

Abstract
Objective
To evaluate the role of hepatocellular and extrahepatic apoptosis during the evolution of Acetaminophen-induced acute liver failure (AALF).
Design & Setting
A prospective observational study in two tertiary liver transplant units.
Patients
88 patients with AALF were recruited. Control groups included patients with non-AALF (n=13), non-hepatic multi-organ failure (MOF, n=28), chronic liver disease (CLD, n=19) and healthy controls (HC, n=11).
Measurements
Total and caspase-cleaved cytokeratin 18 (M65 and M30 measured on admission and sequentially on day 3, 7 and 10 following admission. Levels were also determined from hepatic, portal vein and systemic arterial blood in seven patients undergoing transplantation. Protein arrays of liver homogenates from AALF patients were assessed for apoptosis-associated proteins and histological assessment of liver tissue was performed.
Main Results
Admission M30 levels were significantly elevated in AALF and NALF patients compared to MOF, CLD and healthy controls. Admission M30 levels correlated with outcome with AUROC of 0.755 (0.639-0.885, p<0.001). Peak levels in ALF patients were seen on admission then fell significantly but did not normalize over ten days. A negative gradient of M30 from the portal to hepatic vein was demonstrated in AALF patients (p=0.042) at the time of liver transplant. Analysis of protein array data demonstrated lower apoptosis-associated protein and higher catalase concentrations in AALF liver compared to controls (p<0.05). Explant histological analysis revealed evidence of cellular proliferation with an absence of histological evidence of apoptosis.
Conclusions
Hepatocellular apoptosis occurs in the early phases of human AALF, peaking on day 1 of hospital admission and correlates strongly with poor outcome. Hepatic regenerative/tissue repair responses prevail during the later stages of ALF where elevated levels of M30 are likely to reflect epithelial cell death in extra-hepatic organs.
Keywords: Acetaminophen, Acute Liver Failure, Multi-organ failure, apoptosis, caspase, M30
Introduction
Acute Liver Failure (ALF) is a rare but devastating clinical syndrome caused by the sudden loss of function and viability of a critical mass of hepatocytes. Liver cell death leads directly to the jaundice, coagulopathy and encephalopathy that are the clinical hallmarks of this condition which carries a transplant-free survival of less than 50% (1).
Acetaminophen overdose is the commonest cause of ALF in the USA and UK accounting for 42% and 57% of cases respectively (2, 3). Controversy exists regarding whether hepatocellular death occurs as a result of necrosis, apoptosis or a combination of both processes (4). Experimental models of acute liver injury indicate that the majority of hepatocytes die via necrosis, as the sudden and comprehensive loss of mitochondrial function does not permit cells to execute the ATP-dependent process of apoptosis (5, 6). There is however evidence that when cellular ATP levels are preserved, hepatocytes can undergo apoptosis, with the induction of cellular caspases (7-9). Studies in human acetaminophen-induced ALF (AALF) have demonstrated the presence of caspase-cleaved cytokeratin-18 fragments in patient’s sera suggesting cellular apoptosis. However a recent study failed to demonstrate active caspase-3 in AALF patients and thus concluded apoptosis was not a significant phenomenon in this condition (10).
Following the initial toxic insult, the course of acetaminophen poisoning is characterised by activation of innate immune responses that propagate acute liver injury. This is characterized by elevations in pro-toxicant mediators, recruitment and activation of immune effectors (e.g. NK/NKT cells, neutrophils) that perpetuate liver injury via a number of mechanisms including TNF-α and Fas-mediated heaptocyte apoptosis (11). These responses are likely to be major determinants in not only the progression but eventual outcome in human AALF, The resolution phase of AALF is difficult to replicate in animal models, yet increasing our understanding of the pathophysiological processes, involved during the evolution of ALF, from acute liver injury to multiple organ dysfunction is of pivotal importance (12).
Circulating concentrations of apoptosis and necrosis markers, based on caspase-cleaved and intact cytokeratin-18 (CK-18) have been proposed as biomarkers of disease severity and may fulfill an important role in risk stratifying patients with AALF (13, 14) (15). However, since published studies show conflicting results regarding the prognostic utility of these markers the temporal changes in CK-18 based markers and the contribution of non-hepatic epithelial tissues require further evaluation.
This study aims to assess the role of apoptosis during the clinical course of AALF. We report the results of serum markers of apoptosis and necrosis on admission to hospital in a large cohort of AALF patients; present longitudinal data on the progression of these markers through the first ten hospital days including the assessment of hepatic apoptosis at the time of liver transplantation.
