

SUMMARY
We measured half-lives of 21,248 mRNA 3′ isoforms in yeast by rapidly depleting RNA polymerase II from the nucleus and performing direct RNA sequencing throughout the decay process. Interestingly, half-lives of mRNA isoforms from the same gene, including nearly identical isoforms, often vary widely. Based on clusters of isoforms with different half-lives, we identify hundreds of sequences conferring stabilization or destabilization upon mRNAs terminating downstream. One class of stabilizing element is a polyU sequence that can interact with poly(A) tails, inhibit the association of poly(A)-binding protein, and confer increased stability upon introduction into ectopic transcripts. More generally, destabilizing and stabilizing elements are linked to the propensity of the poly(A) tail to engage in double-stranded structures. Isoforms engineered to fold into 3′ stem-loop structures not involving the poly(A) tail exhibit even longer half-lives. We suggest that double-stranded structures at 3′ ends are a major determinant of mRNA stability.
INTRODUCTION
mRNA levels are regulated at the levels of synthesis, posttranscriptional processing, and degradation. In S. cerevisiae, genome-wide measurements of mRNA stability indicate that mRNA half-lives can range over more than two orders of magnitude (Holstege et al., 1998; Wang et al., 2002). Stabilities of individual transcripts as well as functionally related groups of mRNAs can be regulated in response to environmental conditions such as rapid shifts in carbon sources and cellular stress (Vasudevan and Peltz, 2001; Munchel et al., 2011). Coordinated stability control of functionally related categories of transcripts is a mechanism that helps yeast cells adapt to rapidly changing environmental conditions (Cacace et al., 2012).
Most yeast mRNAs are degraded via two evolutionarily conserved pathways (Parker, 2012). The first involves 5′ to 3′ exonucleolytic digestion and is mediated by Xrn1 in the cytoplasm and/or its paralog Rat1 in the nucleus. Alternatively, mRNAs in either compartment can be degraded in the 3′ to 5′ direction by the multisubunit exosome complex. In both instances, mRNA decay is initiated via dissociation of Pab1, the major poly(A)-binding protein in yeast, and cytoplasmic deadenylation of poly(A) tails, predominantly by the Ccr4-Pop2-Not complex and (to a lesser extent) by the poly(A)-specific nuclease Pan2-Pan3. Deadenylated transcripts can then be directly degraded by the exosome or, following the removal of the 5′ cap, by either Xrn1 or Rat1.
Transcript stability can be regulated by sequence-specific RNA-binding proteins, microRNAs (miRNAs), and by RNA secondary structure. For example, proteins binding to AU-rich elements modulate the stabilities of mRNAs that encode proteins involved in oncogenic transformation and stress (Hitti and Khabar, 2012). In a related mechanism, miRNAs interact directly with specific sequences in 3′ UTRs via base-pairing and induce destruction of the targeted mRNAs (Eulalio et al., 2009). Secondary structure in some viral RNAs is associated with increased stability, possibly by impeding the digestion of poly(A) tails by deadenylases (Brown et al., 2012). Triple helix formation involving the poly(A) tail and other RNA elements in viral genes is associated with increased stability (Mitton-Fry et al., 2010; Brown et al., 2012; Tycowski et al., 2012).
mRNA stability is typically assayed by blocking transcriptional initiation and measuring the RNA levels remaining at various times after the block. In yeast, the use of a temperature-sensitive RNA polymerase (Pol) II (mutated Rpb1 subunit) has permitted half-life measurements of the majority of genes. However, this approach is problematic because half-lives are measured in cells subjected to a heat shock and then incubated at a nonoptimal temperature. Drugs that disrupt Pol II-driven transcription also affect stress response and other biological pathways. Half-lives have also been calculated from direct measurements of mRNA synthesis upon a pulse of the metabolic precursor 4-thio-uracil and estimates of absolute mRNA levels (Miller et al., 2011). Half-lives determined by these methods were in modest or poor agreement overall and at the level of individual genes.
Most genes in yeasts and higher eukaryotes give rise to an extraordinarily large number of different mRNA isoforms (Moqtaderi et al., 2013; Pelechano et al., 2013). In yeast, this extraordinary heterogeneity is mediated through extensive variation of alternative 5′ and 3′ ends, whose utilization is largely independent (Pelechano et al., 2013). The numerous alternative 3′ ends (i.e., cleavage and polyadenylation sites) are present in varying abundance, with major species rarely making up more than half of all transcripts in any given gene (Moqtaderi et al., 2013).
Previous work has also shown that genes with multiple mRNA isoforms sometimes exhibit different half-lives depending on environmental conditions (Mayr and Bartel, 2009; Ulitsky et al., 2012). In a few instances, these stability differences mapped to 3′ end elements, some of which were demonstrated to be targets of stability-influencing RNA-binding proteins (Allen et al., 2013). However, the extent to which mRNA isoform stability plays a role in the overall level of nuclear gene expression regulation remains unknown. Here, we combine conditional depletion of Pol II and direct RNA sequencing (DRS) (Ozsolak et al., 2009) to measure mRNA isoform stability on a genome-wide scale in S. cerevisiae. We identify mRNA stabilizing and destabilizing elements on a genome-wide scale and provide evidence for a new role of the poly(A) tail and secondary structure in mRNA stability.
RESULTS
Half-Life Measurements Using the “Anchor-Away” Approach
To measure mRNA isoform half-lives in yeast, we used the “anchor-away” approach (Haruki et al., 2008) to perform conditional depletion of Rpb1, the largest Pol II subunit, from the nucleus (Fan et al., 2010). Rapamycin addition causes rapid export of the desired protein (tagged with FRB) from the nucleus into the cytoplasm, where it is stably anchored via FKBP-Rpl13A. In the absence of rapamycin, the strain expressing the Rbp1-FRB fusion has no apparent growth defect when compared to the parental strain expressing wild-type (WT) Rpb1. Addition of rapamycin causes a rapid drop in Pol II occupancy at coding regions, typically with <15% of the initial occupancy level after 20 min (Figure 1A). RNA isolated from cells prior to and at 20 min intervals after rapamycin treatment was subjected to DRS (Ozsolak et al., 2009). Half-lives of mRNAs were calculated by a nonlinear least-squares model, taking into account the limited transcription that occurs shortly after rapamycin addition.
Figure 1. Half-Lives of Yeast mRNAs.
