

Abstract
The lysosomal enzyme alpha-L-iduronidase hydrolyzes terminal iduronic acid from heparan sulfate and dermatan sulfate, and is an essential step in GAG degradation. Mutations of its gene, IDUA, yield a spectrum of mucopolysaccharidosis (MPS) type I clinical disorders. The IDUA mutation, c.712T>A (p.L238Q) was previously noted as a mild mutation. In a longitudinal study of MPS brain structure and function (Lysosomal Disease Network), we found this mutation in 6 of 14 Hurler-Scheie syndrome patients in the age range of 15 to 25 years. We hypothesized that L238Q, when paired with a nonsense mutation, is significantly more severe than other missense-nonsense combinations.
Methods
Of 6 patients with a L238Q mutation, the L238Q allele was paired with a nonsense mutation in 4 patients, paired with a deletion in 1, and with a splice site mutation in another. This group was compared to 6 Hurler-Scheie patients closely matched in age and mutation type. IQ, and other neuropsychological tests were administered as part of the protocol. Medical history was compiled into a Physical Symptom Score (PSS). Assessment of IQ, attention, memory, spatial ability, adaptive function and psychological status were measured.
Results
No group differences were found in mean age at evaluation (17.8 and 19.0 years), duration of ERT, or PSS. By history, all were reported to be average in IQ (4/6 with documentation) in early childhood. All (100%) of the L238Q group had a psychiatric history and sleep problems compared to none (0%) of the comparison group. Significant differences were found in depression and withdrawal on parent report measures. IQ was lower in the L238Q group (mean IQ 74) than the comparison group (mean IQ 95; p < 0.016). Attention, memory, and visual-spatial ability scores were also significantly lower. Three occurrences of shunted hydrocephalus, and 4 of cervical cord compression were found in the L238Q group; the comparison group had one occurrence of unshunted hydrocephalus and two of cord compression.
Discussion
The missense mutation L238Q, when paired with a nonsense mutation, is associated with significant, late-onset brain disease: psychiatric disorder, cognitive deficit, and general decline starting at a later age than in Hurler syndrome with a mutation-related rate of GAG accumulation and its pathologic sequelae. This particular genotype-phenotype may provide insight into the genesis of psychiatric illnesses more broadly. Consideration of methods for early, brain-targeted treatment in these patients might be considered.
Keywords: Alpha-L-iduronidase, Mucopolysaccharidosis (MPS), L238Q, Hurler-Scheie syndrome, Nonsense mutation, Missense mutation, Intelligence Quotient (IQ), Hurler Syndrome
Introduction
We describe a missense mutation, L238Q, which in mucopolysaccharidosis type I when combined with a nonsense mutation, deletion, or splice site, produces a relatively severe form of Hurler-Scheie syndrome. We document its severity by comparing six patients to with a group with other missense mutations.
Mucopolysaccharidosis type I (MPS I) is an autosomal recessive lysosomal disease due to the deficiency of alpha-L-iduronidase enzyme (IDUA). IDUA hydrolyzes terminal iduronic acid from heparan sulfate and dermatan sulfate, and is an essential step in glycosaminoglycan degradation. Mutations of its gene, IDUA, yield a spectrum of mucopolysaccharidosis (MPS) type I clinical disorders historically distinguished as one of three phenotypes: severe Hurler syndrome, attenuated Scheie syndrome, or the intermediate disorder Hurler-Scheie syndrome. Over 100 mutations have been identified on the IDUA gene located on chromosome 4p16.3.
No biochemical or clinical criteria have been established that reliably distinguish among the three clinical syndromes, or that accurately predict clinical outcomes. Because no agreed upon biochemical or clinical criteria distinguishes the Scheie from the Hurler-Scheie syndrome phenotypes, they frequently are viewed as a spectrum of severity and described as the attenuated form of MPS I [1, 2]. Although homozygosity for null mutations (producing no IDUA protein) such as from nonsense mutations W402X and/or Q70X can be used to define MPS IH, there are innumerable potential genotypes associated with more attenuated phenotypes thus limiting the ability to prognosticate among these.