Materials and Methods
Patient selection and sample collection
The study was approved by a local ethics committee and patients or their nominee gave written informed consent within 24 hours of presentation to Kings College Hospital and the E Garner King General Systems Intensive Care Unit, University of Alberta, Canada.
AALF cohort
Sixty two non-consecutive patients presenting to King’s College Hospital with a clinical history of excess acetaminophen ingestion, deranged liver function tests and contemporaneous or subsequent development of ALF (as defined by a jaundice to encephalopathy time of <28 days (16)) were initially recruited. Clinical and demographic data, including age, sex, biochemistry, arterial blood gas, physiology and APACHE score, were collected from patient at the time of blood sampling. Patient outcomes were recorded as spontaneous survival, death or liver transplantation, with poor outcome defined as death or transplantation. Blood samples were collected in lithium heparin tubes from patients within 48 hours of admission. Blood was centrifuged at 13,000rpm for 10 minutes at 4oC for separation of Plasma. Plasma aliquots were stored at −80 oC pending analysis. Fifteen patients with AALF were also recruited from the E Garner King General Systems Intensive Care Unit, University of Alberta, Edmonton, Canada.
Control cohorts
Pathological control cohorts were recruited from patients at Kings College Hospital with blood sampling and plasma isolation performed as above. Control groups included: 13 patients with ALF of a non-acetaminophen aetiology (NA-ALF) (Viral n=3, seronegative n=5, non-acetaminophen DILI n=5), 19 patients with stable chronic liver disease (CLD) recruited from outpatient clinics; 11 healthy controls (HC), 28 intubated and ventilated general intensive care patients with multi-organ failure (MOF).
Sequential levels
In a sub-cohort of 26 (15 from Edmonton, 11 from the UK) patients with AALF plasma samples were drawn on days 1, 3 , 7 and 10 following admission to delineate the time course of total and cleaved caspases following the initial liver injury.
Regional levels
In seven AALF patients undergoing liver transplantation, blood was collected intraoperatively from the portal vein (PV), hepatic vein (HV) and a systemic artery (the renal artery) (SA) prior to PV clamping. Blood samples were immediately dispensed into lithium-heparin tubes, centrifuged and plasma stored as above.
Tissue collection
Liver explant tissue was collected from six AALF patients at the time of transplantation. Fresh tissue blocks were dissected immediately following removal of the liver and flash frozen in liquid nitrogen before storing at −80 oC until required. Control liver tissue was obtained from donor liver in which consent for research was in place.
Total and cleaved-cytokeratin 18 quantification
Plasma levels of un-cleaved cytokeratin-18 (M65) and caspase-cleaved cytokeratin-18 (M30) were quantified with commercially available ELISAs (Peviva AB, Bromma Sweden), previously validated in clinical trials and used according to manufacturer’s instructions as described elsewhere (17).
Apoptosis proteome array
AALF and control liver tissue was lysed by disruption in lysis buffer. Protein content was determined using modified Lowry protein determination assay according to Schacterle and Pollack, which is based Lowry OH et al. (18, 19). Samples were diluted with phosphate buffered saline to give a normalised final concentration of 250μg protein per 125μl per membrane. The focused proteome array, Proteome ProfilerTM Array Human Apoptosis Array Kit (R&D Systems Inc. MN USA) was performed according to manufacturer’s instructions. Membranes were exposed to an autoradiography cassette for 6 minutes. Film was scanned and analysed in imageJ (http://rsbweb.nih.gov/ij/). Following inversion and subtraction of background exposure, the integrated density of each dot was measured and averaged between duplicates.
Histology
H&E stained liver explant tissue was examined by an expert histopathologist (AQ) for evidence of apoptotic bodies. Double epitope immunohistochemistry was performed using antibodies to Ki-67 and Hep Par-1 and Ki 67 and CK-19.
Statistical analysis
Data were assessed for normality using the D’Agostino-Pearson test and expressed as mean (standard deviation) median (range) contingent on results with log-transformation on non-normally distributed data. Comparison was by one-way analysis of variance (ANOVA), with Tukey’s post hoc test applied for multiple comparison correction. Four time point sequential data were analysed using Repeat measures ANOVA. Paired t-testing on log-transformed data was performed on measurements across organ beds. Apoptotic protein array profiling was analysed by Student’s t test. The prognostic significance of apoptosis markers was assessed by Receiver Operating Characteristic (ROC) Curve analysis using the DeLong method for ROC curve comparison and defining cutoff by Youden Index. Statistical significance was defined at the 95% level. Data were analysed in MedCalc v 12.2.1 (MedCalc software, Mariakerke, Belgium).