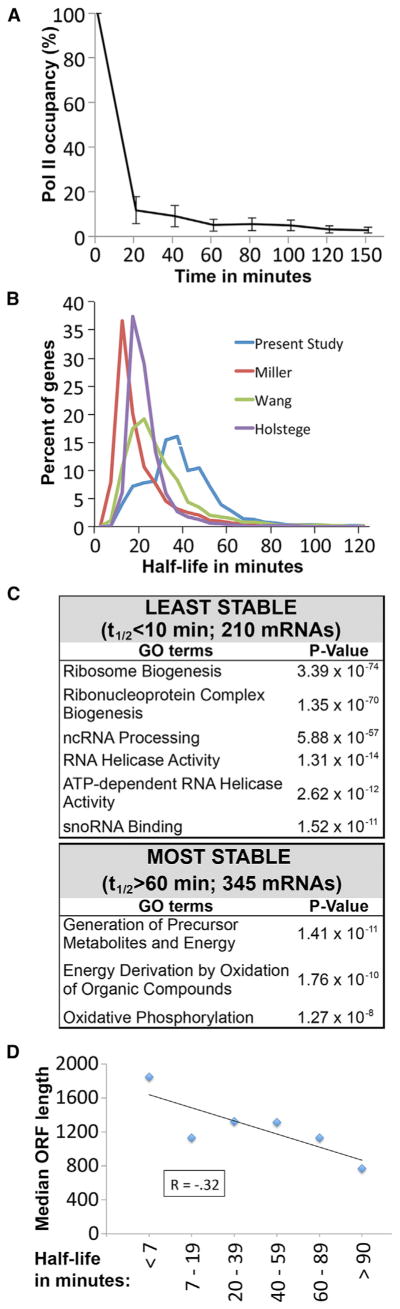
(A) Average Pol II occupancy (mean ± SEM) at 35 genes during the time course (minutes after rapamycin addition) as determined by ChIP.
(B) Distribution of the mRNA half-lives in the present study compared to distributions from three previous studies.
(C) GO categories of genes with unusually short or long half-lives.
(D) Half-life is inversely correlated with ORF length. Stabilities for the 1 kb group are shorter than predicted, most likely due to the many short-lived ribosomal protein genes.
Because yeast mRNAs display a remarkable degree of 3′ end heterogeneity (Wu et al., 2011; Moqtaderi et al., 2013; Pelechano et al., 2013), we grouped reads from all 3′ isoforms of each gene and calculated mRNA half-lives for 4,947 (85%) protein-coding genes (Table S1 available online). The distribution of mRNA half-lives, calculated from two independent experiments, is shown in Figure 1B. The median half-life is 32 min, which is longer than observed previously using different methods (Holstege et al., 1998; Wang et al., 2002; Grigull et al., 2004; Miller et al., 2011). Gene-by-gene comparisons of the various half-life measurements reveal a modest correlation (Figure S1), presumably due to differences in strain backgrounds, growth conditions, and experimental methods. In this regard, we note that the anchor-away approach does not involve switching cells to stress conditions (unlike temperature shifts), and it permits half-life measurements under virtually any environmental condition.
Although the vast majority of yeast mRNAs are relatively short lived compared to the generation time of the cells, 47 genes (1%) give rise to very stable transcripts (half-lives >2 hr). Gene ontology (GO) analysis reveals that genes with mRNA half-lives ≥60 min function primarily in oxidative phosphorylation. In contrast, genes giving rise to mRNAs with the shortest half-lives (<10 min; 210 genes) are generally involved in ribosome biogenesis, mRNA processing, and related aspects of mRNA and rRNA metabolism (Figure 1C). Of particular interest, 20% (10 of 50) of the most unstable mRNAs encode DEAD/DEAH-containing mRNA helicases, many of which are involved in rapid adaptation to external stimuli.
mRNA Stability Is Correlated with Transcript Length
In mammalian cells and bacteria, transcript length positively correlates with turnover (Feng and Niu, 2007). Although previous genome-wide mRNA analyses in S. cerevisiae failed to demonstrate any link between transcript length and half-life (Wang et al., 2002; Feng and Niu, 2007), we observe a negative correlation between transcript length and stability in yeast (Spearman coefficient of −0.32; p = 3.91 × 10−6). Genes expressing the shortest-lived mRNAs are on average more than twice as long as those expressing the most stable mRNAs (Figure 1D). Taken together with the genome-wide correlation analysis, this indicates that yeast mRNAs follow a similar length-dependent instability pattern as observed with transcripts of other species.
Very Similar Isoforms Can Have Different Half-Lives
By combining half-life measurements with genome-scale mapping of polyadenylation sites, it is possible to determine the half-lives of multiple isoforms from the same gene. We computed half-lives for 21,248 isoforms having a minimum of 100 sequence reads at steady state (Table S2). Strikingly, individual isoforms originating from the same genes often show remarkable variation in half-lives. Even closely spaced (separated by <3 bp at 3′ ends) isoforms, 95% of which have similar half-lives, can exhibit considerable variation in half-lives. We identified 519 examples of such near-identical transcripts whose half-lives varied by ≥2-fold, including 259 examples of adjacently terminating transcripts. Among neighboring isoform pairs with different half-lives, the upstream transcript is more stable in 28% of the cases, and the downstream transcript is more stable in 72%. Importantly, because half-life determinations involve the analysis of multiple, internally controlled samples, these examples reflect physiological reality, not experimental artifact.
To address how a single 3′ nucleotide (nt) can influence mRNA stability, we examined nt distributions around pairs of mRNA iso-forms arising from adjacent polyadenylation sites. When all mRNA isoforms are considered, regardless of spacing, the final residue before the poly(A) tail is distributed in keeping with the general nt composition of 3′ UTRs (Figure 2A). For all pairs of iso-forms arising from adjacent polyadenylation sites a and b, the final residue of isoform b is distributed similarly (Figure 2B, left). For adjacent site pairs in which b is at least 2-fold destabilized relative to a, the nt frequencies at the b position are statistically indistinguishable from those in the overall population of adjacent pairs (p = 0.24; Figure 2B, second group from the left). Strikingly, when the isoform b is >2-fold stabilized, there is a strong overrepresentation of U residues at the b position (p = 6.4 × 10−20) (Figure 2B, third group from the left). If we increase the stabilization cutoff to ≥3-fold, >90% of the b transcripts terminate with a U and none with a G (p = 3.3 × 10−6) (Figure 2B, rightmost group). Thus, for a small subset of adjacent transcripts, termination in a U results in increased stability.
Figure 2. Increased Prevalence of U Residues at the Termini of Stabilized Isoforms Arising from Closely Spaced Poly(A) Sites.

(A) Control distribution of residues at the 3′-most position before the poly(A) tail in the overall population of mRNA isoforms.
(B) Distributions of the 3′-most residue of the b isoform in adjacent a,b isoform pairs ending 1 nt apart for all b isoforms, and for isoform pairs where b is more or less stable than a.
(C) Frequency distributions of residues at the last five positions of closely spaced isoforms. Sequence logos represent the distribution of the 3′-most residues in b isoforms terminating 1, 2, or 3 nt downstream of a isoforms. Positions with arrows have statistically significant differences in distribution relative to all isoforms of equal spacing. p values were calculated with a chi-square test.
See also Figure S2.
