

Abstract
The human adult articular chondrocyte is a unique cell type that has reached a fully differentiated state as an end point of development. Within the cartilage matrix, chondrocytes are normally quiescent and maintain the matrix constituents in a low-turnover state of equilibrium. Isolated chondrocytes in culture have provided useful models to study cellular responses to alterations in the environment such as those occurring in different forms of arthritis. However, expansion of primary chondrocytes in monolayer culture results in the loss of phenotype, particularly if high cell density is not maintained. This chapter describes strategies for maintaining or restoring differentiated phenotype by culture in suspension, gels, or scaffolds. Techniques for assessing phenotype involving primarily the analysis of synthesis of cartilage-specific matrix proteins as well as the corresponding mRNAs are also described. Approaches for studying gene regulation, including transfection of promoter-driven reporter genes with expression vectors for transcriptional and signaling regulators, chromatin immunoprecipitation, and DNA methylation are also described.
Keywords: Chondrocyte, Type II collagen, Aggrecan, Monolayer culture, Suspension culture, Alginate, Agarose, PolyHEMA, Three-dimensional scaffolds, Collagen scaffolds, Transfections, Luciferase reporter plasmids, Adenovirus-mediated expression, Chromatin immunoprecipitation assay
1. Introduction
The mature articular chondrocyte embedded in the cartilage matrix is a resting cell with no detectable mitotic activity and a very low synthetic activity (1). The markers of mature articular chondrocytes are type II collagen (COL2A1), other cartilage-specific collagens IX (COL9) and XI (COL11), and the large aggregating proteoglycan aggrecan (ACAN) (Table 1). Chondrocytes also synthesize a number of small proteoglycans such as biglycan and decorin and other specific and nonspecific matrix proteins both in vivo and in vitro. As the single cellular constituent of adult articular cartilage, chondrocytes are responsible for maintaining the cartilage matrix in a low turnover state of equilibrium. In normal adult articular cartilage, the turnover of collagen occurs with a half-life of greater than 100 years (2, 3). The half-life of aggrecan subfractions is in the range of 3–24 years, whereas the glycosaminoglycan constituents on the aggrecan core protein are more readily replaced (4). Furthermore, normal chondrocyte metabolism in situ occurs in low oxygen tension and is remote from a vascular supply. Thus, it is not surprising that changes in expression of these cartilage matrix constituents occur when the chondrocytes are isolated and placed in monolayer culture, where they increase synthetic activity by several orders of magnitude.
Table 1.
Proteins synthesized by mature chondrocytes
| Collagens |
| Type II |
| Type IX |
| Type XI |
| Type VI |
| Type XII |
| Type XIV |
| Type XVI |
| Type XVII |
| Proteoglycans |
| Aggrecan (with link protein and hyaluronic acid) |
| Versican |
| Perlecan |
| Biglycan |
| Decorin |
| Asporin |
| Fibromodulin |
| Lumican |
| PRELP (proline/arginine-rich and leucine-rich repeat protein) |
| Chondroadherin |
| Lubricin, superficial zone protein (SZP), or proteoglycan (PRG)-4 |
| Other noneollagenous proteins (structural) |
| Cartilage oligomeric matrix protein (COMP; thrombospondin-5) |
| Thrombospondin-1 and -3 |
| Cartilage matrix protein (matrilin-1); matrilin-3 |
| Fibronectin |
| Tenascin-C |
| Cartilage intermediate layer protein (CILP) |
| Other noneollagenous proteins (regulatory) |
| S-100 |
| Chondromodulin-I (SCGP) and -II |
| Glycoprotein (gp)-39, YKL-40 |
| Matrix Gla protein (MGP) |
| CD-RAP (cartilage-derived retinoic acid-sensitive protein) |
| Bone morphogenetic proteins (BMP) 2, 7, 13 (GDF-6), 14 (GDF-5) |
| Transforming growth factor β |
| Membrane-associated proteins |
| Integrins (α1β1, α2β1, α3β1, α5β1, α6β1, α10β1, αvβ3, αvβ5) |
| Anchorin CII (annexin V) |
| CD44 |
| Syndecan-3 |
| Discoidin domain receptor-2 (DDR2) |
The collagens, proteoglycans, and other noncollagenous proteins in the cartilage matrix are synthesized by chondrocytes at different stages during development and growth of cartilage and the mature articular chondrocyte may have a limited capacity to maintain and repair some of the matrix components, particularly collagens. Regulatory proteins are secreted by the chondrocytes and may be stored in the matrix. Membrane-associated proteins permit specific interactions with extracellular matrix proteins
Primary cultures of articular chondrocytes isolated from various animal and human sources have served as useful models for studying the mechanisms controlling responses to growth factors and cytokines (see, for review, refs. 5–7). Early attempts to culture chondrocytes were frustrated by the tendency of these cells to “dedifferentiate” in monolayer culture and their inability to proliferate in suspension culture where cartilage-specific phenotype could be maintained (8–10). High-density monolayer cultures maintain the cartilage-specific phenotype until they are subcultured, although gene expression of type II collagen is generally more labile than that of aggrecan (11–13). Monolayer chondrocytes maintain a rounded, polygonal morphology (Fig. 1), but there may be a progressive change to a fibroblast-like morphology with passage of time, especially after subculture with acquisition of some but not all characteristics of the fibroblast phenotype, such as type I and type III collagen gene expression. This dedifferentiation can be accelerated by plating the cells at low densities or by treatment with cytokines such as interleukin-1 (IL-1) (14, 15) or retinoic acid and appears to be associated with the increased expression of genes involved in cell proliferation, such as cyclin D1 (16). Furthermore, the substrate on which the chondrocytes are plated can influence the differentiation capacity of articular chondrocytes (17).
Fig. 1.
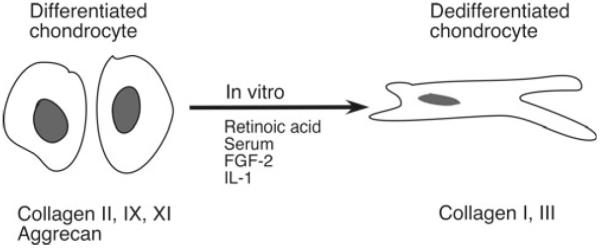
Schematic representation of the “switch” from the differentiated to the dedifferentiated chondrocyte phenotype that occurs during culture and in response to certain cytokines and growth factors in vitro. The extracellular matrix genes that are differentially expressed are indicated.
Since the stability of the phenotype of isolated chondrocytes is critically dependent on cell shape and cell density (18, 19), high-density micromass cultures are useful if sufficient numbers of chondrocytes can be isolated (20, 21), particularly for studying proteoglycan biosynthesis (22). It is also possible to expand the cultures through a limited number of subcultures and “redifferentiate” the cells in fluid or gel suspension culture systems, where the chondrocytes regain morphology and the cessation of proliferation is associated with increased expression of cartilage-specific matrix protein. Culture systems that support chondrocyte phenotype include suspension culture in spinner flasks (23), in dishes coated with a nonadherent substrates (24–26), in pellets or micromasses (27, 28), and in three-dimensional matrices such as collagen gels (29), agarose (12, 30, 31), alginate (32, 33), or collagen sponges (34, 35). Serum-free defined media of varying compositions, but usually including insulin, have also been used, frequently in combination with the other culture systems mentioned above (36).
The use of chondrocytes of human origin has been problematical, since the source of the cartilage cannot be controlled, sufficient numbers of cells are not readily obtained from random operative procedures, and the phenotypic stability and proliferative capacity in adult human chondrocytes are lost more quickly upon expansion in serial monolayer cultures than in cells of juvenile human (11) or embryonic or postnatal animal origin (37, 38). Alternatively, explant cultures of human, but usually bovine, articular cartilage where the chondrocytes remain encased within their own extracellular matrix have been used as in vitro models to study cartilage biochemistry and metabolism (39). However, many experimental manipulations are done more easily using isolated chondrocytes. Later studies focused on adult human articular chondrocytes as target cells for immortalization, using immortalizing antigens such as SV40-TAg (40–42), human papilloma virus type 16 (HPV-16) early function genes E6 and E7 (43), and telomerase (44). Strategies that maintain high cell density and decrease cell proliferation must also be applied to immortalized chondrocyte cell lines, since stable integration of immortalizing genes disrupts normal cell cycle control but does not stabilize expression of the type II collagen gene (45, 46).
This chapter focuses on strategies for isolation, culture, and characterization of isolated human articular chondrocytes and also describes approaches for using different chondrocyte culture systems for evaluating chondrocyte phenotype and studying the regulation of chondrocyte functions.
2. Materials
2.1. Isolation and Culture of Human Chondrocytes
Growth medium for chondrocytes: Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12 1:1 mixture, with l-glutamine (Mediatech, Inc. Manassas, VA). Add 10% fetal calf serum (FCS) immediately before use. Selection of lots of FCS that maintain chondrocyte phenotype (see Note 1) is recommended.
Phosphate-buffered saline (PBS) Ca2+- and Mg2+-free (Mediatech, Inc. Manassas, VA).
Trypsin–EDTA solution: 0.05% trypsin and 0.02% EDTA in Hank's balanced salt solution without Ca2+and Mg2+ (Gibco Invitrogen, Carlsbad, CA). FCS and trypsin–EDTA are stored at −20°C, but should not be refrozen after thawing for use.
Serum substitutes for experimental incubations: Nutridoma-SP (Roche Applied Science, Indianapolis, IN) is provided as sterile concentrate (100×, pH 7.4; storage at 15–25°C protected from light). Dilute 1:100 (v/v) with sterile DMEM/Ham's F-12, without FCS, and use immediately. ITS + Universal Culture Supplement Premix (BD Biosciences, San Jose, CA); provided as a concentrated stock sterile aqueous solution (storage at 2–8°C). Dilute the stock solution 1:100 (v/v) in serum-free DMEM/Ham's F-12 to give final concentrations of 12.5 μg/mL human insulin, 12.5 μg/mL human transferrin, and 12.5 μg/mL selenious acid for experimental purposes.
- Enzymes for cartilage digestion:
-
(a)Pronase (Roche Applied Science, Indianapolis, IN): Prepare freshly in DMEM/Ham's F12 with 10% FCS at a final concentration of 1 mg/mL and sterilize by filtration using a 0.22-μm filter.
-
(b)Collagenase P (Roche Applied Science, Indianapolis, IN): Prepare freshly in DMEM/Ham's F12 with 10% FCS at a final concentration of 1 mg/mL and sterilize by filtration through a sterile 0.22-μm filter.
-
(a)
2.2. Suspension and Three-Dimensional Culture Systems for Chondrocytes
Agarose-coated dishes (25): Weigh out 1 g of high melting point agarose in an autoclavable bottle and add 100 mL of dH2O to make a 1% solution. Autoclave with cap tightened loosely, allow to cool to approximately 55°C, and pipette quickly into culture dishes (1 mL/3.5-cm well of 6-well plate, 3 mL/6-cm dish, or 9 mL/10-cm dish). Allow the gel to set at 4°C for 30 min and wash the surface 2 or 3 times with PBS. Plates may be used immediately or wrapped tightly with plastic or foil to prevent evaporation and stored at 4°C.
Poly-HEMA (poly-2-hydroxyethyl-methacrylate)-coated dishes (26): Prepare a 10% (w/v) solution by dissolving 5 g of poly-HEMA (Sigma-Aldrich, St. Louis, MO) in 50 mL of ethanol in a sterile capped bottle or centrifuge tube. Leave overnight at 37°C with gentle shaking to dissolve polymer completely. Centrifuge the viscous solution for 30 min at 2,000 × g to remove undissolved particles. Dilute the stock solution to 1% (1 mL of 10% polyHEMA in 9 mL ethanol). Layer the 1% poly-HEMA solution on dishes at 0.3 mL/well of 6-well plate or 0.9 mL/6-cm dish and leave with lids in place to dry overnight in a tissue culture hood. Expose open dishes to bactericidal ultraviolet light for 30 min to sterilize.