Results
Elevation of cytokeratin-18 fragments on admission in AALF, NALF and MOF
The cohort characteristics are given in Table 1. Participants from the UK and Canada were matched for age, sex, liver and organ failure severity as shown in Table 1. Elevated levels of caspase-cleaved and total CK-18 were demonstrated on admission blood samples in both ALF patient cohorts. The levels of caspase-cleaved (M30) CK-18 were significantly higher in AALF and NALF than in APACHE-matched MOF control patients, those with CLD and healthy controls (p<0.001, one way ANOVA on log transformed data, (Figure 1)). Patients with MOF and no liver disease had levels of both M30 and M65 intermediate between the ALF cohort and patients with chronic liver disease or healthy controls (p<0.001, one way ANOVA on log transformed data, Figure 1). Using the ratio of M30 to M65 (an ‘apoptosis index’) failed to demonstrate a difference between ALF and MOF patients. Patients with CLD showed the lowest levels of this index (p=0.004, one way ANOVA on log transformed data, data not shown).
Table 1. Comparison of UK and Canadian cohorts of patients with AALF demonstrating matching of demographics and liver and organ severity.
The non to these units and/or fluid resuscitation prior to inclusion. This may also explain the difference in M65 levels.
| Centre | |||
|---|---|---|---|
| Variable | Canadian | UK | P value |
| Age (years) | 39 (21-72) | 36 (15-68) | 0.614 |
| Sex (M:F) | 4:14 | 25:38 | 0.278 |
| AST (iU/L) | 2600 (2330-15886) | 4717 (40-17800) | 0.775 |
| Bilirubin (umol/l) | 91 (33-197) | 67 (30-237) | 0.129 |
| Creatinine (umol/l) | 142 (47-757) | 262 (47-478) | 0.339 |
| INR | 4.1 (2.1-10) | 6.8 (1.5-15) | 0.188 |
| Lactate (mmol/l) | 6.0 (3.1-27) | 3.6 (1.3-18) | 0.051 |
| pH | 7.32 (7.04-7.42) | 7.39 (7.0-7.5) | 0.717 |
| Platelets (×109/l) | 52 (13-243) | 75 (20-146) | 0.386 |
| WCC (×109/l) | 13.8 (3.6-24) | 10.6 (4.7-20.8) | 0.161 |
| APACHE II | 22 (4-37) | 22 (5-33) | 0.512 |
| HE Grade | 2 (1-4) | 3 (1-4) | 0.057 |
| M30 | 6776 (257-31754) | 3636 (50-22361) | 0.107 |
| M65 | 32361 (2297-74525) | 8194 (50-71193) | 0.003 |
| M65/M30 | 4.41 (0.29-75.2) | 2.73 (.49-63.7) | 0.135 |
Figure 1. One-way ANOVA of total and cleaved cytokeratin-18.
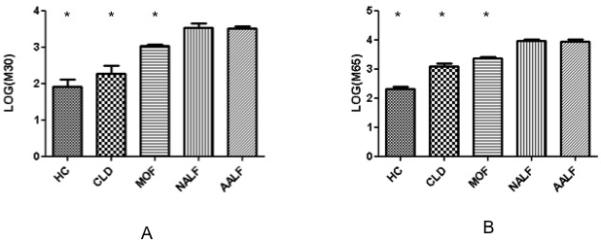
Data are presented as mean (SEM) of log transformed data with Tukey’s post hoc test for further comparison.
A) M30 levels demonstrate significant group differences with levels in AALF significantly higher than in HC, CLD or MOF (*p<0.05 for comparison with AALF group). M30 levels in the MOF patients were intermediate and significantly higher than those in the HC and CLD groups and lower than the AALF patients.
B) M65 levels demonstrate similar pattern of differences, with levels highest in AALF and NALF patients, intermediate in MOF and CLD and lowest in HC. Levels of this cell death associated marker were significantly higher in AALF than the non-liver failure control groups (*p<0.05 for comparison with AALF group).
Prognostic utility of circulating levels of M30 and M65
Levels of both M30 and M65 on admission correlated with poor outcome, as defined by death or transplantation. The performance of these assays as prognostic markers was assessed by ROC analysis. Admission M30 proved to have the greatest prognostic accuracy in this cohort with an AUROC of 0.755 (0.639-0.885, p<0.001), sensitivity of 89% and specificity of 61% with a cut-off of 2718. The ‘apoptosis index’ of M30/M65 (AUROC 0.690 (0.553-0.826, p=0.006) sensitivity 56%, specificity 78%, cut-off <2.1) outperformed the M65 assay alone (AUROC 0.569 (0.421-0.718, p=0.359) sensitivity 86%, specificity 42%) (p=0.02 for comparison, DeLong method) (see Figure 2).
