherence; Middle Aged; Models, Biological; Nonlinear Dynamics; Oligopeptides; pharmacokinetics; therapeutic use; Organophosphonates; therapeutic use; Pharmacogenetics; Pyridines; pharmacokinetics; therapeutic use; Ritonavir; pharmacology; therapeutic use; Young Adult
Keywords: Atazanavir, Adherence, Pharmacogenetics, Population pharmacokinetics
INTRODUCTION
Atazanavir (ATV) is a potent HIV-1 protease inhibitor (PI) used as a principal component of combined antiretroviral therapy (cART) for first line HIV treatment since 2003(1, 2). Its pharmacologic characteristics allow for once daily dosing either as a 300 mg capsule boosted with ritonavir (100 mg) or 400 mg capsule administered with food. In addition to the convenience of once daily dosing, the advantage of atazanavir over other available PIs lies in its favorable safety profile with respect to the dyslipidemia risk (3).
Despite its favorable pharmacologic properties allowing for once daily dosing, atazanavir pharmacokinetics is associated with high variability between- and within-patients, posing a challenge for assessing and predicting not only individual exposures but also the target concentration–time profile needed for optimal pharmacotherapeutic management (4–6). Potential sources of large between- and within- patient variability can be multiple: a) the food effects on oral bioavailability and absorption, b) drug-drug interactions, c) variability in ritonavir boosting, d) a large array of plausible pharmacogenetics interactions e) adherence variability. Additionally, high atazanavir concentrations have been associated with important toxicities, for example hyperbilirubinaemia. In patients with HIV virus sensitive to atazanavir, the current targets for the therapeutically optimal range of trough concentrations are at a lower boundary of 150 ng/mL, the minimum effective concentration (MEC) for successful viral suppression as reported in the US Department of Health and Human Services guidelines for antiretroviral therapy, and an upper boundary of 850 ng/mL, defined as a concentration threshold for bilirubinemia (4, 7).
The human cytochromes P450 3A4/3A5 (CYP3A5) are implicated in the hepatic metabolism of atazanavir (8, 9). CYP3A5 is mainly expressed in the liver and has a strong genetic basis (10). Low expression of CYP3A5 is found in homozygous carriers of the CYP3A5*3 allele, whereas homozygous and heterozygous carriers of the CYP3A5*1 allele exhibit high expression of CYP3A5 (10). The decreased (26%) atazanavir CL in CYP3A5 non-expressors has been recently reported in a study with healthy volunteers (11). In addition, there are known polymorphisms associated with UGT1A1, and functional deficiencies result in the accumulation of unconjugated bilirubin in the serum although there is variability in the phenotypic expression of patients carrying genetic polymorphism. Rotger et al, showed that individuals with the UGT1A1*28 allele may develop jaundice when exposed to atazanavir or indinavir (12).
Variable adherence to the prescribed therapy adds an additional general level of complexity in maintaining optimal ARV drug treatment. This topic has been in wide discussion in the last decade (13, 14). Both, poor outcome of treatment and selection of resistant viral strains, have been linked to suboptimal adherence (15–20). Additionally, variable adherence is most likely one of the major contributors to the observed pharmacokinetic variability within and even between patients especially for long term treatments (21–24). Different methods have been proposed for measuring adherence. However in order to assess and quantify the impact of adherence on dynamic systems, such as time courses of the viral load, bilirubin concentrations or PI pharmacokinetics, the adherence pattern needs to be known as well. A precise assessment of adherence may be performed with Medication Event Monitoring System (MEMS)(21, 25, 26), which records exact times of bottle opening for drug intake in combination with informative study design that allows for precise quantification of individual drug exposure and variability.
Undoubtedly, the interplay between individual pharmacokinetics, pharmacogenetics and adherence is complex and of critical importance to maintain atazanavir concentrations within the optimal therapeutic range, which would in turn minimize the incidence of therapeutic failure, development of drug-resistance and development of other toxicities Several previous studies developed population pharmacokinetic models for atazanavir (4–6). These studies reported high unexplained between subject variability in atazanavir PK parameters and suggested that adherence, pharmacogenetics and variability in ritonavir exposure are likely responsible for that but none of these studies had adherence measured via MEMS. Therefore, our study focused on elucidating these relationships in a data-driven quantitative manner. Our aims were: (i) to describe the population PK of atazanavir using accurate patient dosing-histories, (ii) to investigate how different assumptions involved in maximizing the accuracy of dosing-history data may impact the population PK analysis outcomes, (iii) to quantify pharmacogenetic effects of different relevant single nucleotide polymorphism (SNPs) on atazanavir pharmacokinetics, (iv) to establish a joint ritonavir—atazanavir pharmacokinetic model to include the boosting mechanism and (v) to estimate realistically variables linked to effectiveness of cART(24, 27), using the established model, such as cumulative time and the number of trough samples below MEC during 24 weeks of trial.
RESULTS
Patients and data
The 35 patients included were 6 females and 29 males with a median age of 36 years (range 24–66).Median viral load (VL) and CD4 count at inclusion were 23,200 copies/mL (range, 100–457,000) and 436 cells/mm3 (range, 197–573) respectively. Clinical and safety results of the trial were presented elsewhere (28). In brief, only 5 of the 35 patients did not achieve undetectable VL(<40 cp/mL) at W24, with a range of 47 to 152 cp/ml, one patient experienced a severe hyperbilirubinemia (grade 4) with a high atazanavir trough concentration and discontinued ritonavir and one patient experienced a transient cytolytic hepatitis (grade 4) of unknown origin.
In total, 272 atazanavir and 245 ritonavir concentrations from the 35 patients followed during a six-month period were available for the analysis. Raw atazanavir data are shown in Figure 1, where a full profile can be observed as well as trough values at different occasions. Inspection of the raw data indicated high variability in the absorption phase as well as substantial within-subject variability in the trough samples. Median observed maximal (Cmax) and trough concentrations (Ctrough) of atazanavir at W4 were 4,021 ng/mL (range, 1,903–2,907), and 304 ng/mL (range, 40–2,366), respectively.
Figure 1.