Agarose: Autoclave 2% (w/v) low gelling temperature agarose in dH2O, cool to 37°C, and dilute with an equal volume of 2× DMEM containing 20% FCS either without cells or with a chondrocyte suspension.
- Alginate (Keltone LVCR, NF; ISP Alginates Inc., San Diego, CA, USA). Low-viscosity (LV) alginate is used generally. Request LVCR for more highly purified preparation:
-
(a)Prepare 1.2% (w/v) solution of alginate in 0.15 M NaCl.
-
(b)Dissolve alginate in a 0.15 M NaCl solution, heating the solution in a microwave until it just begins to boil. Swirl and heat again two or three times until the alginate is dissolved completely. (Caution: Do not autoclave.) Allow the solution to cool to about 37°C and filter-sterilize. Filtering when warm permits the viscous solution to pass through the filter.
-
(c)Prepare 102 mM CaCl2 and 0.15 M NaCl solutions in tissue culture bottles and autoclave.
-
(d)Prepare 55 mM Na citrate, 0.15 M NaCl, pH 6.0, filter-sterilize, and store at 4°C. Make fresh weekly.
-
(a)
- Three-dimensional (3D) scaffolds: Several types of scaffolds are available commercially, including the following:
-
(a)Gelfoam® (Pharmacia & Upjohn, Kalamazoo, MI), sterile absorbable collagen sponge, purchased as sponge-size 12–7 mm (box of 12).
-
(b)BD™ Three-Dimensional Collagen Composite Scaffold (BD Biosciences): Contains a mixture of bovine type I and type III collagens and is provided as 3D scaffolds with 48-well plates.
-
(c)BD™ Three-Dimensional OPLA® Scaffold (BD Biosciences): Contains a synthetic polymer synthesized from d,d,-l,l-polylactic acid and is provided as 3D scaffolds with 48-well plates.
-
(a)
Recovery of cells from scaffolds: Cell lysis solution: 0.2% v/v Triton X-100, 10 mM Tris-HCl, pH 7.0, 1 mM EDTA.
Collagenase solution: 0.03% (w/v) Collagenase (bacterial, clostridiopeptidase A; Worthington Biochemical Corp., Freehold, NJ) in Hank's balanced salt solution (HBSS).
2.3. Analysis of Matrix Protein Deposition and Synthesis
2.3.1. Alcian Blue Staining
2.5% Glutaraldehyde (diluted from 50% solution; Sigma-Aldrich, St. Louis, MO) in 0.4 M MgCl2 and 25 mM sodium acetate, pH 5.6.
Alcian blue 8GX (Sigma-Aldrich, St. Louis). Dissolve in the 2.5% glutaraldehyde solution to give final concentration of 0.05%. Filter the solution through Whatman paper (or coffee filter).
Washes: 3% acetic acid solutions without and with 25 and 50% ethanol.
2.3.2. Collagen Typing
l-(5-3H)proline (1 mCi/mL; specific activity >20 Ci/mmol) at 25 μ Ci/mL in serum-free culture medium supplemented with 50 μ g/mL ascorbate and 50 μ g/mL β-aminopropionitrile fumarate (β-APN). Filter-sterilize 10× solution of ascorbic acid and β-APN (5 mg of each dissolved in 10 mL of serum-free culture medium), dilute in medium at 1/10 (v/v) to give the volume required for the incubation, and add 25 μL of (3H) proline per mL using a sterile pipette tip.
Pepsin–acetic acid solution: Dissolve 2 mg of pepsin in 1 mL dH2O, then add 58 μL of glacial acetic acid per each mL of solution and cool on ice.
- Reagents for SDS-PAGE (47) and autoradiography:
-
(a)Gel sample buffer (GSB): 0.1 M Tris–HCl, pH 7.6, 3% (w/v) SDS, and 16% (v/v) glycerol.
-
(b)Loading dye: 1% (w/v) Bromophenol blue (BPB; sodium salt; Sigma).
-
(c)Tris–glycine/SDS–5% gradient polyacrylamide or 7–15% gradient gels and Laemmli buffer system (47).
-
(a)
2.3.3. Proteoglycan Synthesis
Biosynthetic labeling of proteoglycans: (35S)sodium sulfate (2 mCi/mL; specific activity >1,000 Ci/mmol). Add to culture medium at 50 μCi/mL.
7 M urea.
DE52 columns, 3.5 cm × 12 cm containing 4 mL of DEAE-cellulose (Whatman, Hillsboro, OR).
Guanidine extraction buffer: 4.0 M guanidine HCl, buffered with 50 mM sodium acetate, and containing 10 mM disodium EDTA. Add immediately before use 100 mM 6-aminocaproic acid, 2.5 mM benzamidine HCl, 5 mM N-ethylmaleimide, and 0.25 mM phenylsulfonyl fluoride (PMSF) from 100× stock solutions in absolute ethanol. The 2× guanidine extraction buffer is prepared at twice the concentrations above.
SDS-PAGE: Precast Tris–glycine SDS–polyacrylamide 4–20% gradient gels (Bio-Rad). Use the GSB and BPB solutions for loading the samples and the Laemmli buffer system (47) (see Subheading 2.3.2, items 3–5).
2.3.4. Immuno cytochemistry
Fixative: 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4. Dissolve 8 g of paraformaldehyde in 150 mL dH2O in Ehrlenmeyer flask on hot plate in fume hood (do not exceed 65°C). Add ~2 mL of 1 N NaOH while stirring, and stir until solution is clear. Let solution cool for 15 min. Add 250 mL of 0.2 M cacodylate buffer, pH 7.4, and adjust pH if necessary.
2.3.5. Western Blotting
Reagents for SDS-PAGE (47) and autoradiography. Tris–glycine/SDS–polyacrylamide gels at polyacrylamide concentration appropriate for the size of matrix protein to be analyzed (see Subheadings 2.3.2 and 2.3.3).
Nylon-supported nitrocellulose membranes, Immobilon-P, 0.45-μm (Millipore, Billerica, MA).
Transfer buffer: 25 mM Tris–HCl, pH 7.6, 192 mM glycine, 10–20% (v/v) methanol.
Tris-buffered saline/Tween (TBST): 20 mM Tris–HCl, 137 mM NaCl, 0.1% (v/v) Tween-20, pH 7.6. Add 5% (w/v) nonfat dry milk (Carnation) as required.
Primary antibody: Dilute in TBST containing nonfat dry milk (BD Biosciences, San Jose, CA) or bovine serum albumin (Santa Cruz Biotechnology, Santa Cruz, CA) according to the supplier's instructions.
Horseradish peroxidase (HRP)-conjugated secondary antibody.
Enhanced chemiluminescence (ECL) substrate for HRP enzyme (GE Healthcare Biosciences, Pittsburgh, PA or Thermo Fisher Scientific, Rockford, IL among others).
2.3.6. Antibodies
Antibodies that detect human collagens and proteoglycans that are specific to cartilage are available commercially from Southern Biotechnology Associates, Inc. (Birmingham, AL; http://south-ernbiotech.com), IBEX Technologies, Inc. (Montreal, QC, Canada; http://www.ibex.ca/), and Chemicon International (Temecula, CA; http://www.chemicon.com). Some antibody preparations are useful for developing quantitative ELISA assays.
2.4. Analysis of mRNA
RNA extraction kit: The TRIzol® reagent (Invitrogen, Carlsbad, CA) or RNeasy® Plus Mini Kit (QIAGEN Inc., Valencia, CA) are suitable for extraction of total RNA from chondrocytes.
Sterile RNase-free solutions, polypropylene tubes, and other materials.
2.5. Analysis of Transcriptional Modulation
2.5.1. Luciferase Reporter Assays
EndoFree Plasmid Maxi Kit (QIAGEN Inc., Valencia, CA).
Lipid-based transfection reagent such as Lipofectamine™ PLUS™ Reagent (Invitrogen, Carlsbad, CA) or FuGENE 6 (Roche Applied Science, Indianapolis, IN).
Serum-free culture medium for transfections: Opti-MEM (Invitrogen, Carlsbad, CA) or DMEM/F-12 (test for optimal transfection efficiency).
Coomassie (Bradford) Protein Assay Reagent (Thermo Fisher Scientific, Rockford, IL).
Firefly and Renilla Luciferase Assay Systems (Promega, Madison, WI) or Dual-Luciferase Reporter Assay (Promega). When overexpressing Firefly luciferase with a second reporter gene, Passive (Promega) or Reporter (Promega) Lysis buffers are recommended.
Adenovirus producer cell line: 293 (ATTC CRL 1573; transformed primary human embryonic kidney).
2.5.2. siRNA-Mediated Knockdown
Lipid-based transfection reagent such as Lipofectamine™ PLUS™ Reagent (Invitrogen, Carlsbad, CA), Lipofectamine™ 2000 Reagent (Invitrogen, Carlsbad, CA) or DharmaFECT® transfection reagents (Thermo Fisher Scientific, Rockford, IL).
siRNA of interest and nontargeting siRNA (Thermo Fisher Scientific, Applied Biosystems/Ambion).
Antibiotic- and Serum-Free DMEM/F12.
2.5.3. Chromatin Immunoprecipitation (ChIP) Assays
ChIP-IT™ Express Chromatin Immunoprecipitation Kit (Active Motif, Carlsbad, CA #53008). The following reagents/materials are not provided in the Kit.
Formaldehyde cross-linking reagent: for preparing the formaldehyde Fixation Solution, add 0.54 mL of 37% formaldehyde (Sigma-Aldrich, St. Louis, MO) to 20 mL of cell culture medium per 150-mm × 25-mm plate (BD Biosciences, San Jose, CA).
Dounce Homogenizer (Kimble Chase Life Science, Vineland, NJ).
Enzymatic shearing: for preparing a working stock of the Enzymatic Shearing Cocktail, dilute the provided Enzymatic Shearing Cocktail 1:100 in a 50% solution of glycerol (Thermo Fisher Scientific, Rockford, IL) in nuclease-free water (QIAGEN Inc., Valencia, CA).
ChIP-grade antibodies.
QIAquick PCR Purification Kit (QIAGEN Inc., Valencia, CA) for DNA purification.
2.5.4. DNA Methylation Analysis
Methyl Detector™ Bisulfite Modification Kit (Active Motif, Carlsbad, CA) for bisulfite conversion of unmethylated cytosines to uracils.
Methylation-sensitive restriction enzyme/s (New England Biolabs, Ipswich, MA).
AllPrep DNA/RNA/Protein Mini Kit (QIAGEN Inc., Valencia, CA) for simultaneous purification of DNA and RNA.
CpGenome Universal Methylated DNA (Millipore, Billerica, MA).
Illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare Biosciences, Pittsburgh, PA).
5-(Aza-deoxycytidine)aza-dC (Sigma-Aldrich, St. Louis, MO) and trichostatin A (Sigma-Aldrich, St. Louis, MO) for inhibition of DNA methyltransferase and histone deacetylase activities, respectively.
3. Methods
The methods described below outline (1) the isolation of human chondrocytes from cartilage and their primary culture in monolayer and after passage, (2) suspension culture systems for maintaining chondrocyte phenotype, analysis of the (3) synthesis and (4) mRNA expression of cartilage-specific matrix proteins, and (5) analysis of transcriptional modulation by transfection of regulatory DNA sequences, knockdown by siRNA transfection of trans-acting factors, chromatin immunoprecipitation assays, and DNA methylation analysis.
3.1. Isolation and Culture of Human Chondrocytes in Monolayer
Human adult articular cartilage is obtained, after Institutional Review Board approval, from the knee joints or hips after surgery for joint replacement or reconstruction or due to trauma, or at autopsy, and dissected free from underlying bone and any adherent connective tissue.