Figure 2. Performance of the M30 and M65 serum cell death assays in predicting poor outcome as defined by death or transplantation.
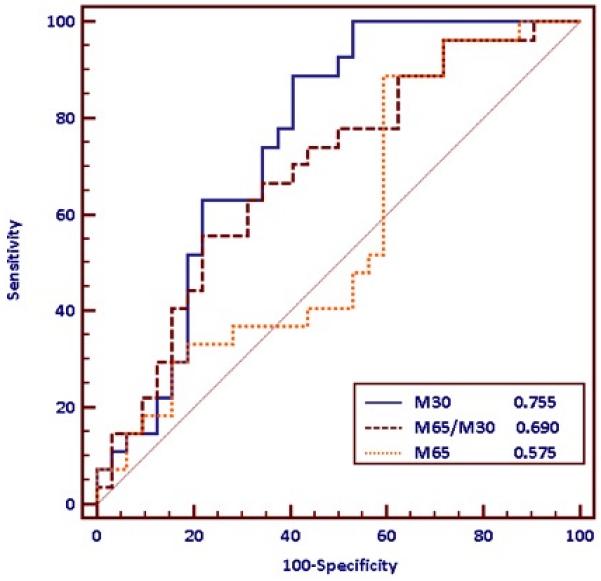
The M30 assay (AUROC 0.755 (0.639-0.885, p<0.001), sensitivity 89%, specificity 61%, cut-off 2718) and ratio of the two markers M30/M65 (AUROC 0.690 (0.553-0.826, p=0.006 ) sensitivity 56%, specificity 78%, cutoff <2.1) outperformed the M65 assay (AUROC 0.569 (0.421-0.718, p=0.359) sensitivity 86%, specificity 42%) in the prediction of which patients would experience poor outcomes based on admission blood samples. (p=0.02 for comparison, DeLong method)
Clinical correlates of hepatic and extra-hepatic organ failure with markers of apoptosis
Neither M30 nor M65 levels correlated with serum AST (Spearman rank correlation 0.091, p=0.650 and −0.020, p=0.917 respectively) or INR (rho=0.269, p=0.193 and rho=-0.164, p=0.412) in this cohort. Bilirubin was negatively correlated with M30 (−0.569, p=0.002) but showed no correlation with M65 (−0.233, p=0.242). Arterial lactate strongly correlated with M30 (0.507, p=0.002) but not M65 (0.050, p=0.799). We found that grade of encephalopathy only trended to correlate with M30 (0.351, p=0.072) and did not correlate with M65 (0.288, p=0.129).
The most consistent set of positive correlations within the AALF cohort was with APACHE II score, (M30-APACHE II, rho =0.524, p=0.012; M65-APACHE II, rho=0.154, p=0.464; M30/M65-APACHE II rho=0.598, p=0.002). Serum creatinine correlated positively with M65; (rho=0.585, p=0.017) but not M30 (rho=0.483, p=0.068) Haemotological indices of sepsis such as white cell count or platelet count did not correlate with total or cleaved CK18.
Temporal changes in M30 over the first 10 days of hospital admission
The levels of total and caspase-cleaved CK18 were greatest on the first hospital day and declined rapidly over the subsequent 10 days (p<0.001 for comparison of day 1 with all other days). At day 7 and 10 M65 and M30 levels were still elevated in comparison with healthy controls (p<0.001) despite resolving liver failure.
Regional levels of cell death markers in late stage AALF
To assess the regional concentrations of M30 and M65 in patients with established ALF, we measured the levels of total and cleaved CK-18 in 7 patients at the time of transplantation. These demonstrated higher levels of M30 in the portal vein (PV) compared to the hepatic vein (HV) (geometric mean ratio of values 0.82 (95%CI 0.67-0.99, p=0.042, paired t-test on log transformed data, Figure 3) with no gradient between HV and systemic arterial levels (geometric mean ratio 1.04 (0.84-1.28), p=0.647). Comparing M65 across the liver showed no significant differences (geometric mean 0.97 (0.64-1.30, p=0.573) (figure 3).
Figure 3. Average levels of M30 and M65 on days 1, 3,7 and 10 of hospital admission.

Peak levels of both markers are seen on the first hospital day before falling significantly. Levels of both markers at day 10 remain elevated above levels seen in healthy control populations.