Several attempts in the past have been made to identify mutations that have specificity with regard to severity or phenotypic characterization. In addition to W402X and Q70X, several mutations such as P533R have been noted to present variably as severe and mild. The IDUA mutation, c.712T>A (p.L238Q) has been previously noted as a mild mutation by Yogalingam et al [3], both severe and moderate by Chandar & Mahalingam [4] and intermediate by Saito et al [5]. In our longitudinal study of MPS brain structure and function, we unexpectedly found that it occurred in 6 of 57 patients recruited longitudinal study of brain structure and function in MPS disorders at the University of Minnesota with associated central nervous system (CNS) difficulties [6]. Our goal was to delineate the severity of this specific mutation and to characterize its phenotype especially with regard to organ system and particularly CNS involvement. Based on these observations, we hypothesized that L238Q, when paired with a severe mutation such as a nonsense mutation, produces a severe form of Hurler-Scheie syndrome with progressive CNS disease.
Methods
Subjects
Subjects were identified among 14 MPS I participants with Hurler-Scheie syndrome in the age range of 15 to 25 years who had DNA analysis in the study “Longitudinal Studies of Brain Structure and Functions in MPS Disorders” an NIH supported study which is a part of the Lysosomal Disease Network (LDN) U54-NS065768. Of these patients 6 had the L238Q mutation from four families (two sets of siblings were included) paired with a nonsense mutation, deletion or splice site. One patient (#7), had the L238Q mutation paired with a missense mutation (E182K); this milder patient was analyzed separately. A comparison group was comprised of the 6 patients between ages 15 to 25 years who did not have the L238Q mutation. Specifics of their mutations can be found in Table 2.
Table 2.
Description of L238Q and Comparison Groups
| Patient Number | Mutation of L238Q Group | Mutation type | Age | Sex | Years of Treatment | Numbers of Surgery | Hydro-cephalus | Shunt Placement | Spinal Cord Compression | Psychiatric problem | Sleep Problem | PSS (Physical Symptom Score) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | L238Q/63delC | Missense/Deletion | 18.6 | F | 5.9 | 6 | Absent | Absent | Present | Present | Present | 10 |
| 2 | L238Q/W402X | Missense/Nonsense | 18.3 | M | 9.1 | 7 | Present | Present | Present | Present | Present | 13 |
| 3 | L238Q/W402X | Missense/Nonsense | 16.5 | M | 7.6 | 6 | present | Absent | Present | Present | Present | 11 |
| 4 | L238Q/W402X | Missense/Nonsense | 19.8 | F | 9.7 | 8 | Present | Present | Absent | Present | Present | 12 |
| 5 | L238Q/W402X | Missense/Nonsense | 17.8 | F | 0.3 | 7 | Present | Present | Present | Present | Present | 14 |
| 6 | L238Q/Int3-2a>g | Missense/SpliceSite | 16.7 | M | 10.4 | 4 | Absent | Absent | Absent | Present | Present | 9 |
| Mutation of Comparison Group | ||||||||||||
| 7 | G256R/W402X | Missense/Nonsense | 15.1 | M | 6.9 | 6 | Absent | Absent | Absent | Absent | Absent | 8 |
| 8 | G265R/A327P | Missense/Missense | 22.3 | M | 12.5 | 8 | Absent | Absent | Absent | Absent | Absent | 10 |
| 9 | Q380R/Q70X | Missense/Nonsense | 12.7 | M | 7.0 | 7 | Absent | Absent | Absent | Absent | Absent | 10 |
| 10 | P533R/P533R | Missense/Missense | 25.5 | M | 7.0 | 12 | Absent | Absent | Absent | Absent | Absent | 14 |
| 11 | R89W/W402X | Missense/Nonsense | 19.0 | M | 9.4 | 8 | Present | Absent | Present | Absent | Absent | 11 |
| 12 | W402X/Int11-7c>t | Nonsense/SpliceSite | 20.9 | F | 9.3 | 10 | Absent | Absent | Present | Absent | Absent | 12 |
With the exception of one patient, measurements from the first visit of our longitudinal study were used. For one L238Q patient, we are using data from the third visit; at the first visit, this patient was too distraught to complete the testing; for the second visit there was also some missing data. At the third visit, after she was treated with antidepressant medication we were able to complete a full battery of tests.