Place slices of cartilage in a 10-cm dish containing serum-free DMEM/F12. Cut cartilage into small pieces using a scalpel blade.
Add pronase solution (~25 mL per 10 g of tissue) and incubate at 37°C for 30 min with slow agitation.
Remove pronase solution and wash twice with PBS.
Add collagenase P solution (~25 mL per 10 g of tissue) and incubate at 37°C overnight (16–18 h) for articular cartilage and up to 48 h for costal cartilage, until the cartilage matrix is completely digested and the cells are free in suspension (see Note 2). Break up any clumps of cells by repeated aspiration of the suspension through a 10-mL pipette or a 12-cc syringe without a needle.
Transfer cell suspension to a sterile 50-mL conical polypropylene tube passing the suspension through a 40–70 μm filter (see Note 2). Centrifuge (5 min, 400 × g) to pellet cells and discard supernatant by aspiration.
Wash the cell pellet twice with PBS and once with DMEM/F12 containing 10% FCS, resuspending cells each time and centrifuging.
Resuspend the final pellet in DMEM/Ham's F-12 containing 10% FCS, perform cell count with a Coulter counter or hemocytometer (dead cells can be recognized by Trypan blue exclusion), and bring up to volume with culture medium to give 1 × 106 cells/mL. For monolayer culture, plate cells at 5 × 104/cm2 (see Table 2), in dishes or wells containing culture medium, and agitate without swirling to distribute the cells evenly. Incubate at 37°C in an atmosphere of 5% CO2 in air with medium changes after 2 days and every 3 or 4 days thereafter (see Note 3). Primary cultures of adult articular chondrocytes before and after passaging are shown in Fig. 2a, b, respectively.
Preparation of subcultured cells (see Note 4): Remove culture medium by aspiration with a sterile Pasteur pipette attached to a vacuum flask and wash with PBS. Add trypsin–EDTA (1 mL/10-cm dish) and incubate at room temperature for 10 min with periodic gentle shaking of dish and observation through microscope to assure that cells have come off the plate. If significant numbers of cells remain attached, continue the incubation for a longer time (≥20 min) or at a higher temperature (37°C) and/or scrape the cell layer with a sterile plastic scraper or syringe plunger. Repeatedly aspirate and expel the cell suspension into the plate using a 5- or 10-mL pipette containing culture medium, and then transfer to a sterile conical 15- or 50-mL polypropylene tube. Perform cell counts or determine the split ratio required (usually 1 dish into 2, or 1:2, for adult articular chondrocytes, or 1:5 for more rapidly growing, denser juvenile chondrocytes). Distribute equal volumes of the cell suspension in dishes or wells that already contain culture medium, rocking plates back-and-forth (not swirling) immediately after each addition to assure uniform plating density (see Note 5).
For experiments, plate cells in dishes or wells at a concentration of 1–2.5 × 104 cells/cm2, depending upon the experimental approach, in DMEM/Ham's F-12 containing 10% FCS. When the cultures are confluent, remove the growth medium and add fresh medium with additives appropriate for test conditions. If it is important that the cells be quiescent, as for analyzing serum-responsive genes, add test agent without changing medium or change to serum-free medium containing a serum substitute (see Note 6), such as Nutridoma-SP or ITS+, followed 1–24 h later by the test agent of interest. Continue incubation at 37°C for short-term time courses of 0.25–24 h or longer time courses of several days.
Table 2.
Culture vessel area vs. chondrocyte number required for plating density of ~2.5 × 104 cells/cm2
| Diameter | Area (cm2) | No. of cells plated |
|---|---|---|
| 16-mm well (24-well) | 2 | 50,000 |
| 2.2-cm well (12-well) | 3.8 | 100,000 |
| 3.5-cm well (6-well) | 10 | 250,000 |
| 6-cm plate | 28 | 750,000 |
| 10-cm plate | 79 | 2 × 106 |
Fig. 2.

Morphology of human articular chondrocytes grown in monolayer culture on plastic. Chondrocytes were isolated from articular cartilage and cultured in DMEM/F12 containing 10% FCS until confluent and then subcultured and grown again to confluence. (a) Primary chondrocytes display the characteristic cobblestone morphology. (b) Passaged chondrocytes display features of a dedifferentiated phenotype, including a portion of cells with fibroblast-like morphology.
3.2. Three-Dimensional Culture Systems for Chondrocytes
Chondrocytes in monolayer culture are susceptible to loss of phenotype during prolonged culture and particularly after subculture. Thus, it is necessary to use culture conditions that maintain differentiated chondrocyte features or that permit redifferentiation (see Note 7). Freshly isolated chondrocytes may be cultured immediately in suspension, where they do not proliferate, or they may be expanded in monolayer culture and placed in suspension culture after several passages.
3.2.1. Suspension Cultures on Agarose- or PolyHEMA-Coated Dishes (See Note 8)
Trypsinize monolayer cultures, spin down cells, wash with PBS, centrifuge, and resuspend in culture medium containing 10% FCS at 1 × 106 cells/mL.
Transfer chondrocyte suspension to dishes that have been coated with 1% agarose or with 1% polyHEMA and culture for 2–4 weeks. The cells first form large clumps that begin to break up after 7–10 days and eventually form single-cell suspensions.
Change the medium weekly by carefully removing the medium above settled cells while tilting the dish, centrifuging the remaining suspended cells, and replacing them in the dish after resuspension in fresh culture medium.
To recover cells for direct experimental analysis, for redistribution in agarose- or polyHEMA-coated wells, or for culture in monolayer, transfer the cell suspension to 15- or 50-mL conical tubes, gently washing the agarose surface at least twice with culture medium to recover remaining cells, and spin down and resuspend cells in an appropriate volume of culture medium for plating or in extraction buffer for subsequent experimental analysis.
3.2.2. Suspension Culture Within Agarose
Precoat plastic tissue culture dishes with cell-free 1% agarose in culture medium (0.5 mL/3.5-cm, 1.5 mL/6-cm, or 4.5 mL/10-cm dish) and allow to gel at room temp. Add the same volume of 1% agarose in medium containing chondrocytes at a density of 1–4 × 106 cells/mL of gel, incubate at 37°C for 20–30 min to allow the cells to settle, and leave at room temp until the agarose forms a gel. Add culture medium containing 10% FCS and incubate at 37°C with medium changes every 3–4 days.
After incubations with test reagents and/or radioisotopes in minimal volumes of appropriate culture medium, the whole cultures may be stored frozen or medium and gel treated separately. For subsequent analysis, add appropriate guanidine extraction buffer directly to the gel (for proteoglycan or RNA extraction) or whole cultures may be adjusted to 0.5 M acetic acid, treated with pepsin, and neutralized, as described below for analysis of collagens. To remove agarose and debris, the samples are centrifuged in a high-speed centrifuge at >10,000 × g at 4°C.
3.2.3. Alginate Bead Cultures (See Note 9)
Trypsinize several 10-cm plates and wash the cells with PBS. Determine the cell count with a hemocytometer and pellet the cells.
Resuspend the pellet in a 1.2% solution of alginate in 0.15 M NaCl at a concentration of 1–4 × 106 cells/mL. Slowly express the alginate suspension in a dropwise manner through a 10-cc syringe equipped with a 22-gauge needle into a 50-mL polypropylene centrifuge tube containing 40 mL of 102 mM CaCl2. Allow the beads to polymerize in the CaCl2 solution for 10 min and wash twice with 25 mL of 0.15 M NaCl. The alginate beads should not be washed in PBS, as they will become cloudy.
Resuspend the beads at 7–15 beads per mL in growth medium supplemented with 25 μg/mL Na ascorbate (see Note 10) and decant to a culture dish or flask. Culture in DMEM/F12 with medium changes every 3 days, carefully pipetting the spent culture medium from the top of the settled beads.
At the end of the culture period (one to several weeks), add the appropriate guanidine extraction buffer or centrifuge at 500 × g for 10 min to recover the chondrocytes with pericellular matrix.
Alternatively, to recover cells from alginate, carefully aspirate the medium from the cultures and wash twice with PBS. Depolymerize the alginate by adding three volumes of a solution of 55 mM Na citrate/0.15 M NaCl and incubate at 37°C for 10 min. Aspirate the solution over the surface of the dish several times to dislodge adherent cells (the cells are sticky) and transfer the suspension to a 50-mL centrifuge tube. Because the solution is quite viscous, centrifuge the cells at 2,000 × g for a minimum of 10 min to completely pellet the cells. Wash the cells twice with PBS before using them for further analysis.
3.2.4. Culture on 3D Scaffolds (See Note 11)
Gelfoam®: Use sterile scalpel blade to cut into pieces of 1 × 1 × 0.5 cm3 and place in wells of sterile 6-well plates. Inoculate by dropping 50 μL of growth medium containing 106 cells on each sponge. Place in incubator for 1.5–2 h, then add 100 μL medium and culture for an additional 1–3 h. Add medium to cover and continue incubation overnight or longer.
BD™ 3D Collagen Composite or OPLA® Scaffolds: Place scaffolds (0.5 cm3) in the 48-well plates provided, in 96-well plates, or other plate as required. Seed scaffolds by dropping 100 μL of growth medium containing 1–5 × 104 cells. Incubate for 1 h, add 150 μL of medium to each scaffold, and incubate for 1.5–3 h. Add medium as required for further culture and experimental conditions.
- Recovery of cells from scaffolds for analysis:
-
(a)Prepare cell lysates for DNA analysis using 250–500 μL of cell lysis solution (0.2% v/v Triton X-100, 10 mM Tris–HCl, pH 7.0, 1 mM EDTA) per scaffold in 1.5-mL tube. Freeze samples at −70°C and subject to two freeze–thaw cycles, thawing at room temperature for 45–60 min. Break up scaffolds with pipette tip, centrifuge, and transfer lysates to fresh tubes. The cell lysates may be analyzed using the Picogreen Assay Kit according to the manufacture's protocol (Molecular Probes).
-
(b)Recover cells for RNA extraction and other analyses by treatment of minced scaffolds with 0.03% (w/v) collagenase in HBSS for 10–15 min at 37°C. Collect cells by centrifugation, wash with PBS, and add appropriate extraction buffer to the final pellet.
-
(a)
3.2.5. Pellet Cultures
Recover the cells using trypsin/EDTA; determine the cell count and spin down to pellet the cells.
Resuspend the pellet to reach a density of 5 × 105 cells per 500 μL of DMEM/Ham's F-12 with10% FCS supplemented with 50 μg/mL ascorbic acid to distribute in centrifuge tubes.
Pellet by centrifugation at 1,000 × g for 5 min at 4°C. Remove the medium, add fresh complete medium, and repeat the centrifugation step.
Remove the medium, add 1 mL of fresh complete medium and incubate at 37°C for 1–4 weeks, depending on the experimental purposes.
Change the medium every second day adding 500 μL of fresh complete medium.
If stimulation with test agents is required, the medium volume can be reduced to 100 μL and pellets can be incubated at various time points in the presence of the test agent or vehicle in a dose- and time-dependent manner.
Harvest the pellets for RNA and protein extraction (28, 48). For immunohistochemical analysis, the pellets are embedded in OCT, snap-frozen, and stored at −80°C till use (Fig. 3). Supernatants can be stored for analyses of secreted products.
Fig. 3.

Three-dimensional culture of articular chondrocytes. After 3 weeks in culture, pellets were embedded in OCT, snap frozen and stored at −80°C. Frozen sections were fixed in PBS/PFA for 5 min and stained with Toluidine blue to detect proteoglycan deposition.
3.3. Analysis of Matrix Protein Synthesis
Chondrocyte culture models are used to examine the effects of cytokines and growth/differentiation factors on the synthesis of chondrocyte phenotypic markers by staining the glycosaminoglycans with Alcian blue, characterizing the collagens and proteoglycans synthesized, and perform immunohistochemistry using specific antibodies against these proteins.