Protein array and histological analysis of apoptosis in late stage AALF
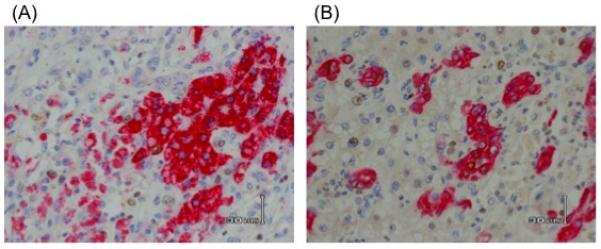
As our data suggested that the hepatic epithelial bed might not be the primary source of total or cleaved cytokeratin-18 by the time of transplantation, we compared a broad panel of proteins associated with apoptosis and tissue repair/regeneration from the explant tissue of 6 patients with AALF and 6 controls. This suggested that catalase (p=0.05) was higher in AALF liver tissue and SMACDiablo (p=0.05), HIF-1α (p=0.05) and cIAP-2 (p=0.02) were lower in AALF than control liver (see Figure 4 A and B). Tissue was examined histologically from 6 explants for AALF. Explants were characterised by evidence of centrilobular necrosis with marked inflammatory infiltrate. Regenerative activity was demonstrated by double-epitope immunohistochemistry showing co-staining of cells for the hepatocyte marker Hep Par-1 and Ki67, a marker of cellular proliferation (see figure 6A). Biliary epithelial proliferation was also demonstrated by co-staining for CK-19 and Ki-67 (figure 6B). A comprehensive assessment of the hepatic microenvironment in late stage AALF has recently been published by us and demonstrates evidence of resolution and regeneration (12). There was an absence of apoptotic bodies or cellular morphology suggestive of apoptosis.
Figure 4. Regional changes in total (M65) or cleaved CK-18 (M30).
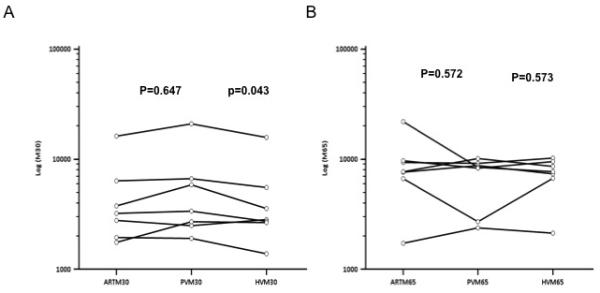
At the time of transplantation for AALF, blood was sampled from the portal vein, hepatic vein and a systemic artery of 7 patients. Levels of M30 and M65 were quantified to determine whether there were regional differences in the concentration of these cell death markers in established AALF.
A) M30 levels show a significant reduction across the liver, between the portal and hepatic veins, with intermediate levels found in systemic arterial blood.
B) M65 levels are not significantly different in the three vascular beds sampled.
Figure 6. Representative sections showing double epitope immunohistochemistry on hepatic explant tissue from AALF showing (A) co-staining for the hepatocyte marker Hep Par-1 and the proliferative marker Ki67 (B) co-staining of biliary epithelial cells with the epithelial marker CK-19 and proliferative marker Ki-67.
Discussion
Here we demonstrate evidence that hepatocellular apoptosis is present during the early phases of human AALF. Our data highlights that during the later stages of AALF, there is evidence of hepatic regenerative/tissue repair processes and an absence of hepatic apoptosis. The persistent elevations in circulating levels of M30 are thus likely to reflect apoptosis in non-hepatic epithelial beds, potentially as a consequence of multiorgan dysfunction.
Our results demonstrate that serum markers of apoptosis and necrosis are markedly elevated on admission to hospital in patients with AALF compared to healthy controls. As the CK-18 based assays are non-organ specific, we included a control group of critically ill patients with matched APACHE II scores in order to determine whether the observed elevation in CK-18 markers was a non-specific phenomenon. We are able to demonstrate that markers of both apoptosis and necrosis in AALF exceed those in critically ill controls, suggesting a hepatocellular origin of a significant proportion of the observed circulating CK18. The finding of hepatocellular apoptosis in human AALF conflicts with evidence from many animal studies which suggest cell death is almost exclusively by necrosis (10, 20). A possible explanation, proposed by Antoine et al (8), is that animal studies are performed on fasted animals in whom ATP is depleted, therefore inhibiting the active process of apoptosis. Evidence from cell culture work and in vivo data in fed animals suggest that if ATP is maintained, hepatocyte apoptosis can be observed in acetaminophen poisoning (7, 8). In clinical practice, the non-fasted status of patients at time of overdose and in-hospital standard care with nutritional support and phosphate supplementation may sustain hepatocellular ATP and permit the execution of apoptosis (21, 22). Further work is necessary to evaluate the impact of phosphate supplementation on hepatocellular death in acute liver injury.