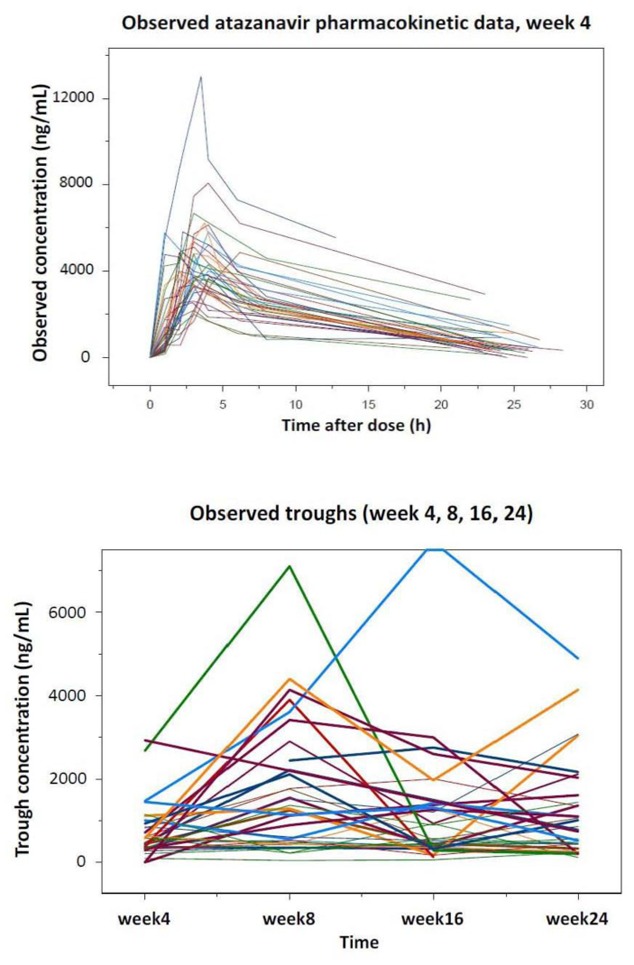
Observed atazanavir concentration, full profile at week 4 (top) and troughs at various occasions (bottom)
Adherence data
Statistical analysis of raw adherence data indicated almost perfect adherence to medications in the studied population with a majority of the prescribed doses were taken as prescribed: median 100% (range 50–100) and median 99.7% (range 51–100)for atazanavir and ritonavir, respectively. The percentage of doses taken on time (±3 hours) was somewhat lower: median 88.2% (range 31.7–100) and median 86.6% (range 30.4–100) for atazanavir and ritonavir, respectively. The deviations from MEMS recorded intake was reported only twice. Similar adherence behavior was observed for tenofovir and emtricitabinepill. The dose-intensity heat maps showing adherence dynamics (incidence and amount of dose intake versus time for each patient) are shown in Figure 2.
Figure 2.
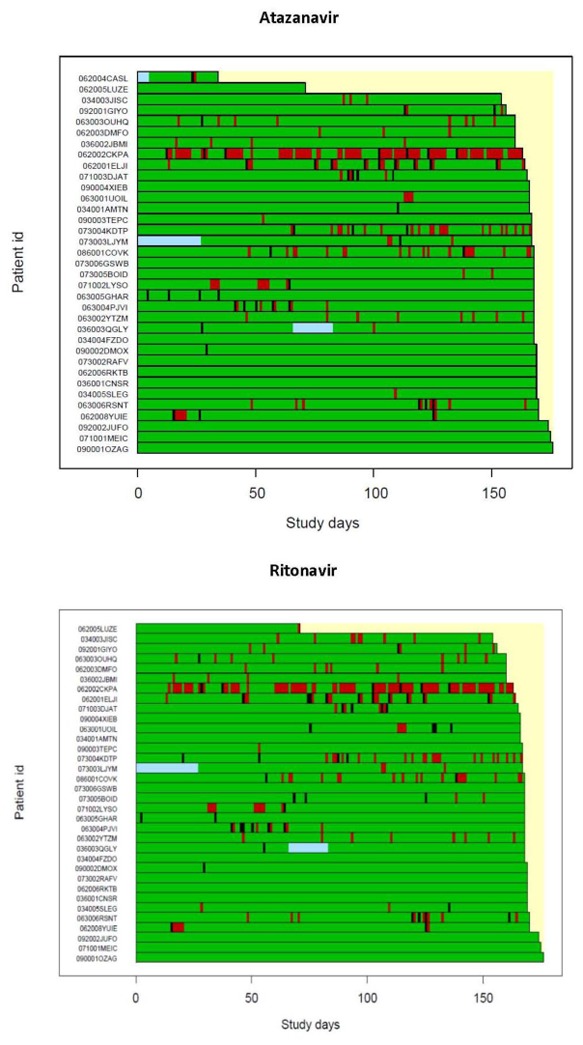
Adherence patterns in the study population for atazanavir (top) and ritonavir (bottom). Green fields indicate correct dose intake, red bars represent days with missed doses, black bars indicate overdosing and blue fields are missing data because of absence of MEMS (patients CASL had atazanavir MEMS data for 4 weeks only). Each patient is shown as a horizontal bar.
Population PK model for atazanavir
A one-compartment model described atazanavir disposition well. The transit compartment absorption model substantially improved the observed data fit compared to all investigated absorption models. When compared to the second best absorption model (first order with a lag time), the transit model decreased −2 log likelihood by 64 points (p<10−14), and also explained an additional 11% of residual variability at the cost of two additional parameters. The comparison of model fits with both the transit and the first order absorption model with a lag time are shown in Figure 3. The proposed transit compartment model was parameterized in terms of the following parameters: number of transit compartments (NN), mean transit time (MTT) which is the average time for a molecule to reach the absorption site, and absorption rate constant (ka).
Figure 3.
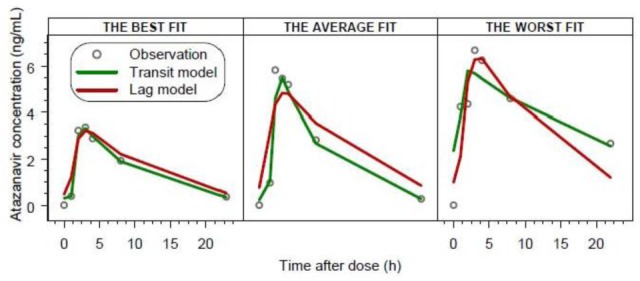
Comparison of individual model fits with the transit compartment model and first order absorption model with a lag time for three patients. The best, average and worst fits were chosen based on the median value of absolute individual weighted residuals.
Impact of adherence assumptions on the outcome of the population PK analysis
With respect to population PK data analysis, the assumption that all patients are at SS gave rise to significant quantifiable inter-occasion variability in CL/F (26.5% CV). The second analysis (AMEMS) in which patient actual MEMS dosing-history were used, led to a negligible within-subject variability estimate indicating that MEMS data explained this variability term entirely. However, a biased estimate of the volume of distribution was observed, thereby potentially affecting the prediction of individual patient’s drug exposures. Numerical difficulties during the estimation procedure were also experienced. In the gold standard analysis, a small proportion of the records of time of dose intake (<5%) were found to be discordant compared to the self-reported dose intake time, therefore excluded from the analysis. That gold standard analysis (AGOLD) based on adjusted dosing histories only, led to the negligible within-subject variability in CL; parameter estimates were reliable, and the estimation procedure was stable. The parameter estimates from the three different analyses are shown in the Table I.