3.3.1. Alcian Blue Staining
Monolayer cultures: Remove culture medium from confluent cultures that have been incubated in the presence of 50 μg/mL ascorbic acid for at least 4 h and wash with PBS. Add Alcian blue/glutaraldehyde solution at room temperature for several hours, remove excess stain by washing with 3% acetic acid, and store cultures in 70% ethanol for subsequent examination by light photomicrography.
- Alginate bead cultures:
-
(a)Using a 25-mL pipette, transfer five beads to a 12 cm × 15 cm tube, and wash twice with 2 mL PBS. Add 1 mL Alcian blue stain and 50 μL of a 50% glutaraldehyde solution. Store for 24 h at 4°C.
-
(b)Aspirate the stain from the beads, and wash twice with 2 mL of 3% (v/v) acetic acid. Destain the beads with rocking at room temp for 5 min sequentially with 2 mL of each of the following solutions: (1) 3% acetic acid, (2) 3% acetic acid–25% ethanol, and (3) 3% acetic acid–50% ethanol. Store the beads in 70% ethanol.
-
(c)Before photographing the stained beads, gently flatten beneath a glass coverslip, taking care not to disrupt the alginate matrix. Alternatively, the beads may be embedded and sectioned prior to photography.
-
(a)
3.3.2. Collagen Typing
Biosynthetic labeling of collagens (see Note 12): Remove serum-containing culture medium, wash with serum-free medium, and add (3H)proline at 25 μCi/mL for a further 24 h in serum-free culture medium supplemented with 50 μg/mL ascorbate and 50 μg/mL β-APN (or without β-APN to retain collagen in the pericellular matrix). Remove culture medium and store at −20°C. Wash cell layer with PBS and solubilize by adding equal volumes of serum-free culture medium and 1 M ammonium hydroxide (an aliquot may be analyzed for DNA).
Collagen typing: To analyze pepsin-resistant collagens, add pepsin–acetic acid solution to equal volume of either labeled culture medium or solubilized cell solution for 16 h at 4°C, lyophilize, redissolve in 2× SDS sample buffer, and neutralize with 1 μL additions of 2 M NaOH to titrate the color of the from yellow–green to blue (but not to violet). To analyze procollagens and fibronectin, add 2× SDS sample buffer containing 0.2% β-ME to an equal volume of the culture medium. Heat samples to boiling for 10 min and load on SDS gels (5% acrylamide running gels or 7–15% gradient gels) that include a radiolabeled rat tail tendon collagen standard in one lane. Perform delayed reduction with 0.1% β-ME on pepsinized samples to distinguish α1(III) from α1 (I or II) collagens. Absence of the α 2(I) collagen band generally indicate the absence of type I collagen synthesis. In cultures containing a mixture of type I and type II collagen, definitive identification of these collagens requires Western blotting using specific antibodies (see Subheading 3.3.5).
3.3.3. Proteoglycan Synthesis
Monolayer cultures: Aspirate the culture medium and wash the cell layer with PBS. Add DMEM/F12 containing 10% FCS supplemented with (35S)sulfate at 50 μCi/mL and 25 μg/mL ascorbate. Incubate at 37°C for 18 h. Remove the conditioned medium to a 15-mL polypropylene tube and wash the cell layer three times with PBS.
Add an equal volume of 7 M urea to the medium. Mix and count 10 μL in liquid scintillation counter. Pass up to 4 mL through DEAE-Cellulose (DE52, Whatman) column, which has been preequilibrated with 7 M urea, to remove unincorporated cpm. Elute proteoglycans with 2 mL of 4 M guanidine extraction buffer. To 500 μL of column eluate, add two volumes of 100% ethanol and precipitate PGs for 2 h at −20°C. Spin at 10,000 × g for 20 min at 4°C. Wash final pellet with 70% ethanol.
Add 4 M guanidine extraction buffer to cell layer (e.g., 2 mL/25-cm2 flask) and extract at 4°C for 24 h with rocking. Transfer extract to a 2-mL screw-cap microcentrifuge tube and spin at 10,000 × g for 20 min at 4°C to pellet particulate material from the sample. Remove supernatant to a fresh 2-mL tube and count 10 μL in a liquid scintillation counter. (Also, 10–50 μl aliquots may be taken at this point for DNA analysis.) Take 250 μL for precipitation of PGs by addition of three volumes of 100% ethanol at −20°C for 2 h (or overnight for alginate extracts), spin at 10,000 × g for 20 min at 4°C, and wash final pellet with 70% ethanol. Dry pellet at room temp for 30 min.
Alginate cultures: Aspirate the medium from a culture containing 50 beads in a 25-cm2 flask cultured on end. Add 4 mL of growth medium supplemented with (35S)sulfate at 50 μCi/mL and 25 μg/mL ascorbate. Incubate at 37°C for 18 h. Remove the conditioned medium to a 12-mL polypropylene tube and wash the beads three times with 5 mL of PBS (5 min/wash).
Medium extraction and purification: Perform as described for monolayer cultures in Subheading 3.3.3, step 2.
Extraction from alginate beads and purification: Transfer the beads (to a 15-mL polypropylene tube), extract radiolabeled PGs with 2 mL of 4 M guanidine extraction buffer at 4°C for 24 h with rocking. Transfer extract to a 2-mL screw-cap microcentrifuge tube and proceed as described for monolayers in Subheading 3.3.3, step 3.
Sulfate incorporation into proteoglycans: Dissolve ethanol-precipitated pellet in 100 μL of 1× GSB. Count 3 μL in a liquid scintillation counter and calculate the cpm incorporated, after accounting for dilutions, against the concentration of DNA in the cell extract. The (35S)sulfate incorporation may also be determined after passing the guanidine extracts over Sephadex G-25M in PD 10 columns and eluting under dissociative conditions by scintillation counting.
SDS-PAGE analysis: Take a volume of cell or medium extract in GSB that corresponds to 0.25 μg of DNA in the cell extract and add DTT to a final concentration of 0.5 mM and 1% Bromophenol blue to a final concentration of 0.1%. Heat 5 min at 100°C and store remaining sample at −20°C. Electrophorese ~20,000 cpm on a 4–20% polyacrylamide gradient gel. Fix the gel in acetic acid/methanol for 1 h, dry, and expose to film at −80°C. Visualize radiolabeled proteoglycans by autoradiography, as shown in Fig. 4 (49–51) (see Note 13).
Fig. 4.
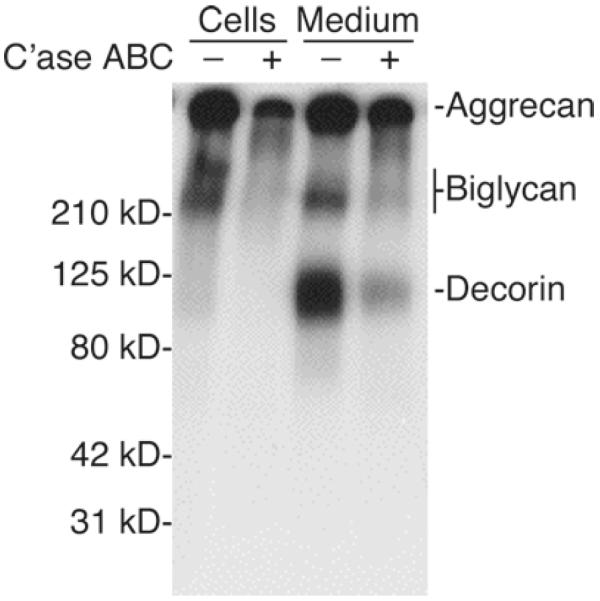
SDS-PAGE analysis of proteoglycans synthesized by human articular chondrocytes. Primary cultures were grown to confluence in DMEM/F12 containing 10% FCS. The cultures were incubated with (35S)sulfate in the presence of 25 μg/mL ascorbate during the final 18 h. Radiolabeled proteoglycans were purified by ethanol precipitation of 4 M guanidine extracts of cell monolayers or by DEAE ion-exchange chromatography and ethanol precipitation of 7 M urea extracts of the medium prior to SDS-PAGE on a 4–20% gradient gel. Equal volumes of cell and medium extracts representing equivalent proportions of the total culture were loaded on the gel. Molecular weight standards are indicated to the left of the panel. The migration of aggrecan, biglycan, and decorin is indicated to the right. Treatment with chondroitinase (C'ase) ABC resulted in the expected loss of sulfation.
3.3.4. Immuno cytochemistry
Plate cells in plastic Lab-Tech 4-chamber slides (Nunc, Inc. Naperville, IL) at 6 × 104 cells/chamber in culture medium containing 10% FCS. Add 25 μg/mL ascorbic acid with the first medium change and daily thereafter (see Note 10).
When the cultures have reached confluence, add the desired test reagents. At the end of the incubation period, carefully wash the chambers three times with PBS and fix the cells with 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4, for 2 h at 4°C.
Rinse twice with 0.1 M cacodylate buffer. Add antibodies that recognize human type II collagen, aggrecan, etc. (see Subheading 2.3.6) to different chambers at concentrations recommended by the supplier. Incubate separate chamber slides with chondroitinase ABC for 30 min at 37°C prior to addition of monoclonal antibodies to expose epitopes.
Visualize the staining by incubation with a gold-conjugated secondary antibody (Auroprobe LM, Amersham) followed by silver enhancement (e.g., IntenSE Kit, Amersham).
3.3.5. Western Blotting
Plate cells in 6-well tissue culture plates at a density of 2.5 × 105 cells per well in culture medium containing 10% FCS. Add 25 μg/mL of ascorbic acid with the first medium change and daily thereafter (see Note 10). When the cultures have reached confluence, add the desired test reagents.
At the end of the incubation period, wash the cell layers with 3 mL/well of PBS. Add 1 mL of trypsin–EDTA to each well, and incubate at room temperature for ≤10 min to allow the cells to detach. Add serum-containing medium, transfer the suspension to a 15-mL centrifuge tube, and wash twice with 5 mL of PBS, pelleting cells by centrifugation at 1,000 × g.
Dissolve each pellet with 20 μL of GSB, electrophorese on a Tris–glycine/SDS–10% polyacrylamide precast gel, and transfer to Immobilon-P membrane by electroblotting with a transfer current of 100 V for 1 h.
Block membrane with 10 mL TBST containing 5% nonfat powdered milk or BSA (Blocking buffer) at 4°C for 1 h with rocking, and wash three times (10 min/wash) in 10 mL TBST.
Incubate the membrane with the antibody at the appropriate dilution in 5 mL of TBST in Blocking buffer for 1 h at room temperature with rocking.
Wash the membrane as in step 4, and incubate with an HRP-conjugated secondary antibody at the appropriate dilution in TBST or Blocking buffer.
Wash the membrane and detect bound antibody by enhanced chemiluminescence according to the manufacturer's protocol and expose to film for autoradiography.
3.4. Extraction and Analysis of RNA
Recovery of cells for extraction: For monolayer cultures, lyse cells directly on the culture dish by adding TRIzol® or RLT Plus lysis buffer reagents directly onto the 12 or 6-well plates. Depolymerize the alginate cultures as described in Subheading 3.2.3, step 3. Transfer cell suspension to sterile, RNase-free polypropylene tube of appropriate size, centrifuge at 1,000 × g at 4°C, washing two or three times with ice-cold PBS.
Extract total RNA using TRIzol® reagent, RNeasy® Plus Mini Kit, or other method as preferred, according to the manufacturer's instructions. If using the RNeasy® Plus Mini Kit, the use of QIAshredder columns is advisable to obtain a better homogenized lysate.
Resuspend RNA in nuclease-free water and determine the RNA concentration in a spectrophotometer. The final preparations should give yields of approximately 10 μg of RNA per 1 × 106 cells with the appropriate A260/A280 ratio of approximately 2.0. It is advisable to determine the integrity of the isolated RNA by analyzing the apparent 28S:18S rRNA ratio (should be ~2:1) in a denaturing agarose gel electrophoresis, or by the use of microfluidics-based technology.