Procedures
Molecular diagnosis was performed by Sanger sequencing of all exomes of the IDUA gene at the Gene Therapy and Diagnostics Lab, University of Minnesota. DNA was isolated from leukocyte pellets, PCR-amplified, and then sequenced with BigDye® Terminator (Life Technologies). The reactions were sequenated on an ABI 3130 Avant sequencer (Life Technologies) with subsequent analysis on Sequencer software (Gene Codes Corporation).
Medical and treatment histories were obtained from the patient or parents on case report forms developed for the study. Clinical files of patients were also reviewed to check accuracy and obtain additional information. A scoring system was devised to quantify abnormalities across six features, which were scored by a system of frequency and severity. The overall summary score is based on scores from four medical domains (skeletal/orthopedic, vision, hearing, and cardio-respiratory), the number of surgical procedures and presence of hydrocephalus (Table 1 shows the MPS Physical Symptom Scale). We have found this severity score to be reliable and to correlate with daily living skills and quality-of-life (unpublished data).
Table 1.
MPS Physical Symptom Scale.
| Feature | Score | Description |
|---|---|---|
| Skeletal/Orthopedic | 0 | No Orthopedic Symptom |
| 1 | 1 – 2 Symptoms | |
| 2 | 3 – 4 Symptoms | |
| 3 | 5 – 6 Symptoms or Cord Compression | |
| Orthopedic symptoms include Limited Range of Motion, Kyphosis, Scoliosis, Hip Dysplasia, Knock Knee and High Arch Foot | ||
| Vision | 0 | No Visual impairment or Symptom |
| 1 | Mild Corneal Clouding or Glaucoma or Cataract | |
| 2 | Moderate Corneal Clouding or Both Glaucoma and Cataract | |
| 3 | Severe Corneal Clouding or Retinal Degeneration | |
| Hearing | 0 | None |
| 1 | Mild Hearing Loss | |
| 2 | Moderate Hearing Loss | |
| 3 | Severe Hearing Loss | |
| Cardio-respiratory | 0 | None |
| 1 | 1 – 2 Cardiac or Respiratory Symptoms | |
| 2 | 3 – 4 Symptoms or presence of Sleep Apnea | |
| 3 | 5 – 6 Symptoms or history of Cardiac Surgery | |
| Cardiac and Respiratory symptoms include Murmur, Hypertension, Valve Disease, Cardiac Surgery, Chronic Nasal Discharge/Obstruction, Tonsils/Adenoids, Respiratory Infection/Reactive Airway Disease and Sleep Apnea | ||
| Hydrocephalus | 0 | Absent |
| 1 | Hydrocephalus Without Shunt | |
| 2 | Hydrocephalus With Shunt | |
| 3 | Revision of Shunt | |
| Number of Surgeries | 0 | No Surgery |
| 1 | Less than 4 Surgeries | |
| 2 | 4 to 8 Surgeries | |
| 3 | More than 8 Surgeries | |
| Total Score = 0–18 | ||
To measure neurobehavioral functions we use WASI (Wechsler Abbreviated Scale of Intelligence) [7], for Full Scale IQ, TOVA-errors of omission (Test of Variables of Attention) [8], to assess attention, BVMT (Brief Visual Spatial Memory Test) [9], to assess nonverbal memory and learning, either CMS (Children’s Memory Scale) [10], or WMS (Wechsler Memory Scale) [11], to measure memory for stories, and the JLO (Judgment of Line Orientation) [12] to assess spatial perception. VABS-II (Vineland Adaptive Behavior Scales, Second Edition) [13] Composite score was used to evaluate the full spectrum of adaptive behavior. To assess current psychological status BASC-PRS and BASC–SRP (Behavior Assessment System for Children-Parent Rating Scale and Self-Report of Personality) [14] were used. However, we had only four of six parents of patients in the comparison group complete the BASC-PRS. Five of six in each group completed the BASC-SRP (older patients completed an adult inventory which we did not include in our statistical analysis). For the BASC-PRS we included the withdrawal scale, depression and atypical behavior scales. Based on our previous clinical observations these scales were most sensitive in adolescents with MPS disorders. Only the depression scale was included for the BASC-SRP.