Table 1.
Comparison of atazanavir model parameters obtained with three different analyses with respect to dosing histories assumptions. Relative standard errors, expressed in percentages, are given in parenthesis (except for full MEMS where they were not obtained).
| PK Parameters | Ass: SS assumption | AMEMS: Full MEMS | AGOLD: Gold standard |
|---|---|---|---|
| CL/F (L/h) | 7.2 (4.4) | 6.8 | 6.9 (8.1) |
| V/F (L) | 79.2 (8.9) | 102 | 81.1 (6.8) |
| ka (h−1) | 2.7 (26.8) | 5.6 | 3.2 (42.1) |
| MTT (h) | 1.3 (11.2) | 1.5 | 1.35 (11) |
| NN | 17.3 (46.7) | 8 | 11.5 (26.4) |
| BSV (CL/F) * | 47.4 (28.7) | 44.3 | 40.2 (32.7) |
| WSV (CL/F) * | 26.5 (16.9) | <1 | <1 |
| BSV (V/F) * | 30.0 (43.2) | 61.4 | 30.1 (28.5) |
| BSV (ka) * | 73.5 (47.2) | 120 | 78.4 (73.2) |
| BSV (MTT) * | 47.4 (28.7) | 40.1 | 45.2 (31.8) |
| RV (week 4) * | 18.7 (16.9) | 27.7 | 19.4 (15.5) |
| RV (> week 4) * | 38.0 (33.1) | 47.4 | 43.4 (10.6) |
Note: All BSV, WSV and RV are expressed as CV (%).
BSV=Between-subject variability, WSV=Within-subject variability, RV=Residual variability, MTT=mean transit time, NN=number of transit compartments, ka=absorption rate constant
Pharmacogenetics and covariates
Pharmacogenetic data analysis revealed a statistically significant (p<0.01) effect of the CYP3A5 polymorphism on the atazanavir CL. Patients with at least one copy of the CYP3A5*1 allele (n=12) exhibited 42% increased oral CL compared with patients with both CYP3A5*3 allele. None of other demographic and genetic covariates deemed to be important. The visual predictive check for the final model stratified for different CYP3A5 genotypes is shown in Figure 4.
Figure 4.
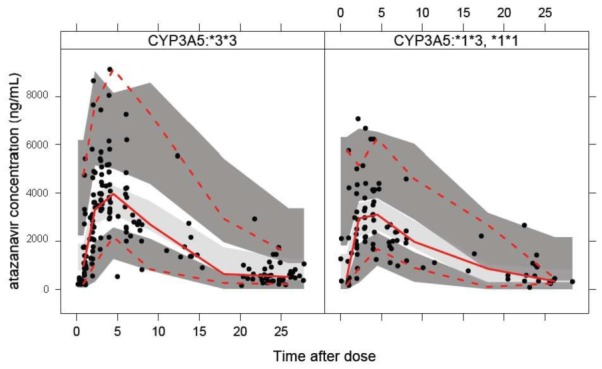
Visual predictive check for the final atazanavir model stratified for different CYP3A5 genotypes: homozygote *3*3 shown in the left panel and heterozygote *1*3 and homozygote *1*1 shown in the right panel. Red solid line indicates observed data median, red dotted lines are 95% observed percentiles, grey shaded area is the simulated median with uncertainty and dark grey shaded areas are simulated 95% percentiles with uncertainty.
Joint ritonavir-atazanavir model
Ritonavir concentration time profiles were best described with the one compartment model. Absorption was described well with the transit compartment absorption model. Parameter estimates of the final ritonavir PK model are given in the Table 2. Ritonavir exposure (AUC0-24) defined as dose divided by individual CL was a sufficient driver to explain the boosting mechanism of atazanavir CL. The estimate of unboosted atazanavir CL, e.g. in the absence of ritonavir, was rather high (16 L/h). Ritonavir proved to be an efficient booster exhibiting maximal inhibitory effect of 98% with AUC50of 6,230 ng.h/mL, which was on the lower end of the observed ritonavir exposure in this study (7th percentile). The schematic view of the proposed model is shown in Figure 5. The increase in atazanavir CL for the carriers of the CYP3A5*1 allele was 28% after inclusion of ritonavir-boosting mechanism. The parameter estimates for the final PKPG model are given in the Table II.
Table 2.
Model parameter estimates for the final ritonavir (left column) and joint ritonavir-atazanavir (right column) PK-PG model using a gold standard analysis. Relative standard errors, obtained by nonparametric bootstrap (n=100) and expressed in percentages, are given in parenthesis.
| Ritonavir | Atazanavir | |
|---|---|---|
| CL/F (L/h) | 10.3 (9) | 16.0** (64) |
| βCYP3A5*1 | - | 1.28 (59) |
| AUC50 of RTV (ng.h/mL) | - | 6,230 (91) |
| Emax | - | 0.98 (3) |
| V/F (L) | 87.9 (11) | 81.1 (6) |
| ka (h−1) | 3.9 (58) | 3.3 (146) |
| MTT (h) | 1.1 (16) | 1.36 (14) |
| NN | 14.4 (95) | 11.5 (29) |
| BSV (CL/F) * | 38.5 (33) | 31 (28) |
| BSV (V/F) * | 37.3 (50) | 31 (33) |
| Correlation (CL/F-V/F) | 0.74 (45) | - |
| BSV (ka) * | 248 (161) | 81 (133) |
| BSV (MTT) * | 49.3 (77) | 44 (39) |
| RV week 4 (%) * | 40.8 (8) | 19.4 (16) |
| RV > week 4 (%) * | 40.8 (8) | 42.3 (10) |
BSV=Between-subject variability, RV=Residual variability,
(CL/F)0=atazanavir CL in absence of ritonavir, MTT=mean transit time, NN=number of transit compartments, ka=absorption rate constant, AUC50 – ritonavir exposure needed for half maximal boosting effect, Emax – maximal effect of ritonavir boosting, βCYP3A5*1 – factor of increase in atazanavir CL with at least one copy of CYP3A5*1 allele, RV=residual variability
Figure 5.
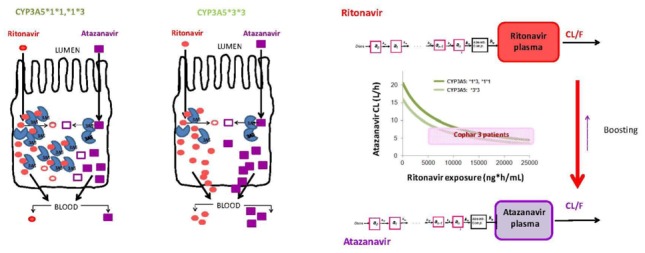
Schematic view of: left, effect of CYP3A5 genotype on ritonavir - atazanavir first pass effect; right, final linked ritonavir – atazanavir model. CYP3A5*3*3 are low expressors and CYP3A5*1*3, *1*1 are high expressors.