Store at −20°C in nonself-defrosting freezer or at −80°C.
Analyze mRNAs by Northern blotting (15), semiquantitative RT-PCR (42, 52), or real-time PCR (53) by published methods. Type II collagen gene expression is relatively stable in primary cultures of human chondrocytes, at least through the first 12 days of culture (11, 54).
3.5. Analysis of Gene Transcription
Transfection studies are performed to analyze the DNA sequences and transcription factors involved in the regulation of gene expression using plasmid vectors, in which the expression of reporter genes such as CAT (55) or luciferase (42, 54, 56, 57) is driven by cis-acting regulatory sequences, such as those regulating type II collagen gene (COL2A1) transcription (Fig. 5). Coexpression of wild type or dominant-negative mutants of transcription factors, protein kinases, and other regulatory molecules, mediated by plasmid or adenoviral vectors may be performed to further dissect the mechanisms involved and to identify candidate trans-acting factors. The relative contribution of those factors in the transcriptional control of downstream target genes can be then addressed by (1) knockdown of the trans-acting factors, (2) chromatin immunoprecipitation assay of protein–DNA interactions in vivo within specific regulatory elements, and (3) epigenetic analysis of mechanisms such as the DNA methylation status of regulatory sequences (54, 58–60).
Fig. 5.
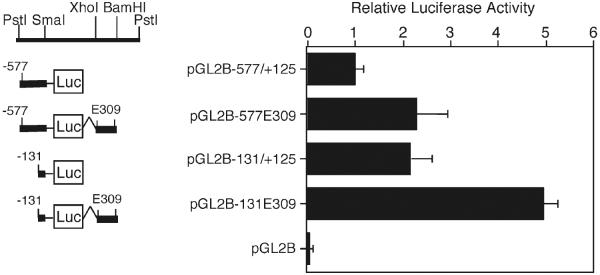
Expression of the COL2A1 promoter human articular chondrocytes. Luciferase reporter constructs containing COL2A1 sequences were transfected in primary chondrocytes using LipofectAMINE+ and incubated in DMEM/F-12 containing 1% FCS for 24 h prior to harvest for luciferase assay. The promoter constructs, pGL2B−577/+125, and pGL2B−131/+125 without or with the E309 enhancer region (+2,388/+2,696 bp) from intron 1 were compared. Luciferase activity was normalized to the amount of protein and expressed as relative activity to that of untreated cells transfected with pGL2B−577/+125E309. Each value is calculated as the mean ± SD of the results from 3 to 6 wells and results are representative of at least three experiments. Note that the empty vector, pGL2-Basic, expressed at 1–2% of the levels of the COL2A1 promoter constructs.
3.5.1. Transient Transfections Using Luciferase Reporter Plasmids
Prepare plasmids using the EndoFree Plasmid Maxi Kit, according to the manufacturer's instructions, to generate endotoxin-free DNA (see Note 14).
Seed 2.5 × 104 cells/cm2 in 6-, 12- or 24-well tissue culture plates in DMEM/F-12 containing 10% FCS (determine optimal cell number to insure that cultures are greater than 50% confluent at the time of transfection, and assure uniform plating density by rocking plates back-and-forth (not swirling) immediately after seeding the cells (see Note 5)).
Change the medium to fresh growth medium 24 and 3 h before the transfection to insure that the cells are actively dividing. Hyaluronidase may be added during these medium changes to increase the transfection efficiency (see Note 15).
- Prepare lipid/DNA complexes in serum-free DMEM/F-12 or Opti-MEM using LipofectAMINE+ or FuGENE 6, according to the manufacturer's protocol. Prepare in bulk for multiple transfections. Volumes indicated for transfections in 6-well plates. Do not vortex at any step:
-
(a).LipofectAMINE+ : For each well, add 92 μL of serum-free medium to a small sterile polypropylene tube, add 1 μL of plasmid DNA (maximum of 1 μg; see Note 16), and tap gently to mix. Add 6 μL of PLUS reagent, mix, and incubate for 15 min at room temperature. Dilute 4 μL of LipofectAMINE+ reagent into 100 μL of serum-free medium, mix, and add to each reaction mixture. Mix and leave at room temperature for an additional 15–30 min at room temperature.
-
(b)FuGENE 6: For each well, add 96 μL of serum-free medium to a small sterile polypropylene tube, add 3 μL of FuGENE 6 reagent, and tap gently to mix. Add 1 μL plasmid of DNA (maximum of 1 μg) to the prediluted FuGENE 6 reagent and incubate for 15 min at room temperature.
-
(a)
While the lipid–DNA complexes are forming, replace culture medium on cells with serum-free medium to give a final volume of 1 mL. Add the lipid–DNA complex mixture dropwise to the well and incubate for 4 h at 37°C.
Dilute the transfection medium by adding to the wells an equal volume of DMEM/F-12 containing 2% Nutridoma-SP (or 20% FCS) and incubate for 2 h to overnight. Add test agent without medium change and incubate further for 18 h (up to 48 h) (see Note 17).
3.5.2. Cotransfections Using Plasmid Vectors for Expression of Recombinant Proteins
Prepare plasmids as described above.
Titrate each expression vector and its corresponding empty vector, at amounts ranging from 10 to 200 ng per well, against a fixed amount of reporter vector. Equalize the total amount of reporter plus expression plasmid in each well (<1 μ g/well) by adding empty vector and maintain equal volumes (see Note 16).
After cotransfection, incubate the cells for 18–24 h to permit expression of recombinant protein prior to treatment with test reagents.
3.5.3. Adenovirus-Mediated Expression of Recombinant Proteins
Infect the 293 producer cell line with adenoviral vector containing the cDNA encoding the wild-type or mutant protein to be coexpressed and determine the titer (MOI) by standard techniques.
Incubate chondrocytes in DMEM/Ham's F-12 containing 10% FCS for 18 h following transfection of the reporter construct.
Remove medium and wash cells with PBS.
Add 1 mL of serum-free medium containing adenovirus at 1:125 MOI. Incubate at 37°C for 90 min.
Add 1 mL of DMEM/Ham's F-12 containing 20% FCS and continue incubation for 18 h.
Change medium to fresh DMEM/ Ham's F-12 containing 10% FCS or 1% Nutridoma-SP, incubate for 1 h, and treat with test agent for 18 h.
3.5.4. Luciferase Assay
Remove the medium from the cells, rinse with PBS and prepare cell lysates by extraction with 200 μL/well of a 6-well plate of Passive Lysis buffer, which will passively lyse cells without the requirement of a freeze–thaw cycle.
Rock culture plates for 5–10 min. Scrape adherent cells with a policeman and transfer solubilized cells to a 1.5-mL microcentrifuge tube.
Microcentrifuge 5 min at maximum speed at 4°C, transfer supernatant to a clean microcentrifuge tube, and store on ice.
Determine the protein content using the Coomassie (Bradford) Protein Assay Reagent.
Determine luciferase activities using the Luciferase Assay System or equivalent, according to manufacturer's protocol. Mix, manually or automatically, 20 μL of cell lysate with 100 μL of Luciferase Assay Reagent and read in a luminometer. Normalize to the amount of protein (or internal control such as β-galactosidase) and express as relative activity against the empty vector or untreated control. Perform each treatment in triplicate wells and each experiment at least three times to ensure reproducibility and significance.
Renilla luciferase control vectors may be used routinely or as necessary to check the purity of new plasmid preparations or relative activities of mutant and wild type constructs using the Dual-Luciferase Reporter Assay System.
3.5.5. siRNA-Mediated Knockdown
Seed 2.5 × 104 cells/cm2 in 6-well plates (assuring uniform plating density) 24–36 h before transfection.
- For each transfection sample, prepare tubes containing the siRNA:LipofectAMINE+ mix as follows:
-
(a)Dilute the appropriate amount of siRNA (see Note 18) in 125 μL of serum-free DMEM/Ham's F12 containing 7.5 μL of PLUS Reagent. Incubate for 15 min at room temperature.
-
(b)Add 125 μL of serum-free media containing 5 μL of LipofectAMINE, mix by tapping, and incubate for 30 min at room temperature.
-
(c)Add 250 μL of serum-free medium to give a final volume of 500 μL.
-
(a)
Remove complete medium from the 6-well plates and add 500 μL of serum-free medium per well.
Add 500 μL of the siRNA transfection mix on top of the serum-free medium added to the cells.
After overnight (16–18 h) incubation at 37°C, replace the transfection media with fresh serum-free or defined medium.
Incubate the cells at 37°C for the time required to achieve knockdown (KD) of the targeted gene (see Note 18).
When addressing involvement of the KD gene in downstream gene expression, add the testing agents at the optimal concentrations and times and proceed to RNA or protein isolation followed by real-time RT-PCR or Western blotting analysis.
3.5.6. Chromatin Immunoprecipitation Assay
The following is an adaptation with minor modifications of the Enzymatic Shearing ChIP Protocol suggested by ActiveMotif.
Plate 2.5 × 105 cells/plate in 150-cm2 tissue culture plates (see Note 19) in DMEM/F12 containing 10% FCS. If test agents are to be used to compare between conditions, serum-deprivation may be required prior to add the test agents to the culture medium.
For cross-linking, remove media from the cells, add 20 mL of Fixation Solution per plate and incubate 10 min at room temperature with gentle agitation.
Remove Fixation Solution, rinse with PBS, add Glycine-Stop-Fix solution (10 mL per plate), and incubate for 5 min at room temperature with gentle agitation.
Remove Glycine-Stop-Fix solution and rinse with PBS.
Collect cells into a 15-mL Falcon tube by scraping with 2 mL of ice-cold PBS containing protease inhibitors.
Centrifuge the collected cells (10 min, 1,300 × g at 4°C) and discard the supernatant.
Resuspend pellets in 1 mL ice-cold Lysis buffer containing protease inhibitors and incubate on ice for 30 min.
Transfer cells to an ice-cold dounce homogenizer, homogenize on ice (10 strokes) and transfer lysates to clean prechilled 1.7-mL centrifuge tubes.
Centrifuge (10 min, 2,300 × g at 4°C), discard supernatant and resuspend pellets in 350 μL of Digestion buffer. Incubate at 37°C for 5 min.
Add 17 μL of the Enzymatic Shearing Cocktail to the reaction, vortex to mix, and incubate at 37°C for 8 min (see Note 20).
Stop reaction by adding 7 μL of ice-cold 0.5 M EDTA; chill on ice for 10 min and centrifuge at 16,100 × g, 10 min at 4°C.
Collect supernatant (~400 μL) containing the sheared chromatin. Use immediately for immunoprecipitation (see Note 20) or store in aliquots at −80°C (adding protease inhibitors) until use.
For preclearing: incubate from 2 to 5 μg of chromatin (see Note 20) with 25 μL of Protein G Magnetic Beads and 5 μg of normal IgG for 2 h at 4°C in an end-to-end rotator. After 2 h, spin the samples, discard bead pellets, collect supernatants and save 10% for input samples (store at −20°C).
Set up immunoprecipitation (IP) reactions in 1.7-mL siliconized tubes: make two preparations per condition, where the antibody of interest will be used in one IP reaction and a normal IgG in the other. Add ChIP buffer 1 (10–20 μL, depending on the volume of the sheared chromatin), 25 μL of Protein G Magnetic beads and nuclease-free water (up to 100 or 200 μL, depending on the volume of the sheared chromatin) to the precleared chromatin. Add 1–5 μg of antibody and incubate overnight (16–18 h) at 4°C in an end-to-end rotator.
Spin the samples, place the tubes in a magnetic stand and discard the supernatant. Wash twice with ChIP buffer 1 and twice with ChIP buffer 2 (3 min each, room temperature in an end-to-end rotator).