A similar analysis with isoform pairs in which the downstream b isoform terminates at position a +2 or a +3 (Figure 2C) also reveals a strong enrichment of U residues at the a +1 position in cases where b isoform is more stable than a isoform. In addition, there is also a milder enrichment for a U residue at the a position and a slight enrichment for a U (and to a lesser extent C) residue at the b position. The identification of U-enriched sequences at the relatively stabilized termini is consistent with the observed over-representation of U at the 3′-most position of stabilized transcripts. Interestingly, an AA dinucleotide adjacent to the 3′ end is associated with shorter half-life (Figure S2).
Distinct Clusters within an Individual Gene Often Have Different Half-Lives: Genome-wide Identification of mRNA Stabilizing and Destabilizing Elements
More than one-quarter of all genes (826 out of 3,098) with two or more significant isoforms and most genes that contain six or more isoforms display significant half-life heterogeneity (Figures 3 and S3; Table S3). Although individual isoforms within genes can vary tremendously with respect to half-lives, many genes have “clusters” of isoforms with endpoints that extend over a range of neighboring nts and possess similar half-lives. We arbitrarily define a cluster as a group of isoforms occurring over no more than a 30 nt window, with a maximum gap of 10 nt between consecutive members, each possessing a half-life differing by no more than 2-fold from that of any other member. A total of 2,286 clusters consist of at least two isoforms, and 586 clusters consist of three or more isoforms. Because we restricted our analysis to significant isoforms possessing a minimum of 100 sequence reads in steady state, clusters typically contain additional, below-threshold isoforms with similar half-lives. Analysis of 4,033 clusters (including neighboring unclustered single isoforms) reveals that most pairs of consecutive clusters (2,622 or 65%) have similar (within 1.5-fold) half-lives (Table S4), meaning that the intervening sequences between these clusters are neutral with regard to stability.
Figure 3. Variability in mRNA Isoform Half-Lives Is Typical for Many Genes.

Isoforms for a representative subset of genes with at least ten significant mRNA 3′ isoforms are shown as squares from left to right in the order found in the 3′ UTR, with half-lives in minutes shown and also indicated by color. Genes in blue encode 3′ mRNA isoforms varying over more than a 2-fold range from shortest to longest half-life. See also Figure S3 and Tables S2 and S3.
Interestingly, we identified several hundred genes in which different clusters exhibit significant differences in isoform half-lives (Figures 4 and S4). For example, isoforms in the upstream cluster of FCP1 exhibit far greater instability than isoforms belonging to the downstream cluster (Figure 4B). Conversely, isoforms in the upstream cluster of SCW4 are more stable than isoforms in the downstream cluster (Figure 4C). Some genes (e.g., ARG8) contain multiple clusters with wide-ranging half-lives, whereas others (e.g., UTP14) possess distinct clusters with similar half-lives (Figures 4A and 4D). Overall, we identified 560 pairs of consecutive clusters in which the isoforms in the downstream cluster are more stable and 851 examples in which the isoforms in the downstream cluster are less stable (Table S4).
Figure 4. Identifying mRNA Stabilizing and Destabilizing Elements by Half-Life Differences of Adjacent Isoform Clusters.

For each gene, the left panel shows the isoform pattern in steady state, and the right panel shows the pattern after Pol II depletion for the indicated time. The average half-life of each cluster is indicated with a box, and stabilizing (S) and destabilizing (D) elements are marked with lines. All isoform positions are shown relative to the 3′ end of the ORF. See also Figure S4 and Table S4.
Distinct isoform clusters with different half-lives are most simply explained by the existence of mRNA stability elements located between the clusters. Stabilizing elements are defined as increasing the stability of the downstream cluster relative to the upstream cluster, and destabilizing elements are defined as decreasing the stability of the downstream cluster. Thus, our genome-scale analysis identifies 560 stabilizing elements and 851 destabilizing elements. Importantly, unlike the few stabilizing or destabilizing elements described previously that rely on the analysis of mutated derivatives, the large number of elements described here is defined in the context of WT genes.
A PolyU Motif Is an mRNA Stabilizing Element
Using MEME (Bailey and Elkan, 1994), we identified a 20 nt polyU tract in ~10% of the sequences associated with enhanced downstream transcript stability (Figure 5A) but did not find any sequence motifs associated with mRNA destabilizing elements. Individual polyU tracts may contain occasional substitutions, much like poly(dA:dT) promoter elements that inhibit nucleosome formation (Struhl, 1985; Iyer and Struhl, 1995; Raveh-Sadka et al., 2012). Unlike the very short, U-rich stretch present at or near the immediate 3′ ends of some stabilized transcripts (see above and Figure 2), this stability-enhancing polyU motif was present at various locations with respect to the 3′ ends, with a median distance of 20 nt upstream of transcript 3′ ends (Figure 5A). This polyU stability-enhancing element is clearly different in both composition and length from the 10 nt UK(U)8 motifs implicated in zebrafish transcript stabilization and destabilization (Ulitsky et al., 2012). It also differs from the much longer (60–90 nt) herpesvirus family ENE fold that stabilizes viral RNA transcripts via formation of a novel clamp-like structure (Brown et al., 2012).
Figure 5. A PolyU Element Is Present at Stabilizing Sequences and Can Confer Stabilization upon Other Loci.

(A) Sequences of individual stabilizing elements containing the polyU motif with the consensus motif shown below.
(B) Introduction of the polyU element at five out of six neutral inter-cluster sequences stabilizes the downstream isoform cluster. For each gene, the upstream cluster (black lines) is unaffected by the polyU substitution (black square labels) compared to WT (black triangular labels). In all cases except KSH1, the downstream cluster (red lines) is stabilized by the polyU substitution (red square labels) compared to WT (red triangular labels). The decay curves are plotted as lines on a log2 scale with error bars representing the ranges of individual data points. y axis labels show the relative abundance (2y).
(C) The propensity of poly(A) tail sequences to participate in double-stranded structures is related to stabilization. Predicted relative double strandedness of the poly(A) tail of the downstream isoform b is shown as compared to the upstream isoform a (higher in b indicated by black; lower in b indicated by gray) for isoform pairs flanking the 40 most stabilizing (top 1%) intervening sequences, the 40 most destabilizing intervening sequences, or 100 neutral elements.
(D) Predicted double strandedness of the poly(A) tail for pairs of adjacent isoforms exhibiting the greatest half-life differences. p values were calculated with a Fisher’s exact test.
See also Table S6.
The PolyU Element Stabilizes mRNA Transcripts
To test whether the polyU element is an autonomous stabilizing element, we transplanted it into different genes between consecutive clusters with similar average isoform half-lives. In the resulting derivatives, the spacing between the upstream and downstream clusters is not altered, and the number of nt substitutions ranges from 4 to 16. In five of six cases tested by isoform-specific quantitative RT-PCR, the polyU element substantially increases (1.5- to 2.5-fold; average 2-fold) the stability of isoforms terminating in the downstream cluster (Figure 5B). The half-lives of the upstream clusters, as expected, remained unaffected by the substitutions. In the already U-rich APT2 3′ UTR, alteration of only 4 nt to generate a polyU element causes a clear increase in the stabilities of the mRNAs terminating within the downstream cluster (compare red lines for WT and mutant downstream clusters in Figure 5B). Thus, even relatively small changes in 3′ UTR sequences can have great bearing on transcript turnover.