Model Predictions and Simulations
The number of trough samples and the cumulative time below MEC over 24 weeks were computed for each patient (180 trough values for each patient). If the adherence was perfect, none of the trough samples would be below MEC (150 ng/mL), except for one individual who had an extremely high CL and was a CYP3A5*1 carrier, and whose trough values were all below the target level. And therefore similar result holds for the cumulative time below this threshold. However, when the true MEMS dosing histories were used, during the 24 weeks, 4patients (12 %) had more than 10 trough samples below the MEC of 150 ng/mL and 8 patients (24 %) spent more than 100 h below MEC.
DISCUSSION
Today, there are more HIV patients on the planet who do not have the access to powerful protease inhibitors than the ones who do have the access to PIs. For those HIV patients who do have the access to effective PI-based HIV treatment, successful management of the HIV therapy still presents a great challenge both for the physician and the patient. Complex interplay between highly variable drug specific pharmacokinetics, patient adherence, and genetic background of the patient poses difficulties in maintaining the optimal therapeutic levels of the PIs required in order on one hand, to minimize the risk of therapeutic failure associated with the risk of viral resistance, and on the other hand, to prevent drug-related toxicities. A fully integrated pharmacokinetic-pharmacogenetic model with an integrated ritonavir-related boosting mechanism and real time adherence data was developed for atazanavir pharmacokinetics.
In this study, atazanavir disposition was best described by a one compartment model, which is in line with previous modeling studies (4–6). Kile et al found that two compartment model was best fit for atazanavir pharmacokinetics, which was not the case for our data. This is likely due to the design: Kile et al collected very rich data in healthy volunteers (9 samples) while we collected6 samples in patients. It is common that with very rich design, more complex models can be applied. However, we do not anticipate that this would alter any of our major findings.
In the absence of ritonavir-related boosting and pharmacogenetic relationships, apparent atazanavir CL and V were 6.9 L/h and 81.1 L, respectively, resulting in an elimination half-life of 8.1 h, which is consistent with previously reported values (8.2–8.9 h) (4–6). Our work departs from previously published atazanavir models in reporting our evaluation of relevant absorption models. Our study implemented a semi-mechanistic transit compartment model in order to describe highly variable atazanavir absorption. The transit model assumes that the delay in the absorption process is a consequence of the drug passing through chain of linked compartments (steps), before reaching the absorption site (29). Our analysis illustrates that the transit compartment model is an attractive alternative for the modeling of the variable absorption, especially for anti-infective agents (30, 31).
Ritonavir has been developed and used as a pharmacokinetic booster of protease inhibitors (32). Ritonavir is a potent inhibitor of CYP3A4, the primary enzyme responsible for the metabolism of PIs, therefore decreasing the clearance and increasing half life of PIs and providing a basis for increasing the dosing interval to once daily (QD). The most common dose used for boosting is a 100 mg capsule per PI dosing. In our integrated model, the ritonavir-related boosting mechanism of atazanavir CL/F has been introduced. It was best described by the inhibitory Emax model, where the baseline atazanavir CL value in the absence of ritonavir was estimated (16 L/h, which is similar to previous reports(11)) as well as maximal extent of inhibition and ritonavir exposure needed for half maximal effect. Ritonavir is an established and proven potent booster with maximal (98%) inhibition of atazanavir CL, suggesting that a majority of the patients have been maximally boosted. Interestingly, a lower value of ritonavir exposure (6,230 ng.hr/mL) is estimated to be needed for half maximal ritonavir boosting effect. This estimated ritonavir exposure is at the lower end of the observed ritonavir exposures (7th percentile), which suggest potential overdosing of ritonavir in most patients, whereby lower doses of ritonavir could produce the desired boosting effect in order to maintain once daily atazanavir dosing. This result may be of interest considering the fact that 100 mg qd ritonavir has been implicated in increased risk of lipid level increase (33, 34). Furthermore, ritonavir may also be responsible for gastrointestinal adverse events experienced during treatment with PIs, therefore potentially affecting the adherence to PIs and ultimately the success of HIV therapy (35).
Atazanavir is primarily metabolized by CYP3A4/A5 and pharmacogenetic differences are expected to be observed with respect to the bioavailability and clearance of the drug. Even though our study was rather small (35 patients), we have quantified a significant increase in atazanavir CL (28%) in the patients with at least one CYP3A5*1 allele, which is similar to the previously reported values (9). Kile et al reported 26% difference in unboosted atazanavir CL/F in healthy volunteers with different CYP3A5 genotypes. Our study confirms their findings in naïve HIV patients but also provides evidence that this effect is significant in presence of ritonavir and is of same magnitude at steady state.
Adherence to the prescribed therapy has been repeatedly named as the key factor for successful treatment outcome. Importantly, adherence will contribute to the observed pharmacokinetic variability. This fact is critical when drug levels must be maintained within a desired therapeutic window, or specifically in the case of PIs, above minimal effective concentrations. Furthermore, the ultimate goal of all model-based analysis is not only to quantify random variability between subjects, but also to explain these differences by plausible covariates. Ultimately, if almost of the variability in drug response can be explained, it would provide a basis for personalized dosing regimens. However, even-though a large number of demographic and pharmacogenetic covariates are often available for the analysis, only a small portion of the observed variability is explained. We hypothesized that adherence fluctuations would explain the majority of the observed variability. Therefore, we performed a sub-analysis of our data focusing on assumptions surrounding dosing histories. Our first analysis, which is in line with an implicit assumption in most population PK analyses, assumed perfect adherence and that all patients were at steady state. Under the steady-state assumption, the random variability in CL/F was 74%, corresponding to 47% of between-subject variability and 27% within-subject (or between-occasion) variability. Once MEMS adherence data from only “reliable-patients” were included, the between-subject variability in CL/F decreased to 40%, while the within-subject variability became negligible. This finding suggests that almost half of the initially quantified variability was not true variability in clearance, but was attributable to the unwarranted assumption of perfect adherence by all patients. This finding also reminds one that important covariates (e.g., weight, age, pharmacogenetics, etc), do not explain such a large portion of variability. It is consistent however, with the conclusion of Harter and Peck that variability in adherence rivals pharmacokinetics as the leading source of variability in drug response (36).
In our study, we have measured adherence using the MEMS caps, which record the exact time of the bottle opening. In most of the cases, the opening of the drug bottle corresponds to the drug intake; however some deviations to this assumption may occur. To obtain data closest to the truth, we asked subjects to keep a diary where these deviations were recorded. In addition, at each clinic visit, we asked them to report the time of drug intake from the previous day. This information was very helpful since we could compare the MEMS records with the individual’s diary entry, and most of them agreed. A small proportion of the records (<5%) were discordant, leading to identifying these patient records as “unreliable”. Even though the proportion of unreliable data points was small, it was important to exclude them from the analysis, in order to generate a gold standard cohort for comparison, increase model stability and guarantee analysis convergence.