To elute the chromatin: resuspend the washed beads in 50 μL of elution buffer and incubate for 15 min in an end-to-end rotator.
For reverse cross-linking, spin the tubes and add 50 μL of Reverse Cross-linking buffer. Place the tubes on a magnetic stand to pellet the beads. Collect the supernatants and transfer them to clean 0.2-mL PCR tubes. Take input samples (stored at −20°C in step 13), add 88 μL of ChIP buffer 2 and 2 μL of NaCl. Incubate all the samples at 95°C for 15 min in a thermocycler.
Spin briefly and return tubes to room temperature; add 2 μL of Proteinase K, mix, and incubate for 1 h at 37°C in a thermocycler.
At room temperature, spin briefly and add 2 μL of Proteinase K Stop Solution. Purify the DNA using QIAquick columns or phenol/chloroform and proceed to the PCR analysis.
3.5.7. DNA Methylation Analysis
Immediately after isolation of articular chondrocytes, extract DNA and RNA from a portion of chondrocytes to assess the expression and the percentage of DNA methylation of the gene/s of interest (see Note 21).
Only chondrocytes isolated from deep-zone non-OA cartilage will be utilized for the cell culture experiments outlined below.
Seed 1 × 104 cells/cm2 (see Note 22) in DMEM/Ham's F-12 containing 10% FCS.
Separate isolated deep-zone non-OA chondrocytes in five groups as follows (a) noncultured (for RNA and DNA isolation, as already stated in Subheading 3.5.7, step 2), (b) control (without treatment), (c) treated with 5-aza-dC (with a single addition of trichostatin A to facilitate access of 5-aza-dC), and (d) cultured with the test agent/s.
Twice per week, replace culture medium with fresh culture medium containing the different experimental treatments (i.e., 5-aza-dC or test agent/s).
When cells reach confluence (4–5 weeks in culture), harvest RNA and DNA from all the culture groups and proceed to analysis.
Perform real time RT-PCR analysis to address whether long-term exposure to the test agent/s leads to aberrant expression of the genes of interest as compared to the untreated control and the noncultured chondrocytes.
Address the DNA methylation status of the promoter/s of interest (from DNA isolated out of the different experimental groups) by bisulfite modification. Design specific CpG-free PCR primers (http://www.urogene.org/methprimer/) to PCR-amplify the target promoter sequence and clone the PCR products.
Send several colonies for sequencing and select candidate CpG site/s for quantification depending upon the average of the sequencing data.
Quantify the percentage of methylated candidate CpG site/s by digestion with methylation-sensitive restriction enzymes followed by real time PCR analysis (see Note 23). Use fully methylated and nonmethylated (obtained by demethylation of the CpGenome Universal Methylated DNA) DNA as internal controls to adjust for the digestion efficiency and to generate standard curve/s for PCR calculations. Use nondigested DNA as a control for normalization of the respective DNA-digested samples.
4. Notes
Batches of serum should be tested and selected on the basis of the capacity to support expression of chondrocyte-specific matrix gene expression. High capacity to induce cell proliferation is not necessarily associated with the ability to maintain phenotype.
Chondrocytes are quite resilient and tolerate the prolonged incubation times required for complete dissociation of the matrix or the absence of serum for the digestion of other sources such as costal cartilage (15). If the digestion is not complete by the end of the allotted time, then more collagenase solution may be added, or the suspension may be recovered and the fragments left behind for further digestion. These conditions result in suspensions that are essentially single cell, and therefore, it is not necessary to resort to filtration through a nylon mesh, as has been done by others when shorter digestion times are used (12). However, we have introduced here the filtration of the final suspension through a filter that allows the cells to pass through and retains undigested material. These considerations are important for decreasing the loss of chondrocytes during their isolation from valuable human cartilage specimens.
After initial plating of the primary cultures, the chondrocytes require 2–3 days before they have settled down and spread out completely. Culture for approximately 4–7 days is required before reasonable amounts of total RNA may be extracted. Although the cultures may continue to express chondrocyte phenotype (e.g., type II collagen and aggrecan mRNAs) for several weeks, expression of nonspecific collagens I and III may begin as early as day 7 after isolation. Adult articular chondrocytes are strongly contact-inhibited and they may exhibit loss of phenotype within 1–2 weeks of monolayer culture. Juvenile costal chondrocytes continue to express chondrocyte phenotype (e.g., type II collagen mRNA) for several weeks and will form multilayer cultures. After they are subcultured, both types of chondrocytes cease the expression chondrocyte matrix proteins, but this loss of phenotype is reversible, and the cells may be redifferentiated in 3D or suspension culture.
Since chondrocytes adhere strongly to tissue culture plastic, possibly because of the presence of calcium ion-binding glycosaminoglycans in the pericellular matrix and cell membrane, a trypsin–EDTA solution rather than trypsin alone should be used for full recovery of chondrocytes from tissue culture plastic during passaging. It is preferable not to use any antibiotics so that any contamination that arises becomes apparent immediately. If necessary, particularly when antibiotics are used in other cultures in the same incubator or culture facility, standard concentrations of penicillin–streptomycin or other antibiotics that are suggested for fibroblast cultures are acceptable for use in chondrocyte cultures.
Primary chondrocyte cultures should be used for experimental analyses immediately before or just after confluence is reached to permit optimal matrix synthesis and cellular responsiveness. If the cells are not used or subcultured, they may be left at confluence for several weeks with weekly medium changes as long as the volume of the culture medium is maintained. If long-term culture results in the deposition of excessive matrix that is not easily digested with trypsin–EDTA, then a single cell suspension may be obtained by using a dilute solution of collagenase (0.25%) and trypsin (0.25%) in PBS.
The synthetic activities of chondrocytes in monolayer culture are inversely related to proliferative activities. Thus, the expression of genes encoding matrix proteins and their deposition into the extracellular matrix increase compared to cell growth-associated genes. For experiments, the growth medium should not be changed within 3 days before addition of the test agent. Alternatively, the cells should be made quiescent by changing to serum-free medium supplemented with an insulin-containing serum substitute such as Nutridoma-SP or ITS+, followed 18–24 h later by the addition (without medium change) of the test agent of interest. Confluent cultures may tolerate serum-free medium containing 0.3% bovine serum albumin for up to 48 h or longer.
While the growth and maintenance of chondrocytes in primary culture or after subculture requires the use of 10% FCS, the loss of phenotype that occurs under these conditions may be delayed if the cells are plated at four- to tenfold higher density. Since high cell yields are not usually attainable from human cartilage sources, the reversibility of the loss of phenotype may be exploited by expanding the chondrocyte populations in monolayer cultures, redifferentiating the cells in fluid suspension culture and replating them in monolayer immediately before performing the experimental procedure.
After several passages in monolayer, chondrocytes may be redifferentiated by 2 weeks or more of culture in alginate beads or in suspension over agarose or polyHEMA.
The method for culture of chondrocytes in alginate beads has been adapted from previously published methods (32, 33). For long-term alginate cultures, high viscosity alginate may provide more stable beads. Serum at concentrations as low as 0.5%, serum substitutes, or combinations of growth and differentiation factors or hormones have been used successfully, depending upon the experimental protocol, to permit chondrocyte phenotypic expression. Note that articular chondrocytes do not proliferate when cultured in fluid or gel suspension.
Ascorbate, which is required for synthesis and secretion of proteoglycans and collagens, is added daily to alginate or other 3D cultures to permit secretion and deposition of extracellular matrix, particularly when staining techniques are to be used. Add 25 μg/mL of ascorbate during the final 24–72 h of incubation when radiolabeling proteoglycans with 35S-sulfate or collagens with 3H-proline for characterization by SDS-PAGE.
Culture of immortalized chondrocytes in 3D scaffolds is a useful approach for tissue engineering applications. The commercially available methods are recommended because of their ease of use. Published methods are available for fabricating collagen sponges (34) and other 3D scaffolds, where the composition may be manipulated, for example, by using type II collagen and/or adding GAGs and other cartilage-specific matrix components. The biodegradable scaffolds are particularly useful if the cell-seeded scaffolds are to be implanted in animals. For studies entirely in vitro, where incubation periods of more than a few days are required, it is recommended that cultures be performed in wells that fit the size of the scaffolds. Otherwise, the culture surface of the well or dish should be coated with a nonadherent substrate or treated in such a way as to prevent attachment of cells that may migrate out from the sponges. Additional analytical methods have been described using Gelfoam® (61) and BD™ 3D scaffolds (see Web site at http://www.bdbiosciences.com/discovery_labware/Products/tissue_engineering/).
Biosynthetic labeling and immunocytochemistry procedures are readily performed on chondrocytes in a solid suspension system such as alginate, agarose, or collagen gels. Alginate culture may be the method of choice, since the chondrocytes are easily recovered by depolymerization of the alginate with a calcium chelator.
Various methods are available for analysis and characterization of proteoglycans. We have found the described methods to be a convenient and rapid approach for screening the relative amounts and molecular sizes of newly synthesized proteoglycans. Specific antibodies are available for more precise identification by either Western blotting or immunocytochemistry, as described in Subheading 2.3.6.
Although chondrocytes are generally less susceptible than monocyte/macrophages and other immune cells to endotoxin, it is possible that the transfection conditions, the proliferative state of the cells, or other factors may sensitize the cells to low concentrations of endotoxin (62). Endotoxin itself induces and activates transcription factors that are common to inflammatory responses and may thus upregulate or downregulate the promoter of interest, thereby masking the response to a cytokine or growth factor.
Hyaluronidase added before and/or during transfections has been shown to increase transfection efficiencies in chondrocytes (63–65).
The total amount of plasmid to be transfected, including reporter, expression and internal control plasmids, should not exceed 1 μg/well of 6-well plate and the optimal amount for the culture system should be tested empirically. If variable amounts of expression vector, for example, are included, the total amount of plasmid in each well should be equalized by the addition of the empty vector. Wells transfected with appropriate empty vector controls, without and with treatment with test agent, should also be included. Since primary chondrocytes are very difficult to transfect, we use them in definitive experiments in studies that mainly involve the use of immortalized chondrocyte cell lines (54, 56, 57).
The times of incubation following transfection before addition of the test agent may vary according to the cell density and culture condition and should be tested empirically. Nutridoma-SP, ITS+, or other serum substitute may be used for experiments that require quiescent cells for the reasons indicated in Note 6. Note also that test agents should be added without medium change to avoid induction of pathways of interest by serum growth factors or other constituents of the control medium.
The knockdown efficacy (degree of reduction of the targeted gene) depends upon transfection efficiency (successful delivery of the siRNA into the cells is usually cell dependent), efficacy of a given siRNA sequence/s, transcription rate of the gene of interest, and protein stability (time- and/or siRNA concentration-dependent). Therefore, those variables must be considered and empirically addressed when designing RNAi experiments. Transfection efficiencies can be determined with the use of fluorescent dye (e.g., rhodamine or FITC)-conjugated siRNA sequences, which allow for visualization of the positive siRNA-transfected cells as well as determination of siRNA subcellular localization and stability in time-course experiments. Knockdown efficacy can be determined in dose–response and time-course experiments, where different concentrations of the siRNA sequence/s (e.g., from 100 to 10 nM) are delivered into the cells and the knockdown of the targeted gene is addressed at different time-points analyzing mRNA (usually, 24, 48, and 72 h posttransfection) and protein (usually, 48, 72, and 96 h posttransfection) levels as compared with those of mock-treated cells and cells transfected with non-targeting siRNA sequences (58, 59).