McGill et al in a recent study of clinical AALF have demonstrated a lack of active caspase 3 in patient sera, which lead to the conclusion that apoptosis is not a significant mode of cell death in this condition (10). They commented that their ‘abnormal liver test’ group had low acetaminophen levels as they presented to hospital late. If apoptosis is, as our results suggest, a transient and early process, it is possible that active caspase-3 with a short half-life would not be detected in late presenting patients. The apparent conflict between the active caspase-3 data and the consistent demonstration of caspase-cleaved CK18 in human AALF merits further investigation.
The longitudinal data we present shows a rapid decline in markers of both apoptosis and necrosis after admission to hospital. This suggests the initial peak represents cellular loss as a direct effect of the time-limited acetaminophen toxicity in the initiation phase, rather than an ongoing process. The magnitude of the initial apoptotic injury is a clear correlate of patient outcome following overdose as demonstrated by a strong relationship between admission M30 levels and patient outcome. In terms of biomarker utility, our findings of an association between poor outcome and elevated serum markers of apoptosis and necrosis are consistent with the results of a recently published multicentre study also analysing a large cohort of AALF patients (23). Thus, between these two data sets a consensus is emerging that early elevation in serum cell death markers are predictive of poor outcome in AALF.
In the late stages of AALF, while caspase-cleaved and intact CK-18 concentrations fall, they persist in the circulation at levels exceeding those in healthy controls. Results from regional sampling of blood in patients undergoing OLT, demonstrate peak levels of caspase-cleaved CK-18 in the portal vein, with a decrease in concentration across the liver. This suggests that at this late stage of disease the liver is no longer an important source of cytokeratin fragments, but their origin may be epithelial tissue beds of the splanchnic system.
Established AALF is characterised by activation of systemic inflammatory responses manifested through circulatory dysfunction and immune dysregulation that mirror the physiological changes encountered during septic shock (24, 25). Serum markers of apoptosis and necrosis are also elevated in sepsis and in both human and animal models it has been shown that intestinal epithelial cells are particularly vulnerable to apoptosis (26, 27). This finding has been replicated in a number of non-infectious critical illnesses where marked intestinal apoptosis is observed despite a non-gut primary source of tissue injury (28-30). Experimental manipulation of intestinal apoptosis has been demonstrated to improve survival in animals with pneumonia and caecal ligation and puncture (31, 32). These results suggest gut epithelial apoptosis is not a mere epiphenomenon in critical illness but may influence outcome. Further studies investigating intestinal epithelial apoptosis in established AALF are warranted to build on our observations and test the hypothesis that the gut undergoes apoptosis in AALF in a manner comparable to sepsis and other critical illnesses.
Immunohistochemical and proteomic examination of the explanted livers from AALF patients also provides evidence that hepatocellular apoptosis does not continue in late stage disease. Histological assessment of explanted liver tissue shows no evidence of apoptosis in this late stage of liver failure, but tissue repair and regeneration processes. Results from the apoptosis protein arrays suggest that at the time of transplantation, the liver environment in AALF is enriched in catalase, contains a paucity of factors from the intrinsic mitochondrial apoptosis pathways when compared to healthy controls. Catalase is a common cellular scavenger of hydrogen peroxide and peroxynitrite, whose overexpression has been shown to attenuate apoptosis (33-35).
Conclusion
We provide evidence that significant hepatocellular apoptosis occurs in human AALF. It is an early and transient phenomenon likely to reflect the initiation/propagation phases of AALF and correlates with an adverse outcome. Later, in established AALF there is no evidence of ongoing hepatocellular apoptosis, in fact our findings suggest resolution, with a regenerative liver microenvironment. The persistence of markers of apoptosis in the sera of patients with established AALF is likely to represent apoptosis in non-hepatic tissue, particularly the gut, as has been demonstrated in other conditions characterized by an overwhelming systemic inflammatory response.
Supplementary Material
Figure 5. Univariate analysis (Student’s t test) comparing mean (SEM) was performed on proteins highlighted in the multivariate analysis as the most discriminatory between AALF and control liver.
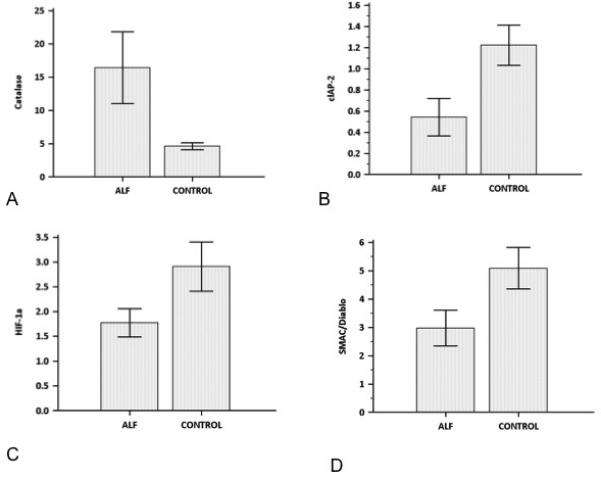
This additional test demonstrated significant differences in normalised concentration (p≤0.05 for all comparisons) between AALF and Control liver for all of the four proteins tested, thus validating the results of the multivariate analysis. A) Catalase B) CIAP-2, D) HIF-1a, D) SMAC/Diablo.