In general, patient adherence in our study was very good. 98% of all atazanavir and ritonavir doses and 86% of the pills were taken by patients as prescribed. However, despite this high adherence rate, we showed that a few patients had atazanavir plasma levels below the target minimal effective concentration during the course of the treatment. Even though the median value of the time below MEC was low (<1% of the dosing interval), some of the patients spent up to 15% of the dosing interval below MEC. More precisely, there were five patients who were, over the treatment course, more than a cumulative 7days with unacceptably low levels of atazanavir. Two of these patients were non-responders at week 24 (out of the 5 non responders). The example of one of these patients is shown in Figure 6 where a full PK profile over 6 months is shown as well as the accumulated time below MEC. If the dosing regimen intake was perfect, none of the patients would be below MEC at any time during treatment. This suggests that even though overall adherence may be high, patients still fall below MEC, and are therefore at risk of treatment failure, if the drug intake is not-timely. This finding accentuates the importance of estimating adherence not only as an aggregate measure over a defined period of time, but also the precise time of each capsule intake. Only when we have this data in hand are we able to search and find plausible answers why treatment may have failed or resistance developed. Two main limitations of the current study are small sample size (35 patients) and the protocol may have induced better adherence (Hawthorne effect).
Figure 6.
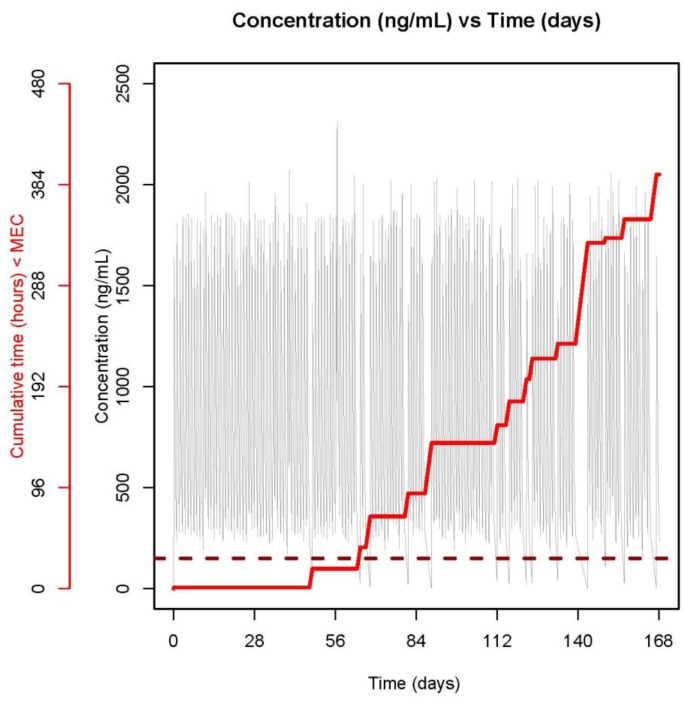
PK profile over 6 months derived based on individual parameter estimates and full dosing histories (grey line) for one patient with VL > 40 cp/mL at week 24. Cumulative time below MEC is shown with a red stair-case line. The target MEC of 150 ng/mL is shown with a dark red broken line.
In conclusion, a full, integrated pharmacokinetic-pharmacogenetic model for atazanavir with incorporated ritonavir-related boosting mechanism and real time adherence was developed for atazanavir pharmacokinetics. With the proposed model, we have delineated and quantified several important aspects of atazanavir-based HIV treatment. We have i) quantified a significant pharmacogenetic effect of CYP3A5 genotype on atazanavir CL, ii) characterized the pronounced ritonavir-related boosting mechanism of atazanavir CL at currently used ritonavir doses, and iii) assessed the large effect of adherence on atazanavir PK variability. The proposed model represents a useful basis for the individualized dosing of atazanavir and provides a precise tool for assessing the important PK-based variables (e.g. time below MEC) for successful HIV-treatment.
METHODS
Clinical trial
The ANRS 134 - COPHAR 3 trial was a multicenter prospective trial conducted in HIV-1-infected naïve patients starting a PI-containing cART treatment. The primary objective of the trial was to study pharmacokinetics/pharmacogenetics of atazanavir given with ritonavir using adherence measured by MEMS in PI treatment-naïve HIV-infected patient.
The trial started and was completed in 2008. It included 35 naïve patients, who were started on treatment containing 300 mg of atazanavir boosted with 100 mg ritonavir and a fixed dose combination of two nucleoside analogs that are co-formulated: tenofovir (300 mg) and emtricitabine (200 mg) and were followed 24 weeks. Reyataz® (atazanavir) and Truvada® (tenofovir and emtricitabine) were kindly provided by Bristol-Myers Squibb and Gilead, respectively. Viruses have to be sensitive to each component of therapy on a genotypic resistance assay performed before entry. No antacids or CYP inducing drugs were allowed during the trial. During the PK visit, drug intake was performed with food administration.
The study was performed according to the Declaration of Helsinki and its amendments. The protocol was approved by the Ethics Committee of Ile de France VII (Le Kremlin Bicetre, France) and all subjects provided written informed consent. The EUDRA CT number is 2007-003203-12.
Study design and Pharmacokinetic data
Patients were sampled during four visits at weeks (W) 4, 8, 16 and 24. During each visit, the patients were asked to report the exact time when the last dose was taken in the previous day. At W4, a trough blood sample was collected first, followed by the dose intake and collection of additional five samples at 1, 2, 3, 4, and 8 hours post-dose. At W8, W16 and W24, only the trough samples were collected. Therefore, each individual had 9 PK samples available over a period of 6 months. Plasma concentrations were determined in the laboratories of the hospitals where the patients were followed by a specific high performance liquid chromatography (HPLC) protocol. The participant laboratories were cross-validated before starting the study. The lower limit of quantification was 50 ng/mL and 25 ng/mL for atazanavir and ritonavir, respectively.
Adherence data
Each patient was supplied with three MEMS capped bottles containing either atazanavir hard-capsules, ritonavir soft-capsules or tenofovir/emitricabine FDC tablets (Truvada®). The exact time of each opening of the cap was recorded. Therefore a full dosing history for the three capsules was available for all the patients. Patients were also requested to note any deviations from MEMS recorded drug intake, for example if a patient would leave for a short holiday and remove the requisite supply of pills in a single cap opening. Additionally, at each clinic visit, patients were asked to report on a self administered questionnaire the time of the previous dose intake, which allowed for comparison with the MEMS recorded time.