ChIP assays are generally limited by the requirement for large cell numbers, the different efficiencies with which different proteins cross-link with their interacting DNA – particularly relevant when addressing binding of transcription factors due to their lower abundance and somewhat dynamic and labile interaction with their target DNA sequences as compared to major chromatin constituents such as histones, the relative amount of a DNA-bound protein of interest within a cell type, and the availability of high-quality and high affinity antibodies against the protein of interest. The indicated number of cells will thus vary depending upon the protein–DNA interaction to be addressed. Therefore, several steps, including cross-linking and shearing, and controls for antibody specificity, must be carefully optimized for each specific assay. The optimization steps required for having a reproducible and specific ChIP assay may be not feasible using primary chondrocytes, given their usually limited availability. In that regard, the use of immortalized chondrocytes may be a useful tool to set the adequate conditions for the specific ChIP assay to be conducted, as it is possible to work with larger amounts of material in a more reproducible manner (54, 58, 59). The assays optimized using the cell lines may then be validated or assessed using primary cells.
Before conducting time- and cost-expensive experiments, several steps should be carefully optimized and empirically tested. Namely, it is advisable to perform small-scale experiments, where different cross-linking and shearing conditions are tested. Prior to performing immunoprecipitation, the sheared chromatin can be reverse cross-linked and analyzed in a 1–1.5% agarose gel (with the conditions described, enzymatic shearing will yield fragments between 250 and 1,000 bp) to ensure optimal shearing, and the yield of DNA can be quantified to use equal amounts of DNA for the immunoprecipitation and comparison of different conditions. Although not always necessary, preclearing or additional BSA-blocking steps may be required to increase the signal-to-noise ratio when using certain antibodies. For PCR analysis of the precipitated DNA, real time PCR analysis is recommended as it permits a more refined and quantitative analysis of the final result. When analyzing PCR products in agarose gels (Fig. 6), the PCR must be stopped in linear stage of amplification. Sequencing of the PCR products is suggested to ensure specific amplification.
Previous studies have shown that there may be zonal variations in the methylation status of certain genes (60). For example, the chondrocytes from the superficial zone of normal cartilage may show increased/aberrant expression of proteases and cytokines and, therefore, chondrocytes from the middle to deep zones are used to assess physiological methylation status and compare with chondrocytes from osteoarthritis patients.
Experimental groups are designed with chondrocytes obtained from the same individual and, therefore, the number of cells may be limiting. More importantly, epigenetic events require long-term analysis and, thus, cells should be plated at low densities so they do not reach confluence quickly and allow for long-term treatment with the testing agent/s and inheritance of the epigenetic changes.
After screening of the methylation status of a given promoter by bisulfite modification and identification of candidate CpG site/s, platforms such as PyroMark Q96 Systems (QIAGEN Inc., Valencia, CA) allow for the quantitative analysis of multiple CpG sites simultaneously, whereas the enzyme-based method detailed here allows for the assessment of one CpG site at the time.
Fig. 6.
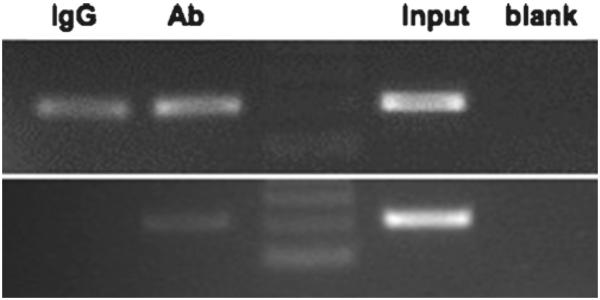
ChIP analysis performed without or with a preclearing step. Chromatin was cross-linked and enzymatically sheared, and after reverse cross-linking of the DNA–protein complexes, the lysates were either (upper panel) used directly for immunoprecipitation without a preclearing step or (lower panel) precleared prior to overnight incubation with the antibody against a protein of interest (Ab1) or normal rabbit IgG (IgG). The precipitated DNA was purified and PCR-amplified, and the PCR products were resolved in a 2.5% agarose gel.
Acknowledgments
Dr. Goldring's research related to this project was supported in part by grants from the National Institutes of Health, AG022021, AR054887, and AR045378, and the Arthritis Foundation. The authors are grateful to Lujian Tan, Haibing Peng, Lii-Fang Suen, James Birkhead, Merrilee Flannery, James Robbins, Makoto Osaki, and Bob Choy for supplying technical expertise and data, and to Dr. Thomas Sculco at The Hospital for Special Surgery and Dr. Benjamin Bierbaum at the New England Baptist Hospital for supplying cartilage samples. We also wish to acknowledge Dr. Helmtrud (Trudy) I. Roach, recently deceased, for her contributions to our understanding of epigenetics in chondrocytes and for transferring the technology to our laboratory.
References
- 1.Goldring MB. Cartilage and Chondrocytes. In: Firestein GS, Budd RC, Harris EDJ, McInnes IB, Ruddy S, Sergent JS, editors. Kelley's Textbook of Rheumatology. 8th Edition Ch. 3. Saunders an imprint of Elsevier, Inc.; Philadelphia: 2009. pp. 37–69. [Google Scholar]
- 2.Maroudas A, Palla G, Gilav E. Racemization of aspartic acid in human articular cartilage. Connect Tissue Res. 1992;28:161–169. doi: 10.3109/03008209209015033. [DOI] [PubMed] [Google Scholar]
- 3.Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–39031. doi: 10.1074/jbc.M006700200. [DOI] [PubMed] [Google Scholar]
- 4.Maroudas A, Bayliss MT, Uchitel-Kaushansky N, Schneiderman R, Gilav E. Aggrecan turnover in human articular cartilage: use of aspartic acid racemization as a marker of molecular age. Arch Biochem Biophys. 1998;350:61–71. doi: 10.1006/abbi.1997.0492. [DOI] [PubMed] [Google Scholar]
- 5.Otero M, Goldring MB. Cells of the synovium in rheumatoid arthritis. Chondrocytes. Arthritis Res Ther. 2007;9:220. doi: 10.1186/ar2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldring MB, Otero M, Tsuchimochi K, Ijiri K, Li Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann Rheum Dis. 2008;67(Suppl 3):iii75–82. doi: 10.1136/ard.2008.098764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldring MB, Marcu KB. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res Ther. 2009;11:224. doi: 10.1186/ar2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtzer J, Abbott J, Lash J, Holtzer A. The loss of phenotypic traits by differentiated cells in vitro. I. Dedifferentiation of cartilage cells. Proc. Natl. Acad. Sci. USA. 1960;46:1533–1542. doi: 10.1073/pnas.46.12.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ham RG, Sattler GL. Clonal growth of differentiated rabbit cartilage cells. J. Cell. Physiol. 1968;72:109–114. doi: 10.1002/jcp.1040720205. [DOI] [PubMed] [Google Scholar]
- 10.Green WT., Jr. Behavior of articular chondrocytes in cell culture. Clin. Orthopaed. Rel. Res. 1971;75:248–260. doi: 10.1097/00003086-197103000-00030. [DOI] [PubMed] [Google Scholar]
- 11.Goldring MB, Sandell LJ, Stephenson ML, Krane SM. Immune interferon suppresses levels of procollagen mRNA and type II collagen synthesis in cultured human articular and costal chondrocytes. J. Biol. Chem. 1986;261:9049–9056. [PubMed] [Google Scholar]
- 12.Aulthouse AL, Beck M, Friffey E, Sanford J, Arden K, Machado MA, Horton WA. Expression of the human chondrocyte phenotype in vitro. In Vitro Cell. Devel. Biol. 1989;25:659–668. doi: 10.1007/BF02623638. [DOI] [PubMed] [Google Scholar]
- 13.Kolettas E, Buluwela L, Bayliss MT, Muir HI. Expression of cartilage-specific molecules is retained on long-term culture of human articular chondrocytes. J Cell Sci. 1995;108(Pt 5):1991–1999. doi: 10.1242/jcs.108.5.1991. [DOI] [PubMed] [Google Scholar]
- 14.Goldring MB. Control of collagen synthesis in human chondrocyte cultures by immune interferon and interleukin-1. J Rheumatol. 1987;14(Spec No):64–66. [PubMed] [Google Scholar]
- 15.Goldring MB, Birkhead J, Sandell LJ, Kimura T, Krane SM. Interleukin 1 suppresses expression of cartilage-specific types II and IX collagens and increases types I and III collagens in human chondrocytes. J Clin Invest. 1988;82:2026–2037. doi: 10.1172/JCI113823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnabel M, Marlovits S, Eckhoff G, Fichtel I, Gotzen L, Vecsei V, Schlegel J. Dedifferentiation-associated changes in morphology and gene expression in primary human articular chondrocytes in cell culture. Osteoarthritis Cartilage. 2002;10:62–70. doi: 10.1053/joca.2001.0482. [DOI] [PubMed] [Google Scholar]
- 17.Barbero A, Grogan SP, Mainil-Varlet P, Martin I. Expansion on specific substrates regulates the phenotype and differentiation capacity of human articular chondrocytes. J Cell Biochem. 2006;98:1140–1149. doi: 10.1002/jcb.20754. [DOI] [PubMed] [Google Scholar]
- 18.von der Mark K, Gauss V, von der Mark H, Muller P. Relationship between cell shape and type of collagen synthesised as chondrocytes lose their cartilage phenotype in culture. Nature. 1977;267:531–532. doi: 10.1038/267531a0. [DOI] [PubMed] [Google Scholar]
- 19.Watt FM. Effect of seeding density on stability of the differentiated phenotype of pig articular chondrocytes in culture. J Cell Sci. 1988;89(Pt 3):373–378. doi: 10.1242/jcs.89.3.373. [DOI] [PubMed] [Google Scholar]
- 20.Kuettner KE, Pauli BU, Gall G, Memoli VA, Schenk RK. Synthesis of cartilage matrix by mammalian chondrocytes in vitro. I. Isolation, culture characteristics, and morphology. J. Cell Biol. 1982;93:743–750. doi: 10.1083/jcb.93.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassleer C, Gysen P, Foidart JM, Bassleer R, Franchimont P. Human chondrocytes in tridimensional culture. In Vitro Cell. Devel. Biol. 1986;22:113–119. doi: 10.1007/BF02623497. [DOI] [PubMed] [Google Scholar]
- 22.Thonar EJ, Buckwalter JA, Kuettner KE. Maturation-related differences in the structure and composition of proteoglycans synthesized by chondrocytes from bovine articular cartilage. J Biol Chem. 1986;261:2467–2474. [PubMed] [Google Scholar]
- 23.Norby DP, Malemud CJ, Sokoloff L. Differences in the collagen types synthesized by lapine articular chondrocytes in spinner and monolayer culture. Arthritis Rheum. 1977;20:709–716. doi: 10.1002/art.1780200211. [DOI] [PubMed] [Google Scholar]
- 24.Glowacki J, Trepman E, Folkman J. Cell shape and phenotypic expression in chondrocytes. Proc. Soc. Exp. Biol. Med. 1983;172:93–98. doi: 10.3181/00379727-172-41533. [DOI] [PubMed] [Google Scholar]
- 25.Castagnola P, Moro G, Descalzi-Cancedda F, Cancedda R. Type X collagen synthesis during in vitro development of chick embryo tibial chondrocytes. J Cell Biol. 1986;102:2310–2317. doi: 10.1083/jcb.102.6.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reginato AM, Iozzo RV, Jimenez SA. Formation of nodular structures resembling mature articular cartilage in long-term primary cultures of human fetal epiphyseal chondrocytes on hydrogel substrate. Arthritis Rheum. 1994;37:1338–1349. doi: 10.1002/art.1780370912. [DOI] [PubMed] [Google Scholar]