Acknowledgments
Financial Support: We gratefully acknowledge the Imperial National Institute of Health Research Biomedical Research Centre for infrastructure support and the King’s College Hospital Research & Development Department and Institute of Liver Studies Histopathology Department for ongoing support and the Medical Research Council UK.
Dr. McPhail has received grant support from Wellcome Trust. Dr. Abeles received NIHR-Doctoral Research fellowship from Oct 2009-Oct 2010 and the fellowship had a travel allowance; he also received an EASL young investigator travel grant 2011 and 2012. Dr. Leslie hold multiple operating grants. Dr. Ma has received funding from the National Institutes of Health, Wellcome Trust and the Howard Hughes Medical Institute. Dr. Shawcross is an advisory board member at Norgine, has given expert testimony for Talbot Walker LLP and Cooperative Legal Services, has received grant support from HEFCE Clinical Senior Lectureship and Royal Society Grant. She has received payment for lectures from Frontiers in Hepatology and was sponsored to attend the European Association for the Study of the Liver 2011 by Bristol Myers Squibb. Dr. Bernal was a case review consultant for the University of Bordeaux in 2011 and received a speaking fee from the Cardiovascular Society of India in 2011. Dr. Dharwan has consulted for Boherenger Ingelham Cytonet, has received grant support from Astellas, payment for lectures from AASLD and EASL, and receives royalties from Oxford University Press. Dr. Heaton is a consultant for Astellas/Novartis, received payment for lectures and payment for the development of educational presentations from Astellas. Dr. Thursz is a board member for the European Association for Study of the Liver, a consultant for Abbott Pharmaceuticals, has received payment for lectures from Gilead, BMS, and Janssen; he has also received travel reimbursements from Merck. Dr. Wendon has consulted for KASE A Kasai-Asai and is on the medical advisory board for Pulsion and Excalenz. Dr. Antoniades has received grant support from EASL Physician Scientist Fellowship and funding from King’s College Hospital R&D Department.
References
- 1.Lee WM. Acute liver failure in the United States. Seminars in liver disease. 2003;23(3):217–226. doi: 10.1055/s-2003-42641. [DOI] [PubMed] [Google Scholar]
- 2.Bernal W, Auzinger G, Wendon J. Prognostic utility of the bilirubin lactate and etiology score. Clinical gastroenterology and hepatology. 2009;7(2):249. doi: 10.1016/j.cgh.2008.09.009. author reply 249. [DOI] [PubMed] [Google Scholar]
- 3.Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42(6):1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 4.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: A tale of two deaths? Hepatology. 2006;43(S1):S31–S44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 5.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134(6):1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicological sciences. 2006;89(1):31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 7.Kon K, Ikejima K, Okumura K, et al. Role of apoptosis in acetaminophen hepatotoxicity. Journal of Gastroenterology and Hepatology. 2007;22(s1):S49–S52. doi: 10.1111/j.1440-1746.2007.04962.x. [DOI] [PubMed] [Google Scholar]
- 8.Antoine DJ, Williams DP, Kipar A, et al. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Molecular medicine. 2010;16(11-12):479–490. doi: 10.2119/molmed.2010.00126. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Williams CD, Koerner MR, Lampe JN, et al. Mouse strain-dependent caspase activation during acetaminophen hepatotoxicity does not result in apoptosis or modulation of inflammation. Toxicology and applied pharmacology. 2011;257(3):449–458. doi: 10.1016/j.taap.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGill MR, Sharpe MR, Williams CD, et al. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. The Journal of clinical investigation. 2012;122(4):1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antoniades C, Berry P, Wendon J, et al. The importance of immune dysfunction in determining outcome in acute liver failure☆. Journal of Hepatology. 2008;49(5):845–861. doi: 10.1016/j.jhep.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Antoniades CG, Quaglia A, Taams LS, et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. 2012;56(2):735–746. doi: 10.1002/hep.25657. [DOI] [PubMed] [Google Scholar]
- 13.Ku NO, Liao J, Omary MB. Apoptosis generates stable fragments of human type I keratins. J Biol Chem. 1997;272(52):33197–33203. doi: 10.1074/jbc.272.52.33197. [DOI] [PubMed] [Google Scholar]