Each patient whose MEMS record agreed with his or hers personal declared record within ±3 hours was considered to have a reliable MEMS dosing history, indicating that the MEMS recorded time was similar to the time of actual drug intake. If a MEMS record did not agree with patient’s personal record, this record was considered less reliable and the dosing history preceding the clinical visit was discarded from the analysis as well as collected sample at the clinical visit, when gold standard approach analysis was used (more details below).
Pharmacogenetic data
Six genetic polymorphisms were studied: MRP2 (rs717620), MRP4 (rs1751034), ORM1 (A721G), CYP3A5 (rs776746), MDR1 (rs1045642) and UGT1A1*28 (rs8175347).All of the genotyping analyses were performed in the same laboratory. The subjects were genotyped by the TaqMan allelic discrimination assay or GeneScan analysis. For both methods, genomic DNA was extracted from peripheral blood mononuclaear cells using the Puregene Kit (Gentra systems, Minneapolis, USA) according to the manufacturer’s protocol. For each polymorphism, departure from Hardy-Weinberg proportions was tested using χ2 test with degrees of freedom equal to the number of observed genotypes minus 1.
Data analysis
i. Development of population model for atazanavir
All data were analyzed using the non-linear mixed effects approach available in NONMEM VI. The First Order Conditional Estimation with interaction (FOCEI) method was employed throughout the analysis. A basic model structure was established using the full PK profile data from W4 and assuming steady state conditions. As atazanavir exhibit highly variable absorption, special attention was paid to the modeling of this process. Several models were investigated, including the zero order absorption models, first order absorption model with lag time, sequential zero and first order model, transit absorption model. The individual parameters were assumed to be log-normally distributed and proportional error was employed for description of residual variability. The model building procedure was guided by the likelihood ratio test, diagnostic plots and internal model validation techniques, including visual and numerical predictive checks.
ii. Adherence modeling
Adherence data were summarized in terms of following statistical quantities: percent (%) of days with correct number of doses taken, % of total prescribed doses taken and % of doses taken within ± 3 hours of the protocol specified dosing time. Adherence data were also explored using a graph where daily adherence behaviors, such as if dose was taken, taken twice or missed, were visualized for each patient and each drug. This plot is often referred to as a “dose intensity gap map”. In order to assess the impact of dosing history assumptions on the outcome of the population analysis, we performed three separate analyses from the entire atazanavir dataset (full PK profile from W4 and three additional trough samples at W8, W16 and W24). The first analysis, based on the assumption of full adherence to the protocol specified dosing history (Ass), included the assumption that all patients were at steady state and that the time of last dose intake as reported by the patient before a PK visit was accurate. The second analysis, a MEMS recorded dosing history analysis (AMEMS),assumed that each dosing time recorded by MEMS was accurate. The third analysis, which we termed the gold standard analysis (AGOLD),used ”reliable” dosing-history data only, which consisted only in concentration data from which MEMS records of time of intake were concordant (within 3 hours) with patients last reported time of dose intake before a PK visit. When MEMS data were missing (no MEMS for some patients sometimes), the corresponding concentration data were not analyzed The previously established basic structural model was extended to include within-subject variability (WSW) in CL and was rerun using three different dosing history data as described above (Ass, AMEMS and AGOLD). Parameter estimates were compared with particular attention to the magnitude of the variability parameters (between-subject, within-subject and residual variability).
iii. Pharmacogenetic and demographic covariate analysis
Six genetic polymorphisms (MRP2, MRP4, MDR1, CYP3A5, UGT1A1,ORM1) were evaluated as possible genetic covariates influencing pharmacokinetic parameters of atazanavir. Potential impact of these genetic markers on atazanavir and ritonavir pharmacokinetics are described in Table 1 of supplementary material. Mutant and heterozygotic alleles were grouped together into one category in order to increase the power of the search. Categorizations in three classes were also tested: wild homozygotes versus heterozygotes versus mutant homozygotes.
Demographic covariates were also available: age, sex and body weight. The covariate search was performed using a stepwise covariate model building procedure described elsewhere (37, 38). The procedure included a forward inclusion step, where parameter-covariate relationships are added to the model in a stepwise manner until no further relationship is statistically significant (p<0.05). Backwards elimination steps followed, where the identified relationships were excluded from the model if they failed to achieve stricter statistical significance (p<0.01), in order to account for the multiple testing.
iv. Development of the population models for ritonavir and the joint ritonavir-atazanavir model
Full dosing history data were available for ritonavir and the gold standard approach was used. Ritonavir concentration time data were modeled using a similar methodology to the one described above for atazanavir.
Once separate ritonavir and atazanavir models were established, the next step involved development of the joint ritonavir-atazanavir model, where the boosting mechanism was introduced. A range of models was evaluated in which atazanavir CL/F was either: (i) a function of ritonavir concentrations, (ii) a delayed response to ritonavir concentrations, or (iii) a function of ritonavir steady state exposure. For the models employing ritonavir concentrations as a driver of the boosting mechanism, both simultaneous and sequential approaches were tested. Steady state ritonavir exposure was computed as ritonavir dose divided by individual CL value. A model for the boosting mechanism utilized a nonlinear inhibitory Emax function, where the maximal boosting effect was estimated as well as AUC50, which is the ritonavir exposure needed for half maximal effect (Equation 1). CL0 represents unboosted atazanavir CL.
v. Model simulations and predictions
The final PK-PG model for atazanavir using the dosing history data from the “gold standard approach” and the model-estimated individual parameters were used to predict different variables linked to effectiveness cART (24, 27). For each patient we estimated, over the 24 weeks of the trial, (i) the number of trough samples below MEC (150 ng/mL) for wild-type viruses (4, 7), and (ii) the cumulative time with concentration below MEC. This information were estimated using first a hypothetical perfect dosing regimen, as if drug was taken every 24h and second using the actual dosing regimen as recorded by MEMS.
CPT Highlights.
What is the current knowledge on the topic?
Population pharmacokinetics of CYP3A substrates ritonavir and atazanavir is well understood. Adherence to medication is a key factor in large concentration variability leading to poor treatment outcome.
What question this study addressed?
The study quantifies the relationship between adherence, CYP3A5 genotype, ritonavir and atazanavir pharmacokinetics in HIV-infected treatment-naïve patients.
What this study adds to our knowledge?
Adherence is the major contributor to variability in atazanavir pharmacokinetics. Oral clearance is increased by 28% in patients with a copy of CYP3A5*1,in ritonavir presence. Optimal boosting effect is achievable by lower ritonavir exposures.
How this might change clinical pharmacology and therapeutics?
The atazanavir therapeutic dose can be individualized based on CYP3A5 genotype. If good adherence is achieved, ritonavir dose can be lowered. Real time adherence allows calculation of cumulative drug exposure during a treatment period. TDM using a single plasma sample can be misleading.