Ability of the Poly(A) Tail to Form Secondary Structures Is Linked to Stabilizing and Destabilizing Elements
The observation that the polyU mRNA stability motif occurs at various distances upstream of cleavage sites suggests that it might function via the formation of secondary structures with poly(A) tails common to mRNAs. In nearly every instance analyzed, the presence of a U-rich stabilizing element is, not surprisingly, predicted to form a very stable stem-loop structure with poly(A) tails (mean ΔG, −12.7 kcal/mol). Similarly, structural modeling of mRNA isoforms from genes containing the transplanted polyU element predicts the formation of stable stem-loops of at least 20 nt at 3′ ends (Table S5).
To assess the role of the poly(A) tail more generally, we compared the predicted folding of downstream mRNA isoforms from three groups: the 40 most stabilizing elements, the 40 most destabilizing elements, and 100 inter-cluster sequences with no effect on isoform stability (Figure 5C). For every mRNA in a group, we compared the poly(A) tail folding of an isoform b terminating downstream (containing the element) and an isoform a terminating upstream (lacking the element). The poly(A) tails of isoforms containing the best stabilizing elements were predicted to be much more likely to fold into stem-loops and other double-stranded structures relative to those of the corresponding upstream-terminating isoforms (Figure 5C). Conversely, the group containing the best destabilizing elements in 3′ UTRs was predicted to have a significantly reduced propensity to fold into U-A stem-loops with the poly(A) tail. These highly significant (p = 4.4 × 10−6), diametrically opposed effects of stabilizing and destabilizing elements on poly(A) tail structure strongly suggest that for a substantial proportion of yeast mRNA isoforms, formation or disruption of poly(A) secondary structure, presumably through U-rich tracts, is an important determinant of stability.
Given the apparent role of the poly(A) tail in mRNA stability and our observation that terminal U residues can affect the stability of near-identical isoforms, we performed a similar structural analysis on isoforms derived from adjacent polyadenylation sites with different stabilities. We analyzed the poly(A) double strandedness of the 40 most stabilized and the 40 most destabilized b isoforms in pairwise comparisons with their 1 nt shorter a counterparts (Figure 5D). The results are in accord with those observed for stabilizing and destabilizing elements (Figure 5C). b isoforms belonging to the most stabilized group are four times more likely to exhibit greater double strandedness than the corresponding a isoforms. Conversely, for the most destabilized b isoforms, it is the a isoform that has a 3-fold higher likelihood of possessing greater double-stranded character (Figure 5D). Thus, even for near-identical isoforms, secondary structure involving the poly(A) tail appears to play an important role in mRNA stability.
An Engineered Stem-Loop Structure in the 3′ UTR Increases mRNA Stability
Because mRNA stability mediated by the polyU element appears to reflect its ability to form stem-loops with the poly(A) tail, we hypothesized that other stabilizing elements might also function by forming stable hairpin or other secondary structures involving 3′ UTRs near mRNA cleavage sites. To test whether secondary structures that exclude poly(A) sequences can confer increased isoform stability, we introduced stable, GC-rich stem-loop structures into two inter-cluster regions whose bounding clusters exhibit similar isoform half-lives. The substitutions in these two unrelated loci are expected to alter the folding of the downstream cluster isoforms by forming stem-loop elements that are predicted to be even more stable than those formed by the polyU elements/poly(A) tail (Table S5). The presence of these GC-rich stem-loops results in an even more dramatic stabilization of the downstream cluster transcripts than that observed for the polyU elements, without affecting the stability of upstream isoforms (Figure 6). These results strongly suggest that a highly stable secondary structure located near isoform 3′ ends, of which a poly(A) secondary structure is but one example, may function as an important determinant of increased mRNA isoform stability.
Figure 6. Introduction of a Stem-Loop-Forming G-C-Rich Element into Neutral Inter-Cluster Sequences Strongly Stabilizes the Downstream Isoform Cluster.

Upstream and downstream cluster decay curves for HXT2 and RPA43 derivatives containing WT sequences (blue triangles), polyU elements (maroon squares), or G-C elements (green circles) are plotted as lines on a log2 scale, with error bars representing the ranges of individual data points. y axis labels show the relative abundance (2y). See also Table S6.
PolyU-Containing Isoforms Exhibit Reduced Pab1, Ski2, and Xrn1 Binding
PolyU-Poly(A) stem-loop formation should decrease single-stranded poly(A) sequences accessible for binding to Pab1, the major poly(A)-binding protein in yeast (Parker 2012). Pab1 binding to poly(A) tails in vivo requires a minimum stretch of ~10–12 consecutive A residues (Sachs et al., 1987) and is highly cooperative. Pab1 binding is implicated in the inhibition of poly(A) tail deadenylation by the Ccr4-Pop2 complex that eventually results in the exonucleolytic degradation of mRNAs by either the exosome or the decapping/Xrn1 complexes (Simón and Séraphin, 2007; Zhang et al., 2013).
Using data from genome-wide mapping of mRNA-binding proteins (Tuck and Tollervey, 2013), we analyzed the binding of Pab1 and other components of the RNA degradation machinery to transcripts derived from natural polyU-containing loci. In nearly all cases, polyU-containing isoforms from downstream clusters exhibit dramatically lower levels of Pab1 binding compared to their upstream counterparts that lack this element (Figures 7A and 7B). Similarly skewed binding preferences to upstream clusters are also observed for Ski2 and Xrn1, two proteins that play important roles in controlling rates of cytoplasmic mRNA turnover (Figures 7A and 7B) (Parker, 2012). In contrast, Pab1, Ski2, and Xrn1 exhibit comparable binding patterns to upstream and downstream clusters at loci lacking polyU elements (Figure 7B). Strong binding preferences of Pab1/Ski2/Xrn1 for upstream clusters in polyU-containing loci are not due to nonspecific decreases in downstream cluster binding (or nonspecific increases in upstream cluster association) because both the mRNA cleavage and processing factor Hrp1 and the Mtr4 helicase (a presumptive nuclear homolog of Ski2; Halbach et al., 2013) exhibit comparable binding to upstream and downstream mRNA cluster isoforms of polyU-containing loci (Figure 7B). The lower Pab1 binding to polyU-containing isoforms strongly supports the hypothesis that polyU elements form stem-loop structures with poly(A) tails in vivo.
Figure 7. Reduced Pab1, Ski2, and Xrn1 Binding to PolyU-Containing Isoforms.

(A) Representative genomic loci exhibiting reduced Pab1, Ski2, and Xrn1 binding to polyU element-containing mRNA isoforms.