Statistical Analysis
Descriptive statistics included the mean, standard deviation, and range, which were tabulated separately for the L238Q and comparison groups. Comparisons between groups used the t-test with unequal variances and Welch degrees of freedom for confidence intervals and P-values. All analyses were conducted using R v2.15.2 [15].
Results
Mutation, demographic and medical data are presented in Table 2. Table 3 summarizes the neurobehavioral data. There were three females in the L238Q group and 1 female in the comparison group. No difference was found in age at evaluation, but the comparison group was more variable. No difference was found in age or years of ERT treatment for the two groups. Furthermore, the MPS Physical Symptom Score was almost identical. Although more surgeries occurred in the comparison group, that was primarily in the oldest two patients, thus reflecting cumulative surgeries with age.
Table 3.
Patient characteristics for L238Q and comparison group. Values expressed are mean (SD) or range where indicated:
| Covariates | L238Q Group | Comparison Group |
|---|---|---|
| Total | 6 | 6 |
| Gender | 3 F, 3 M | 1 F, 5 M |
| Age at visit | 17.8 (1.5) range=15.7–19.8 |
19.0 (4.7) range=12.7–25.5 |
| Age at 1st treatment | 10.6 (4.1) | 10.6 (4.3) |
| Years of treatment | 7.2 (3.7) | 8.4 (2.2) |
| Physical Symptom Score | 11.7 (1.6) range=10–14 |
10.8 (2.0) range=8–14 |
| Number of surgeries | 6.3 (1.4) | 8.5 (2.1) |
| Neuropsychological Measures: standard scores 100±15 | ||
| - WASI IQ | 74 (5.3) | 95 (15.3) |
| - TOVA-errors of omission | 66 (27.0) | 101 (3.7) |
| - BVMT | 64 (14.3) | 84 (16.3) |
| - CMS/WMS | 72 (22.3) | 100 (10.9) |
| - JLO | 64 (12.6) | 82 (24.8) |
| - VABS | 74 (5.8) | 86 (14.7) |
| BASC: T Scores 50±10 | ||
| - Parent; Withdrawal | 61 (5.6) | 48 (7.5)2* |
| - Parent; Depression | 56 (8.6) | 45 (5.9)2 |
| - Parent; Atypicality | 55 (7.0) | 44 (3.9)2 |
| - Self; Depression | 57 (13.8)1 | 45 (2.5)1 |
superscripts indicate number of cases with missing data
IQ was significantly different in the two groups. All six L238Q subjects tested below the average range, while only one comparison subject did. Attention, as measured on the TOVA errors of omission, a measure of vigilance, was significantly different. Memory measures were also significantly different for both BVMT (Brief Visual spatial Memory Test) and for memory in context or memory for stories (CMS/WMS). JLO (Judgment of Line Orientation) for visual spatial perception showed a mean difference with a trend to significance. Notably, the comparison group was quite variable. The VABS-II or Vineland, measuring adaptive skills, did show some differences reflected by a trend, but the differences were not as dramatic as on the cognitive measures.
100% of the L238Q group had some type of psychiatric and sleep problems and none of the comparison subjects did. In a parent report of psychological problems on the BASC-PRS significantly more difficulties with withdrawal, depression, and ‘atypical’ behavior were found in the L238Q group. In the BASC-SRP, we found depression was endorsed by the L238Q patients somewhat more than the comparison group. Four of the six L238Q patients we have historical data of 5 years or more ago indicating that at initial evaluation these children were within the average range and now are not.