Acknowledgments
We thank all the patients who participated in the ANRS 134 -COPHAR 3 trial. We thank Bristol-Myers Squibb and Abbott laboratories for providing atazanavir and ritonavir pure samples for drug assay, respectively. Radojka Savic was financially supported by a Postdoc grant from the Swedish Academy of Pharmaceutical Sciences (Apotekarsocieteten).
Appendix: ANRS 134 - COPHAR 3 study group
Scientific Comittee: A. Barrail-Tran, A. Brunet, M-J Commoy, S. Couffin-Cadiergues, D. Descamps, X. Duval, C. Goujard, C. Le Guellec, F. Mentré, G. Nembot, A-M. Taburet, B. Vrijens.
Clinical centers: Dr Ajana, Dr Aissi, Dr Baclet, Pr Besnier, Dr Bollens, Dr Boulanger, Mme Brochier, Dr Brunet, Dr Chaix, Dr Ciuchete, Dr Ghosn, Pr Duval, Dr Ferret, Mr Ferret, Mme Gaubin, Pr Goujard, Pr Girard, Dr Kouadio, Mme Lupin, Dr Parienti, Mme Parrinello, Dr Poinçon de la Blanchardière, Pr May, Mme Marien, Mme Medintzeff, Mme Metivier, Mme Mole, Pr Molina, Mme Nau, Dr Ouazene, Dr Pintado, Dr Quertainmont, Mme Ramani, Dr Rami, Dr Sellier, Dr Simon, Dr Talbi, Mme Thoirain, Pr Verdon, Pr Trépo, Dr Wassoumbou, Pr Yazdanpanah.
Pharmacological centers: Dr Barrail-Tran, Dr Gagneux, Dr Delhotal, Dr Hoizey, Dr Houdret, Dr Leguellec, Dr Peytavin, Dr Poirier, Dr Sauvageon, Dr Taburet.
Virological centers : Pr André, Dr Soulié, Pr Calvez, Dr Morand-Joubert, Dr Harchi, Dr Bocket, Dr Mourez, Dr Palmer, Dr Pallier, Dr Deschamps, Dr Mazeron, Mme Bolmann, Mr Storto, Mme Thanh Thuy.
Monitoring: G. Nembot, G. Unal, F. Mentré.
Statistical Analysis: F. Mentré, X. Panhard, R. Savic, B. Vrijens
Footnotes
Conflict of interest: None
References
- 1.Piliero PJ. Atazanavir: a novel HIV-1 protease inhibitor. Expert Opin Investig Drugs. 2002 Sep;11(9):1295–301. doi: 10.1517/13543784.11.9.1295. [DOI] [PubMed] [Google Scholar]
- 2.BMS. Summary Product Characteristics. Bristol Myers Squibb; 2005. Reyataz (Atazanavir Sulfate) Capsules. [Google Scholar]
- 3.Goldsmith DR, Perry CM. Atazanavir Drugs. 2003;63(16):1679–93. doi: 10.2165/00003495-200363160-00003. discussion 94–5. [DOI] [PubMed] [Google Scholar]
- 4.Colombo S, Buclin T, Cavassini M, Decosterd LA, Telenti A, Biollaz J, et al. Population pharmacokinetics of atazanavir in patients with human immunodeficiency virus infection. Antimicrob Agents Chemother. 2006 Nov;50(11):3801–8. doi: 10.1128/AAC.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickinson L, Boffito M, Back D, Waters L, Else L, Davies G, et al. Population pharmacokinetics of ritonavir-boosted atazanavir in HIV-infected patients and healthy volunteers. J Antimicrob Chemother. 2009 Jun;63(6):1233–43. doi: 10.1093/jac/dkp102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solas C, Gagnieu MC, Ravaux I, Drogoul MP, Lafeuillade A, Mokhtari S, et al. Population pharmacokinetics of atazanavir in human immunodeficiency virus-infected patients. Ther Drug Monit. 2008 Dec;30(6):670–3. doi: 10.1097/FTD.0b013e3181897bff. [DOI] [PubMed] [Google Scholar]
- 7.Porte CJLl, Back D, Blaschke T, Boucher CAB, Fletcher CV, Flexner C, et al. Updated guideline to perform therapeutic drug monitoring for antiretroviral agents. Reviews in Antiviral Therapy. 2006;2006(3):4–14. [Google Scholar]
- 8.Le Tiec C, Barrail A, Goujard C, Taburet AM. Clinical pharmacokinetics and summary of efficacy and tolerability of atazanavir. Clin Pharmacokinet. 2005;44(10):1035–50. doi: 10.2165/00003088-200544100-00003. [DOI] [PubMed] [Google Scholar]
- 9.Anderson PL, Aquilante CL, Gardner EM, Predhomme J, McDaneld P, Bushman LR, et al. Atazanavir pharmacokinetics in genetically determined CYP3A5 expressors versus non-expressors. J Antimicrob Chemother. 2009 Nov;64(5):1071–9. doi: 10.1093/jac/dkp317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27(4):383–91. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 11.Kile DA, Mawhinney S, Aquilante CL, Rower JE, Castillo-Mancilla JR, Anderson PL. A Population Pharmacokinetic-Pharmacogenetic Analysis of Atazanavir. AIDS Res Hum Retroviruses. 2012 Apr 20; doi: 10.1089/aid.2011.0378. [DOI] [PubMed] [Google Scholar]
- 12.Rotger M, Taffe P, Bleiber G, Gunthard HF, Furrer H, Vernazza P, et al. Gilbert syndrome and the development of antiretroviral therapy-associated hyperbilirubinemia. J Infect Dis. 2005 Oct 15;192(8):1381–6. doi: 10.1086/466531. [DOI] [PubMed] [Google Scholar]
- 13.Blaschke TF. Variable adherence to prescribed dosing regimens for protease inhibitors: scope and outcomes. Curr Opin HIV AIDS. 2008 Nov;3(6):603–7. doi: 10.1097/COH.0b013e32831271c2. [DOI] [PubMed] [Google Scholar]
- 14.Osterberg LG, Urquhart J, Blaschke TF. Understanding forgiveness: minding and mining the gaps between pharmacokinetics and therapeutics. Clin Pharmacol Ther. 2010 Oct;88(4):457–9. doi: 10.1038/clpt.2010.171. [DOI] [PubMed] [Google Scholar]
- 15.Bae JW, Guyer W, Grimm K, Altice FL. Medication persistence in the treatment of HIV infection: a review of the literature and implications for future clinical care and research. AIDS. 2011 Jan 28;25(3):279–90. doi: 10.1097/QAD.0b013e328340feb0. [DOI] [PubMed] [Google Scholar]
- 16.Bangsberg DR, Hecht FM, Charlebois ED, Zolopa AR, Holodniy M, Sheiner L, et al. Adherence to protease inhibitors, HIV-1 viral load, and development of drug resistance in an indigent population. AIDS. 2000 Mar 10;14(4):357–66. doi: 10.1097/00002030-200003100-00008. [DOI] [PubMed] [Google Scholar]