(B) Genome-wide binding of Pab1, Ski2, Xrn1, and Hrp1 to mRNA isoforms in upstream and downstream clusters. All inter-cluster elements ≥30 nt were subdivided into two groups: one containing the previously identified native polyU sequences, and one containing all other elements. The median expression-corrected factor occupancy at upstream (blue bars) and downstream (red bars) clusters was plotted for each group. p values at the bottom of the panel indicate the statistical significance in the differences of the upstream/downstream cluster-binding ratios between the two groups and were calculated by the Mann-Whitney U test.
(C) Pab1 binding to upstream and downstream clusters at WT and transplanted loci. Pab1 occupancy at upstream and downstream mRNA clusters spanning WT loci (HXT2 and RPA43; left), polyU-containing WT (FCP1) or transplanted (HXT2 and RPA43; second from left), GC-rich transplanted (HXT2 and RPA43; second from right), or coding sequence control (PMA1; right) was measured by RIP. Plotted values (mean ± SEM) represent the averages of three independent experiments.
Transplanted PolyU and GC Elements Cause Reduced Pab1 Binding
Because transplantation of polyU and GC-rich stem-loop elements into ectopic loci results in greater stability of downstream cluster mRNA isoforms (Figure 5), we addressed whether these newly stabilized isoforms would also exhibit reduced Pab1 binding. Specifically, by combining RNA immunoprecipitation (RIP) and isoform-specific RT-qPCR, we assayed Pab1 binding to upstream and downstream clusters of alleles containing transplanted elements or their WT counterparts (Figure 7C). As expected, Pab1 occupancy is nearly identical at upstream and downstream clusters of WT HXT2 and RPA43 alleles (Figure 7C, leftmost group), ranging between ~5- and 10-fold over background (defined by an internal fragment within the ~3 kb coding region of the PMA1 gene that is predicted to have no Pab1 binding; Tuck and Tollervey, 2013). By contrast, Pab1 binding is several-fold lower in downstream clusters of the HXT2 and RPA43 derivatives containing transplanted polyU elements, an effect similar in magnitude to that observed at FCP1, which contains a naturally occurring polyU stabilizing element (Figure 7C, second group from left). Transplantation of GC structures into the HXT2 and RPA43 loci also results in a drop in downstream cluster poly(A) binding (Figure 7C, third group from left). Thus, transplanted polyU and GC stabilizing elements are sufficient to cause reduced Pab1 association to downstream isoforms that show increased stability in vivo.
DISCUSSION
A New Method for Measuring mRNA Half-Lives in Yeast and Relationship to Previous Measurements
We describe a new, genome-scale method for measuring half-lives in yeast cells. The anchor-away approach directly blocks transcription by removing an essential subunit of Pol II from the nucleus via an experimental manipulation that has little effect on cell growth in the strain background used, and it permits half-life measurements under essentially any physiological condition. In contrast, half-life measurements using a temperature-sensitive Pol II (Rpb1-1 derivative) are restricted to conditions of elevated temperature and involve a heat shock that affects cell physiology. Furthermore, the Rpb1-1 derivative is a mutated version of the WT protein, strains harboring the rpb1-1 allele exhibit a growth defect even at the permissive temperature, and the mechanistic basis of the transcriptional defect of the Rpb1-1 derivative is unknown. Methods using small molecule inhibitors of transcription also suffer from nonspecific effects on cell physiology and less direct and poorly understood mechanisms of transcriptional inhibition. The anchor-away approach represents a direct measurement of RNA half-lives, in contrast to methods that calculate half-lives from measurements of synthesis rates and estimates of mRNA abundance. Nevertheless, it should be noted that all methods for measuring mRNA half-lives are affected by experimental perturbations and analytical assumptions, and mRNA decay rates can change upon global inhibition of transcription (Haimovich et al., 2013; Sun et al., 2013).
The median half-life in our experiments is 32 min, which is considerably longer than the 11–23 min half-lives measured previously by Holstege et al. (1998), Wang et al. (2002), Grigull et al. (2004), and Miller et al. (2011). When assayed on a gene-by-gene basis, our half-life measurements are in modest agreement with results obtained with the Rpb1-1 approach (R = 0.31–0.56), but not with results obtained via RNA synthesis measurements. Perhaps these differences are due to the nonelevated temperature (as compared to the Rpb1-1 approach) or to the uncertainties of absolute mRNA levels (used in context of RNA synthesis measurements). Differences in growth media and genetic backgrounds might also play a role.
3′ Terminal U Residues Can Affect mRNA Isoform Half-Lives
The prevalence of heterogeneity in mRNA isoform half-lives strongly suggests that isoform stability plays an important role in the overall scheme of regulation of gene expression. There are two distinct types of half-life variability of mRNA isoforms of the same gene. First, as will be discussed below, distinct clusters of mRNA isoforms within a gene can have different half-lives. Second, and very unexpectedly, mRNA isoforms whose 3′ endpoints differ by one or a few nts can also have quite different half-lives. When the mRNA isoforms differ by a single nt and the downstream isoform is stabilized, the 3′ terminal base is typically a U. When the 3′ endpoints of the mRNA isoforms differ by 2–3 nt, U residues near but not exactly at the 3′ end are also associated with longer half-lives.
Although U residues at or near the 3′ end can stabilize mRNAs, the vast majority of near-identical isoforms with U residues at or near the 3′ end have similar half-lives. In addition, the subset of U-ending isoforms that are stabilized does not possess a common sequence motif, strongly suggesting that the increased stability is not a consequence of the creation of a recognition sequence for an RNA-binding factor. Instead, it seems more likely that the stabilizing effects of the 3′ terminal U residues occur only in the context of other features such as secondary structure (see below). Finally, our results suggest that the previously observed U-rich sequences near poly(A) cleavage sites (Zaret and Sherman, 1982; Zhao et al., 1999; Mandel et al., 2008; Moqtaderi et al., 2013) may reflect relative stability of the RNA isoforms in addition to favorable locations for cleavage per se.
Genome-wide Mapping of mRNA Stabilizing and Destabilizing Elements
By analogy with other gene regulatory elements, mRNA stabilizing and destabilizing elements are classically defined by comparing related mRNA derivatives that have different half-lives and either do or do not contain the element. Thus, the numerous examples of physically separate isoform clusters within a single gene that have different half-lives represent a genome-wide identification of mRNA stabilizing and destabilizing elements. Most simply, these elements map to RNA sequences located somewhere between the cluster endpoints, although adjacent sequences located within the cluster may also play a role in determining mRNA half-lives.
mRNA stabilizing and destabilizing elements could be recognized by specific RNA-binding proteins, by RNA sequences within the transcript itself, or by a separate RNA species. In many eukaryotic organisms, miRNAs and other small RNA species interact with 3′ UTRs of mRNAs, but these do not appear to exist in S. cerevisiae. Alternatively, if some of these elements are recognized by specific RNA-binding proteins, one would expect to observe overrepresented sequence motifs in the set of either stabilizing or destabilizing sequences, as compared to control regions that lie between mRNA isoform clusters with similar half-lives. However, with the exception of the polyU stabilizing element (see below), we were unable to find any sequence motif for stabilizing or destabilizing elements. Although hardly conclusive, this finding argues against the idea that these elements are recognized by specific RNA-binding proteins, which in turn implies that RNA-binding proteins may not play a predominant role in differential isoform stability.