- 27.Croucher LJ, Crawford A, Hatton PV, Russell RG, Buttle DJ. Extracellular ATP and UTP stimulate cartilage proteoglycan and collagen accumulation in bovine articular chondrocyte pellet cultures. Biochim Biophys Acta. 2000;1502:297–306. doi: 10.1016/s0925-4439(00)00055-7. [DOI] [PubMed] [Google Scholar]
- 28.Olivotto E, Vitellozzi R, Fernandez P, Falcieri E, Battistelli M, Burattini S, Facchini A, Flamigni F, Santi S, Facchini A, Borzi RM. Chondrocyte hypertrophy and apoptosis induced by GROalpha require three-dimensional interaction with the extracellular matrix and a co-receptor role of chondroitin sulfate and are associated with the mitochondrial splicing variant of cathepsin B. J Cell Physiol. 2007;210:417–427. doi: 10.1002/jcp.20864. [DOI] [PubMed] [Google Scholar]
- 29.Gibson GJ, Schor SL, Grant ME. Effects of matrix macromolecules on chondrocyte gene expression: Synthesis of a low molecular weight collagen species by cells cultured within collagen gels. J. Cell Biol. 1982;93:767–774. doi: 10.1083/jcb.93.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benya PD, Shaffer JD. Dedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell. 1982;30:215–224. doi: 10.1016/0092-8674(82)90027-7. [DOI] [PubMed] [Google Scholar]
- 31.Aydelotte MB, Kuettner KE. Differences between sub-populations of cultured bovine articular chondrocytes. I. Morphology and cartilage matrix production. Conn. Tiss. Res. 1988;18:205–222. doi: 10.3109/03008208809016808. [DOI] [PubMed] [Google Scholar]
- 32.Guo J, Jourdian GW, MacCallum DK. Culture and growth characteristics of chondrocytes encapsulated in alginate beads. Conn. Tiss. Res. 1989;19:277–297. doi: 10.3109/03008208909043901. [DOI] [PubMed] [Google Scholar]
- 33.Hauselmann HJ, Aydelotte MB, Schumacher BL, Kuettner KE, Gitelis SH, Thonar EJ-MA. Synthesis and turnover of proteoglycans by human and bovine adult articular chondrocytes cultured in alginate beads. Matrix. 1992;12:116–129. doi: 10.1016/s0934-8832(11)80053-3. [DOI] [PubMed] [Google Scholar]
- 34.Mizuno S, Allemann F, Glowacki J. Effects of medium perfusion on matrix production by bovine chondrocytes in three-dimensional collagen sponges. J Biomed Mater Res. 2001;56:368–375. doi: 10.1002/1097-4636(20010905)56:3<368::aid-jbm1105>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 35.Xu C, Oyajobi BO, Frazer A, Kozaci LD, Russell RG, Hollander AP. Effects of growth factors and interleukin-1 alpha on proteoglycan and type II collagen turnover in bovine nasal and articular chondrocyte pellet cultures. Endocrinology. 1996;137:3557–3565. doi: 10.1210/endo.137.8.8754787. [DOI] [PubMed] [Google Scholar]
- 36.Adolphe M, Froger B, Ronot X, Corvol MT, Forest N. Cell multiplication and type II collagen production by rabbit articular chondrocytes cultivated in a defined medium. Exp. Cell Res. 1984;155:527–536. doi: 10.1016/0014-4827(84)90212-x. [DOI] [PubMed] [Google Scholar]
- 37.Gerstenfeld LC, Kelly CM, Von Deck M, Lian JB. Comparative morphological and biochemical analysis of hypertrophic, non-hypertrophic and 1,25(OH)2D3 treated non-hypertrophic chondrocytes. Connective Tissue Research. 1990;24:29–39. doi: 10.3109/03008209009152420. [DOI] [PubMed] [Google Scholar]
- 38.Adams SL, Pallante KM, Niu Z, Leboy PS, Golden EB, Pacifici M. Rapid induction of type X collagen gene expression in cultured chick vertebral chondrocytes. Exp. Cell Res. 1991;193:190–197. doi: 10.1016/0014-4827(91)90555-9. [DOI] [PubMed] [Google Scholar]
- 39.Poole AR. Honor Bridgett Fell, Ph.D., D.Sc. F.R.S., D.B.E., 1900–1986. The scientist and her contributions. In Vitro Cell Dev Biol. 1989;25:450–453. doi: 10.1007/BF02624631. [DOI] [PubMed] [Google Scholar]
- 40.Benoit B, Thenet-Gauci S, Hoffschir F, Penformis P, Demignot S, Adolphe M. SV40 large T antigen immortalization of human articular chondrocytes. In Vitro Cell. Dev. Biol. 1995;31:174–177. doi: 10.1007/BF02639430. [DOI] [PubMed] [Google Scholar]
- 41.Steimberg N, Viengchareun S, Biehlmann F, Guenal I, Mignotte B, Adolphe M, Thenet S. SV40 large T antigen expression driven by col2a1 regulatory sequences immortalizes articular chondrocytes but does not allow stabilization of type II collagen expression. Exp Cell Res. 1999;249:248–259. doi: 10.1006/excr.1999.4478. [DOI] [PubMed] [Google Scholar]
- 42.Robbins JR, Thomas B, Tan L, Choy B, Arbiser JL, Berenbaum F, Goldring MB. Immortalized human adult articular chondrocytes maintain cartilage-specific phenotype and responses to interleukin-1 β. Arthritis Rheum. 2000;43:2189–2201. doi: 10.1002/1529-0131(200010)43:10<2189::AID-ANR6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 43.Grigolo B, Roseti L, Neri S, Gobbi P, Jensen P, Major EO, Facchini A. Human articular chondrocytes immortalized by HPV-16 E6 and E7 genes: Maintenance of differentiated phenotype under defined culture conditions. Osteoarthritis Cartilage. 2002;10:879–889. doi: 10.1053/joca.2002.0836. [DOI] [PubMed] [Google Scholar]
- 44.Piera-Velazquez S, Jimenez SA, Stokes D. Increased life span of human osteoarthritic chondrocytes by exogenous expression of telomerase. Arthritis Rheum. 2002;46:683–693. doi: 10.1002/art.10116. [DOI] [PubMed] [Google Scholar]
- 45.Goldring MB. Immortalization of human articular chondrocytes for generation of stable, differentiated cell lines. Methods Mol Med. 2004;100:23–36. doi: 10.1385/1-59259-810-2:023. [DOI] [PubMed] [Google Scholar]
- 46.Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S, Glowacki J, Arbiser JL, Apperley JF. Interleukin-1 beta-modulated gene expression in immortalized human chondrocytes. J Clin Invest. 1994;94:2307–2316. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 48.Olivotto E, Borzi RM, Vitellozzi R, Pagani S, Facchini A, Battistelli M, Penzo M, Li X, Flamigni F, Li J, Falcieri E, Facchini A, Marcu KB. Differential requirements for IKKalpha and IKKbeta in the differentiation of primary human osteoarthritic chondrocytes. Arthritis Rheum. 2008;58:227–239. doi: 10.1002/art.23211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogel KG, Sandy JD, Pogany G, Robbins JR. Aggrecan in bovine tendon. Matrix Biology. 1994;14:171–179. doi: 10.1016/0945-053x(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 50.Robbins JR, Vogel KG. Mechanical loading and TGF-β regulate proteoglycan synthesis in tendon. Arch. Biochem. Biophys. 1997;342:203–211. doi: 10.1006/abbi.1997.0102. [DOI] [PubMed] [Google Scholar]
- 51.Kokenyesi R, Tan L, Robbins JR, Goldring MB. Proteoglycan production by immortalized human chondrocyte cell lines cultured under conditions that promote expression of the differentiated phenotype. Arch. Biochem. Biophys. 2000;383:79–90. doi: 10.1006/abbi.2000.2044. [DOI] [PubMed] [Google Scholar]
- 52.Dell'Accio F, De Bari C, Luyten FP. Molecular markers predictive of the capacity of expanded human articular chondrocytes to form stable cartilage in vivo. Arthritis Rheum. 2001;44:1608–1619. doi: 10.1002/1529-0131(200107)44:7<1608::AID-ART284>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 53.Bau B, Gebhard PM, Haag J, Knorr T, Bartnik E, Aigner T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002;46:2648–2657. doi: 10.1002/art.10531. [DOI] [PubMed] [Google Scholar]
- 54.Peng H, Tan L, Osaki M, Zhan Y, Ijiri K, Tsuchimochi K, Otero M, Wang H, Choy BK, Grall FT, Gu X, Libermann TA, Oettgen P, Goldring MB. ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J Cell Physiol. 2008;215:562–573. doi: 10.1002/jcp.21338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldring MB, Fukuo K, Birkhead JR, Dudek E, Sandell LJ. Transcriptional suppression by interleukin-1 and interferon- g of type II collagen gene expression in human chondrocytes. J. Cell. Biochem. 1994;54:85–99. doi: 10.1002/jcb.240540110. [DOI] [PubMed] [Google Scholar]
- 56.Osaki M, Tan L, Choy BK, Yoshida Y, Auron PE, Cheah KSE, Goldring MB. The TATA-containing core promoter of the type II collagen gene (COL2A1) is the target of interferon-γ-mediated inhibition in human chondrocytes: requirement for Stat1α, Jak1, and Jak2. Biochem. J. 2003;369:103–115. doi: 10.1042/BJ20020928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan L, Peng H, Osaki M, Choy BK, Auron PE, Sandell LJ, Goldring MB. Egr-1 Mediates Transcriptional Repression of COL2A1 Promoter Activity by Interleukin-1beta. J Biol Chem. 2003;278:17688–17700. doi: 10.1074/jbc.M301676200. [DOI] [PubMed] [Google Scholar]
- 58.Ijiri K, Zerbini LF, Peng H, Otu HH, Tsuchimochi K, Otero M, Dragomir C, Walsh N, Bierbaum BE, Mattingly D, van Flandern G, Komiya S, Aigner T, Libermann TA, Goldring MB. Differential expression of GADD45beta in normal and osteoarthritic cartilage: potential role in homeostasis of articular chondrocytes. Arthritis Rheum. 2008;58:2075–2087. doi: 10.1002/art.23504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsuchimochi K, Otero M, Dragomir CL, Plumb DA, Zerbini LF, Libermann TA, Marcu KB, Komiya S, Ijiri K, Goldring MB. GADD45beta enhances Col10a1 transcription via the MTK1/MKK3/6/p38 axis and activation of C/EBPbeta-TAD4 in terminally differentiating chondrocytes. J Biol Chem. 2010;285:8395–8407. doi: 10.1074/jbc.M109.038638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hashimoto K, Oreffo RO, Gibson MB, Goldring MB, Roach HI. DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 2009;60:3303–3313. doi: 10.1002/art.24882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu QQ, Chen Q. Mechanoregulation of chondrocyte proliferation, maturation, and hypertrophy: ion-channel dependent transduction of matrix deformation signals. Exp Cell Res. 2000;256:383–391. doi: 10.1006/excr.2000.4847. [DOI] [PubMed] [Google Scholar]
- 62.Cotten M, Baker A, Saltik M, Wagner E, Buschle M. Lipopolysaccharide is a frequent contaminant of plasmid DNA preparations and can be toxic to primary human cells in the presence of adenovirus. Gene Ther. 1994;1:239–246. [PubMed] [Google Scholar]
- 63.Lu Valle P, Iwamoto M, Fanning P, Pacifici M, Olsen BR. Multiple negative elements in a gene that codes for an extracellular matrix protein, collagen X, restrict expression to hypertrophic chondrocytes. J Cell Biol. 1993;121:1173–1179. doi: 10.1083/jcb.121.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Viengchareun S, Thenet-Gauci S, Steimberg N, Blancher C, Crisanti P, Adolphe M. The transfection of rabbit articular chondrocytes is independent of their differentiation state. In Vitro Cell Dev Biol Anim. 1997;33:15–17. doi: 10.1007/s11626-997-0016-3. [DOI] [PubMed] [Google Scholar]
- 65.Madry H, Trippel SB. Efficient lipid-mediated gene transfer to articular chondrocytes. Gene Ther. 2000;7:286–291. doi: 10.1038/sj.gt.3301086. [DOI] [PubMed] [Google Scholar]