- 17.Bangsberg DR, Perry S, Charlebois ED, Clark RA, Roberston M, Zolopa AR, et al. Non-adherence to highly active antiretroviral therapy predicts progression to AIDS. AIDS. 2001 Jun 15;15(9):1181–3. doi: 10.1097/00002030-200106150-00015. [DOI] [PubMed] [Google Scholar]
- 18.Ekstrand ML, Shet A, Chandy S, Singh G, Shamsundar R, Madhavan V, et al. Suboptimal adherence associated with virological failure and resistance mutations to first-line highly active antiretroviral therapy (HAART) in Bangalore, India. Int Health. 2011 Mar 1;3(1):27–34. doi: 10.1016/j.inhe.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lucas GM. Antiretroviral adherence, drug resistance, viral fitness and HIV disease progression: a tangled web is woven. J Antimicrob Chemother. 2005 Apr;55(4):413–6. doi: 10.1093/jac/dki042. [DOI] [PubMed] [Google Scholar]
- 20.Roge BT, Barfod TS, Kirk O, Katzenstein TL, Obel N, Nielsen H, et al. Resistance profiles and adherence at primary virological failure in three different highly active antiretroviral therapy regimens: analysis of failure rates in a randomized study. HIV Med. 2004 Sep;5(5):344–51. doi: 10.1111/j.1468-1293.2004.00233.x. [DOI] [PubMed] [Google Scholar]
- 21.Vrijens B, Goetghebeur E. The impact of compliance in pharmacokinetic studies. Stat Methods Med Res. 1999 Sep;8(3):247–62. doi: 10.1177/096228029900800305. [DOI] [PubMed] [Google Scholar]
- 22.Vrijens B, Goetghebeur E. Electronic monitoring of variation in drug intakes can reduce bias and improve precision in pharmacokinetic/pharmacodynamic population studies. Stat Med. 2004 Feb 28;23(4):531–44. doi: 10.1002/sim.1619. [DOI] [PubMed] [Google Scholar]
- 23.Vrijens B, Gross R, Urquhart J. The odds that clinically unrecognized poor or partial adherence confuses population pharmacokinetic/pharmacodynamic analyses. Basic Clin Pharmacol Toxicol. 2005 Mar;96(3):225–7. doi: 10.1111/j.1742-7843.2005.pto960312.x. [DOI] [PubMed] [Google Scholar]
- 24.Vrijens B, Tousset E, Rode R, Bertz R, Mayer S, Urquhart J. Successful projection of the time course of drug concentration in plasma during a 1-year period from electronically compiled dosing-time data used as input to individually parameterized pharmacokinetic models. J Clin Pharmacol. 2005 Apr;45(4):461–7. doi: 10.1177/0091270004274433. [DOI] [PubMed] [Google Scholar]
- 25.Blaschke TF, Osterberg L, Vrijens B, Urquhart J. Adherence to medications: insights arising from studies on the unreliable link between prescribed and actual drug dosing histories. Annu Rev Pharmacol Toxicol. 2012 Feb 10;52:275–301. doi: 10.1146/annurev-pharmtox-011711-113247. [DOI] [PubMed] [Google Scholar]
- 26.Vrijens B, Goetghebeur E. Comparing compliance patterns between randomized treatments. Control Clin Trials. 1997 Jun;18(3):187–203. doi: 10.1016/s0197-2456(96)00235-8. [DOI] [PubMed] [Google Scholar]
- 27.Vrijens B, Goetghebeur E, de Klerk E, Rode R, Mayer S, Urquhart J. Modelling the association between adherence and viral load in HIV-infected patients. Stat Med. 2005 Sep 15;24(17):2719–31. doi: 10.1002/sim.2130. [DOI] [PubMed] [Google Scholar]
- 28.Goujard C, Barrail-Tran A, Duval X, Nembot G, Panhard X, Savic RM, et al. Virological Response to Atazanavir, Ritonavir and Tenofovir/Emtricitabine: Relation to Individual Pharmacokinetic Parameters and Adherence measured by Medication Events Monitoring System (MEMS) in Naïve HIV-Infected Patients (ANRS134 trial) Internation AIDS Society; Vienna, Austria: 2010. [Google Scholar]
- 29.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34(5):711–26. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 30.Wilkins JJ, Savic RM, Karlsson MO, Langdon G, McIlleron H, Pillai G, et al. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob Agents Chemother. 2008 Jun;52(6):2138–48. doi: 10.1128/AAC.00461-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zvada SP, Van Der Walt JS, Smith PJ, Fourie PB, Roscigno G, Mitchison D, et al. Effects of four different meal types on the population pharmacokinetics of single-dose rifapentine in healthy male volunteers. Antimicrob Agents Chemother. 2010 Aug;54(8):3390–4. doi: 10.1128/AAC.00345-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill A, van der Lugt J, Sawyer W, Boffito M. How much ritonavir is needed to boost protease inhibitors? Systematic review of 17 dose-ranging pharmacokinetic trials. AIDS. 2009 Nov 13;23(17):2237–45. doi: 10.1097/QAD.0b013e328332c3a5. [DOI] [PubMed] [Google Scholar]
- 33.Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med. 2005 Nov;6(6):421–5. doi: 10.1111/j.1468-1293.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- 34.Collot-Teixeira S, De Lorenzo F, Waters L, Fletcher C, Back D, Mandalia S, et al. Impact of different low-dose ritonavir regimens on lipids, CD36, and adipophilin expression. Clin Pharmacol Ther. 2009 Apr;85(4):375–8. doi: 10.1038/clpt.2008.243. [DOI] [PubMed] [Google Scholar]
- 35.Hill A, Balkin A. Risk factors for gastrointestinal adverse events in HIV treated and untreated patients. AIDS Rev. 2009 Jan-Mar;11(1):30–8. [PubMed] [Google Scholar]
- 36.Harter JG, Peck CC. Chronobiology. Suggestions for integrating it into drug development. Ann N Y Acad Sci. 1991;618:563–71. doi: 10.1111/j.1749-6632.1991.tb27276.x. [DOI] [PubMed] [Google Scholar]
- 37.Lindbom L. Development, Application and Evaluation of Statistical Tools in Pharmacometric Data Analysis. Uppsala: Uppsala University; 2006. [Google Scholar]
- 38.Lindbom L, Pihlgren P, Jonsson N. PsN-Toolkit--a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005 Sep;79(3):241–57. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]