A PolyU Motif Acts as an mRNA Stabilizing Element for a Subset of Yeast Genes and Suggests a New Role for the Poly(A) Tail
The polyU sequence that occurs in ~10% of stabilizing elements functions in unrelated sequence contexts, indicating that the stability information is primarily, and perhaps completely, encoded within the polyU sequence. Multiple lines of evidence suggest that the ability of polyU motifs and poly(A) tails to fold into stable stem-loop structures is important for mRNA stabilization. First, although specific RNA-binding proteins could recognize the polyU element, the most likely candidates, Pub1 and Rrp5, recognize relatively short U-rich stretches found in numerous inter-cluster regions that do not affect mRNA stability. Second, the overwhelming majority (>80%) of polyU stabilizing elements are not found in the data set of known Pub1-binding targets (Duttagupta et al., 2005). Third, Pab1 binding to poly(A) tails of polyU-containing isoforms is sharply reduced, suggesting that the poly(A) tails of these isoforms are less accessible and more likely to be in double-stranded configuration. Fourth, an intercluster sequence within the 3′ UTR of APT2 contains a long poly(U) stretch and a generally U-rich microenvironment, yet it has no detectable effect on isoform stability. Substitution of 4 nt to generate a polyU motif results in a large increase in mRNA isoform stability and a dramatic increase in the predicted stability of the stem-loop structure. More generally, the degree of extended polyU character is strongly linked to mRNA stabilization, whereas the general U-rich nature of sequences between clusters is not.
Our results also suggest that secondary structure involving the poly(A) tail plays a more general role in mRNA stability. Strikingly, as compared to control regions between clusters, poly(A) tails of isoforms containing stabilizing elements are much more likely to fold into stem-loops and other secondary structures, whereas poly(A) tails of isoforms containing destabilizing elements are significantly less likely to form such structures. Even for near-identical isoforms that differ in stability primarily due to U residues at or near the 3′ terminus, secondary structures involving the poly(A) tail are more likely to be formed with stabilized isoforms and less likely to be formed with destabilized isoforms. In addition, the strong decrease of Pab1 binding to polyU-containing isoforms argues that the poly(A) tails of these isoforms are physically shielded from interacting with Pab1, presumably due to the formation of double-stranded structures with poly(A) tails. Taken together, these diametrically opposed structural features of stabilizing and destabilizing elements strongly suggest that formation or disruption of poly(A) secondary structure, presumably through U-rich tracts, is an important determinant of mRNA stability.
Although the reduced Pab1 binding to polyU-containing isoforms is most easily explained by hybridization, and hence reduced accessibility of the poly(A) tail, we do not understand why Pab1 binding is inhibited by the GC-rich stem-loop structure. Perhaps the proximity of the GC-rich stem-loop to the poly(A) tail inhibits cooperative binding of Pab1 to the poly(A) tail and neighboring sequences. Because Pab1 can interact with sequences upstream of cleavage and polyadenylation sites (Simón and Séraphin, 2007; Chritton and Wickens, 2011), any reduction in single-stranded character at the 3′ ends of isoforms and/or poly(A) tails might result in reduced Pab1 binding. It is also possible that the GC-rich elements may be forming higher-order structures such as triple helices that shield the poly(A) tail (Mitton-Fry et al., 2010; Brown et al., 2012; Tycowski et al., 2012). Whatever the mechanism, reduced Pab1 binding is correlated with increased mRNA stability.
A Putative Role for Secondary Structure in Governing mRNA Half-Lives
Although the stabilizing and destabilizing elements have no common sequence motifs other than the stabilizing polyU element found in a subset of 3′ UTRs, it is tempting to speculate that there is a general principle that governs mRNA stability. We suggest that the mRNA stabilizing elements reflect secondary structures between the elements and the poly(A) tail or other sequences within the same RNA (or possibly interactions with a separate RNA in trans). Such gene-specific secondary structure formation would explain why conventional sequence motifs are not observed in mRNA stabilizing or destabilizing elements, with the exception of the polyU motif that interacts with the common poly(A) tail. Conversely, we speculate that destabilizing elements might inhibit stabilizing secondary structures that would otherwise form within an mRNA. In this view, mRNA stability is correlated with the overall ability to form 3′ secondary structures, and stabilizing and destabilizing elements either favor or disfavor overall secondary structure. In this regard, S. cerevisiae 3′ UTRs are much more highly structured than open reading frames (ORFs) and 5′ ends (Kertesz et al., 2010; Wan et al., 2012) and hence are particularly important in mRNA stability. This could be biologically relevant because it would result in preferential stabilization of full-length mRNAs rather than abortive transcripts.
There are two general classes of mechanisms, not mutually exclusive, by which secondary structures might stabilize mRNAs. One possibility is that there are proteins that recognize secondary structures and facilitate recruitment of RNA-degrading activities to the mRNA. Although this mechanism might well hold true for some mRNAs, it is unlikely to be the predominant mechanism by which closely related isoforms possess different half-lives. mRNA secondary structures exhibit a broad variation of structural folds, and this high diversity of binding surfaces makes it unlikely that a single protein (or a family of proteins) can recognize all the different surfaces.
A more likely alternative is that secondary structures act as energetic barriers that block deadenylation of the poly(A) tail and/or hinder the exosome from exonucleolytically degrading the transcript in a 3′ to 5′ direction. Inaccessibility of the poly(A) tail might not only reduce Pab1 binding but also block accessibility of the major cytoplasmic deadenylase Ccr4-Pop2 (and possibly Pan2-Pan3) and subsequent association of degradation factors. By analogy to degradation by the cytoplasmic 5′ to 3′ exonuclease Xrn1 (Muhlrad et al., 1995), exosome-mediated degradation is dependent on disruption of secondary structure to allow single-stranded RNA to fit into the exosome’s catalytic channel (Makino et al., 2013). It is noteworthy that this RNA unwinding function is carried out in the cytoplasm primarily by the Ski2 subunit of the Ski2/Ski3/Ski8 complex (Halbach et al., 2013), which is mostly absent at isoforms stabilized by polyU elements (Figure 7B) (Tuck and Tollervey, 2013). Because exonucleolytic cleavage requires the exosome to protect a relatively large region upstream of the actual cleavage site, secondary structure at or near the 3′ end could inhibit its activity or might prevent its association altogether if the unstructured sequences at the very 3′ ends of the isoforms are of insufficient length. The inhibitory secondary structures could be formed by RNA sequences in the isoform itself, and/or their formation could be increased by interactions with specific RNA-binding proteins or trans-acting RNAs. In either case, the ultimate result would be an increase in relative isoform stability.