- 14.Leers MP, Kolgen W, Bjorklund V, et al. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. The Journal of pathology. 1999;187(5):567–572. doi: 10.1002/(SICI)1096-9896(199904)187:5<567::AID-PATH288>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 15.Kramer G, Erdal H, Mertens HJ, et al. Differentiation between cell death modes using measurements of different soluble forms of extracellular cytokeratin 18. Cancer research. 2004;64(5):1751–1756. doi: 10.1158/0008-5472.can-03-2455. [DOI] [PubMed] [Google Scholar]
- 16.O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273–275. doi: 10.1016/0140-6736(93)91818-7. [DOI] [PubMed] [Google Scholar]
- 17.Cummings J, Ranson M, Butt F, et al. Qualification of M30 and M65 ELISAs as surrogate biomarkers of cell death: long term antigen stability in cancer patient plasma. Cancer chemotherapy and pharmacology. 2007;60(6):921–924. doi: 10.1007/s00280-007-0437-4. [DOI] [PubMed] [Google Scholar]
- 18.Schacterle GR, Pollack RL. A simplified method for the quantitative assay of small amounts of protein in biologic material. Analytical biochemistry. 1973;51(2):654–655. doi: 10.1016/0003-2697(73)90523-x. [DOI] [PubMed] [Google Scholar]
- 19.Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measurement with the Folin phenol reagent. The Journal of biological chemistry. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 20.Gujral JS, Knight TR, Farhood A, et al. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 2002;67(2):322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- 21.Polson J, Lee WM. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41(5):1179–1197. doi: 10.1002/hep.20703. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology. 2002;36(3):659–665. doi: 10.1053/jhep.2002.35069. [DOI] [PubMed] [Google Scholar]
- 23.Antoine DJ, Jenkins RE, Dear JW, et al. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. Journal of hepatology. 2012 doi: 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Rolando N, Wade J, Davalos M, et al. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32(4 Pt 1):734–739. doi: 10.1053/jhep.2000.17687. [DOI] [PubMed] [Google Scholar]
- 25.van der Poll T, Meijers JC. Systemic inflammatory response syndrome and compensatory anti-inflammatory response syndrome in sepsis. Journal of innate immunity. 2010;2(5):379–380. doi: 10.1159/000318190. [DOI] [PubMed] [Google Scholar]
- 26.Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Critical care medicine. 1999;27(7):1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the "motor" of critical illness. Shock. 2007;28(4):384–393. doi: 10.1097/shk.0b013e31805569df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sileri P, Morini S, Sica GS, et al. Bacterial translocation and intestinal morphological findings in jaundiced rats. Digestive diseases and sciences. 2002;47(4):929–934. doi: 10.1023/a:1014733226337. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Wang B, Wu K, et al. Growth hormone downregulated the excessive apoptosis of ileal intestinal epithelial cells in rats during the early course of acute necrotizing pancreatitis. Pancreas. 2002;25(2):205–209. doi: 10.1097/00006676-200208000-00016. [DOI] [PubMed] [Google Scholar]
- 30.Wolf SE, Ikeda H, Matin S, et al. Cutaneous burn increases apoptosis in the gut epithelium of mice. Journal of the American College of Surgeons. 1999;188(1):10–16. doi: 10.1016/s1072-7515(98)00260-9. [DOI] [PubMed] [Google Scholar]
- 31.Coopersmith CM, Chang KC, Swanson PE, et al. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Critical care medicine. 2002;30(1):195–201. doi: 10.1097/00003246-200201000-00028. [DOI] [PubMed] [Google Scholar]
- 32.Coopersmith CM, Stromberg PE, Dunne WM, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287(13):1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- 33.Brezniceanu ML, Liu F, Wei CC, et al. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney international. 2007;71(9):912–923. doi: 10.1038/sj.ki.5002188. [DOI] [PubMed] [Google Scholar]
- 34.Heinzelmann S, Bauer G. Multiple protective functions of catalase against intercellular apoptosis-inducing ROS signaling of human tumor cells. Biological chemistry. 2010;391(6):675–693. doi: 10.1515/BC.2010.068. [DOI] [PubMed] [Google Scholar]
- 35.Yoshioka Y, Kitao T, Kishino T, et al. Nitric oxide protects macrophages from hydrogen peroxide-induced apoptosis by inducing the formation of catalase. Journal of immunology. 2006;176(8):4675–4681. doi: 10.4049/jimmunol.176.8.4675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.