EXPERIMENTAL PROCEDURES
Yeast strain JGY2000, a derivative of BY4741, was constructed by tagging the largest subunit of Pol II (Rpb1) with FRB as described (Fan et al., 2010). The tagged strain displayed no obvious growth defects, and its approximately 90 min doubling time in rich media is indistinguishable from its isogenic precursor (data not shown). Mutant alleles were constructed by the delitto perfetto gene-replacement method (Storici and Resnick, 2006) using oligonucleotides listed in Table S6.
For the time courses, cultures were grown in YPD medium at 30°C to an optical density 600 (OD600) of 0.5. A portion of the cells was harvested at this time point (t = 0), whereas the remaining cultures were treated with 1 μg/ml rapamycin for an additional 20, 40, 60, 80, 100, 120, or 150 min. RNA was isolated from each time point using the hot phenol method and RNeasy kit with DNase I treatment (QIAGEN). For each time point, a separate portion of cells was analyzed by chromatin immunoprecipitation (ChIP) (Fan et al., 2010).
Purified RNA from two separate time courses was subjected to DRS as described (Ozsolak et al., 2009). Increasing amounts of RNA were loaded into the flow cell for the later time points in order to partially offset the reduced overall number of sequence reads obtained from these time points, when the overall mRNA yield is lower. We normalized for the uneven RNA loading in the subsequent analysis of mapped sequence reads by taking into account both cell number and different amounts of RNA loaded at each time point.
To calculate half-lives for entire genes, we identified the time point containing the maximum number of sequence reads for each gene (ti) and eliminated all genes for which the number of reads (Ti) was <100. Most transcripts yielded the maximum number of reads at ti = 0, with a small number of genes peaking at later time points (ti = 20 or ti = 40). Half-lives were calculated as follows. Starting with the time point containing the maximal tags (ti), we looked for the first time point (tf) that contained >10-fold drop in sequence reads from the time point and the number of reads associated with it (Tf). We used the read counts at t = 150 min to calculate half-lives of very stable transcripts whose read counts never dropped below the 10-fold threshold relative to the maximal point. Conversely, we used only the first two data points to calculate half-lives of a few very rapidly turned over transcripts whose read counts dropped >10-fold in the time point (t = 20) after rapamycin addition.
To calculate half-lives (t½), we used the following formula,
where the Microsoft Excel LOGEST function is used to estimate the slope (or goodness of fit) of the points in the time course ranging from initial (Ti, ti) to final (Tf, tf). Correlation coefficients (R2) were very high, with the vast majority of transcripts exhibiting R2 values >0.95. Half-lives for individual isoforms (Ti = 0 ≥ 100) were determined in the same fashion as above. Isoforms analyzed in this study had to possess ≥100 sequence reads at steady state.
RNAs from strains containing polyU or GC-rich structures at ectopic loci were reverse-transcribed at 55°C using oligo(dT)20 and ThermoScript Reverse Transcriptase (Life Technologies). Resulting first-strand cDNAs were quantified by qPCR with upstream gene-specific primers and downstream hybrid primers specific for both poly(A) and cluster sequence (Table S7). Relative isoform abundance was calculated at different time points, and half-lives were computed as described above.
RNA folding was performed with the mFold program (Zuker, 2003). Full-length mRNA isoforms (commencing with the ATG and ending in the specific isoform 3′ end followed by 100 nt of a poly(A) tail) were folded utilizing a 200 nt constraining window. Similar results were obtained when the isoforms were folded using the RNAfold program (Hofacker and Stadler, 2006).
For RIP, Pab1 was tagged with three epitopes of c-Myc protein in strains containing WT, polyU, or GC-rich structures at HXT2 or RPA43, as previously described by Schneider et al. (1995). Tagged strains were grown in YPD to an OD600 of 0.5 and crosslinked with 1% formaldehyde for 15 min. Cell lysis, protein-RNA complex isolation, and DNase I digestion were performed as described by Gilbert and Svejstrup (2006). Protein-RNA complexes were immunoprecipitated with anti-c-Myc antibody (Santa Cruz Biotechnology 9E10) and washed as described by Gilbert and Svejstrup (2006), except that the elution was carried out at 65°C, and not at 37°C. Crosslinks were reversed at 100°C for 10 min, and the RNA was subjected to an additional purification with a QIAGEN miRNeasy kit including on-column DNase I digestion. Reverse transcription of purified RNAs and isoform-specific qPCR were performed as described above.
Supplementary Material
Acknowledgments
We thank Yi Jin, Koon-Ho Wong, Andreas Janzer, and other members of the K.S. laboratory for fruitful discussions and for critical reading of the manuscript. We thank Patrice Milos for her encouragement of this work. This work was supported by grant GM30186 to K.S. from the National Institutes of Health.
Footnotes
ACCESSION NUMBERS
Sequencing data have been deposited into the Gene Expression Omnibus under accession number GSE52286.
Supplemental Information includes four figures and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2013.12.026.
References
- Allen M, Bird C, Feng W, Liu G, Li W, Perrone-Bizzozero NI, Feng Y. HuD promotes BDNF expression in brain neurons via selective stabilization of the BDNF long 3′UTR mRNA. PLoS One. 2013;8:e55718. doi: 10.1371/journal.pone.0055718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Brown JA, Valenstein ML, Yario TA, Tycowski KT, Steitz JA. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENβ noncoding RNAs. Proc Natl Acad Sci USA. 2012;109:19202–19207. doi: 10.1073/pnas.1217338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace F, Paci P, Cusimano V, Germani A, Farina L. Stochastic modeling of expression kinetics identifies messenger half-lives and reveals sequential waves of co-ordinated transcription and decay. PLoS Comput Biol. 2012;8:e1002772. doi: 10.1371/journal.pcbi.1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chritton JJ, Wickens M. A role for the poly(A)-binding protein Pab1p in PUF protein-mediated repression. J Biol Chem. 2011;286:33268–33278. doi: 10.1074/jbc.M111.264572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttagupta R, Tian B, Wilusz CJ, Khounh DT, Soteropoulos P, Ouyang M, Dougherty JP, Peltz SW. Global analysis of Pub1p targets reveals a coordinate control of gene expression through modulation of binding and stability. Mol Cell Biol. 2005;25:5499–5513. doi: 10.1128/MCB.25.13.5499-5513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E. Deadenylation is a widespread effect of miRNA regulation. RNA. 2009;15:21–32. doi: 10.1261/rna.1399509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Moqtaderi Z, Jin Y, Zhang Y, Liu XS, Struhl K. Nucleosome depletion at yeast terminators is not intrinsic and can occur by a transcriptional mechanism linked to 3′-end formation. Proc Natl Acad Sci USA. 2010;107:17945–17950. doi: 10.1073/pnas.1012674107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Niu DK. Relationship between mRNA stability and length: an old question with a new twist. Biochem Genet. 2007;45:131–137. doi: 10.1007/s10528-006-9059-5. [DOI] [PubMed] [Google Scholar]
- Gilbert C, Svejstrup JQ. RNA immunoprecipitation for determining RNA-protein associations in vivo. Curr Protoc Mol Biol. 2006;Chapter 27(Unit 27.4) doi: 10.1002/0471142727.mb2704s75. [DOI] [PubMed] [Google Scholar]
- Grigull J, Mnaimneh S, Pootoolal J, Robinson MD, Hughes TR. Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol Cell Biol. 2004;24:5534–5547. doi: 10.1128/MCB.24.12.5534-5547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimovich G, Medina DA, Causse SZ, Garber M, Millán-Zambrano G, Barkai O, Chávez S, Pérez-Ortín JE, Darzacq X, Choder M. Gene expression is circular: factors for mRNA degradation also foster mRNA synthesis. Cell. 2013;153:1000–1011. doi: 10.1016/j.cell.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Halbach F, Reichelt P, Rode M, Conti E. The yeast ski complex: crystal structure and RNA channeling to the exosome complex. Cell. 2013;154:814–826. doi: 10.1016/j.cell.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Hitti E, Khabar KS. Sequence variations affecting AU-rich element function and disease. Front Biosci (Landmark Ed) 2012;17:1846–1860. doi: 10.2741/4023. [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006;22:1172–1176. doi: 10.1093/bioinformatics/btl023. [DOI] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Iyer V, Struhl K. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995;14:2570–2579. doi: 10.1002/j.1460-2075.1995.tb07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino DL, Baumgärtner M, Conti E. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature. 2013;495:70–75. doi: 10.1038/nature11870. [DOI] [PubMed] [Google Scholar]
- Mandel CR, Bai Y, Tong L. Protein factors in pre-mRNA 3′-end processing. Cell Mol Life Sci. 2008;65:1099–1122. doi: 10.1007/s00018-007-7474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Schwalb B, Maier K, Schulz D, Dümcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dölken L, et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol. 2011;7:458. doi: 10.1038/msb.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitton-Fry RM, DeGregorio SJ, Wang J, Steitz TA, Steitz JA. Poly(A) tail recognition by a viral RNA element through assembly of a triple helix. Science. 2010;330:1244–1247. doi: 10.1126/science.1195858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqtaderi Z, Geisberg JV, Jin Y, Fan X, Struhl K. Species-specific factors mediate extensive heterogeneity of mRNA 3′ ends in yeasts. Proc Natl Acad Sci USA. 2013;110:11073–11078. doi: 10.1073/pnas.1309384110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Decker CJ, Parker R. Turnover mechanisms of the stable yeast PGK1 mRNA. Mol Cell Biol. 1995;15:2145–2156. doi: 10.1128/mcb.15.4.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munchel SE, Shultzaberger RK, Takizawa N, Weis K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell. 2011;22:2787–2795. doi: 10.1091/mbc.E11-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Platt AR, Jones DR, Reifenberger JG, Sass LE, McInerney P, Thompson JF, Bowers J, Jarosz M, Milos PM. Direct RNA sequencing. Nature. 2009;461:814–818. doi: 10.1038/nature08390. [DOI] [PubMed] [Google Scholar]
- Parker R. RNA degradation in Saccharomyces cerevisiae. Genetics. 2012;191:671–702. doi: 10.1534/genetics.111.137265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 2013;497:127–131. doi: 10.1038/nature12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh-Sadka T, Levo M, Shabi U, Shany B, Keren L, Lotan-Pompan M, Zeevi D, Sharon E, Weinberger A, Segal E. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet. 2012;44:743–750. doi: 10.1038/ng.2305. [DOI] [PubMed] [Google Scholar]
- Sachs AB, Davis RW, Kornberg RD. A single domain of yeast poly(A)-binding protein is necessary and sufficient for RNA binding and cell viability. Mol Cell Biol. 1987;7:3268–3276. doi: 10.1128/mcb.7.9.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BL, Seufert W, Steiner B, Yang QH, Futcher AB. Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast. 1995;11:1265–1274. doi: 10.1002/yea.320111306. [DOI] [PubMed] [Google Scholar]
- Simón E, Séraphin B. A specific role for the C-terminal region of the Poly(A)-binding protein in mRNA decay. Nucleic Acids Res. 2007;35:6017–6028. doi: 10.1093/nar/gkm452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Resnick MA. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 2006;409:329–345. doi: 10.1016/S0076-6879(05)09019-1. [DOI] [PubMed] [Google Scholar]
- Struhl K. Naturally occurring poly(dA-dT) sequences are upstream promoter elements for constitutive transcription in yeast. Proc Natl Acad Sci USA. 1985;82:8419–8423. doi: 10.1073/pnas.82.24.8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Pirkl N, Maier KC, Schenk A, Failmezger H, Tresch A, Cramer P. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol Cell. 2013;52:52–62. doi: 10.1016/j.molcel.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Tuck AC, Tollervey D. A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell. 2013;154:996–1009. doi: 10.1016/j.cell.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycowski KT, Shu MD, Borah S, Shi M, Steitz JA. Conservation of a triple-helix-forming RNA stability element in noncoding and genomic RNAs of diverse viruses. Cell Rep. 2012;2:26–32. doi: 10.1016/j.celrep.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, Sive H, Bartel DP. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012;22:2054–2066. doi: 10.1101/gr.139733.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S, Peltz SW. Regulated ARE-mediated mRNA decay in Saccharomyces cerevisiae. Mol. Cell. 2001;7:1191–1200. doi: 10.1016/s1097-2765(01)00279-9. [DOI] [PubMed] [Google Scholar]
- Wan Y, Qu K, Ouyang Z, Kertesz M, Li J, Tibshirani R, Makino DL, Nutter RC, Segal E, Chang HY. Genome-wide measurement of RNA folding energies. Mol Cell. 2012;48:169–181. doi: 10.1016/j.molcel.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu M, Downie B, Liang C, Ji G, Li QQ, Hunt AG. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc Natl Acad Sci USA. 2011;108:12533–12538. doi: 10.1073/pnas.1019732108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Sherman F. DNA sequence required for efficient transcription termination in yeast. Cell. 1982;28:563–573. doi: 10.1016/0092-8674(82)90211-2. [DOI] [PubMed] [Google Scholar]
- Zhang C, Lee DJ, Chiang YC, Richardson R, Park S, Wang X, Laue TM, Denis CL. The RRM1 domain of the poly(A)-binding protein from Saccharomyces cerevisiae is critical to control of mRNA deadenylation. Mol. Genet. Genomics. 2013;288:401–412. doi: 10.1007/s00438-013-0759-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.