Three occurrences of shunted hydrocephalus were found in the L238Q group compared with one non-shunted hydrocephalus in the comparison group; 4 occurrences of cord compression in the L238Q group compared with 2 in the comparison group. 100% of L238Q patients had a history of psychiatric as well as sleep problems and 0% in the comparison group for both problems. By history, all psychiatric problems first emerged in adolescence.
Figure 1. shows the difference between IQ, TOVA, Vineland and memory measures and Figure 2. shows the BASC Parent and Self Report for the L238Q and Comparison group.
Figure 1.

Standard Scores (100 ± 15) on Neuropsychological Tests for the L238Q and Comparison group. Individual scores for each patient are presented with mean and 95% confidence intervals for each group.
Figure 2.
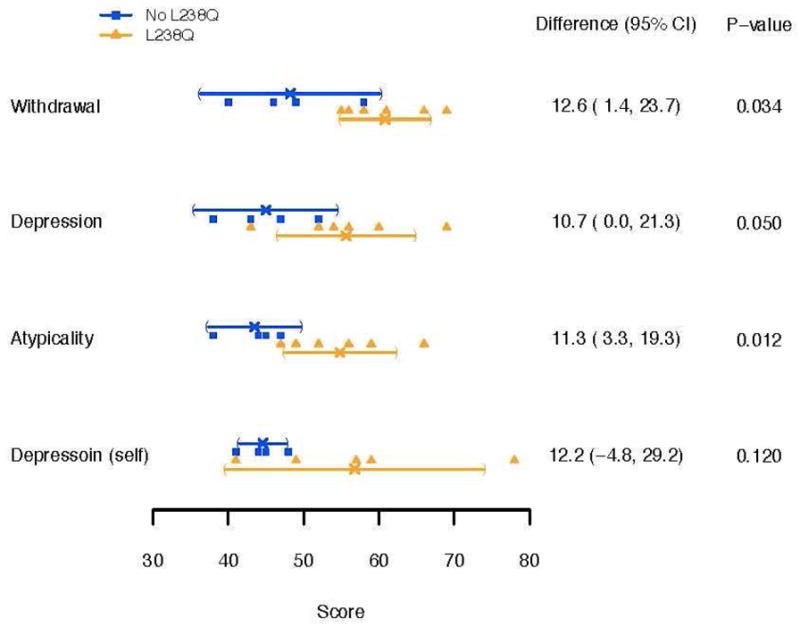
BASC Parent and Self Report for the L238Q and Comparison group. Individual scores for each patient are presented with mean and 95% confidence intervals for each group (T scores (50 ± 10); higher scores are more abnormal).
Case report of L238Q with missense mutation
One additional patient had the L238Q mutation paired with a missense mutation, E182K; her data was examined separately. In contrast to the patients with a second severe mutation her cognitive skills were within normal range, IQ: 104, TOVA - omissions: 107, BVMT: 82, WMS: 100, JLO: 108, VABS-II: 78. Her age at visit was 22.5 years, age at first treatment was12.0 years, and years of treatment was10.5 years. Her MPS Physical Symptom Score was 11, number of surgeries was 8, and she did have spinal cord compression with decompression surgery. She has no psychiatric or sleep problems. Her scores were similar to the comparison group.
Discussion
The missense mutation L238Q, in the biological context of a nonsense mutation, is associated with significant, late-onset brain disease: psychiatric disorder, significant cognitive abnormality, and general decline starting at a later age than is seen in Hurler syndrome. We speculate that there is a mutation-related rate of GAG accumulation and its pathologic sequelae. The rate of accumulation, and the stage in brain development are critical determinants of the neurologic phenotype. In this case, a psychiatric phenotype may be dictated by specific timing of this neuropathic damage, resulting in a distinctive phenotype.
The continuous spectrum of severe to mild clinical phenotypes in MPS I is most likely caused by combinations of different mutant alleles at the IDUA locus [16,17]. Mutations can completely prevent any functional enzyme from forming or can allow for some functional enzyme to be produced, resulting in residual enzyme activity [18,19]. Nonsense mutations result in the complete absence of enzyme protein. Previously identified nonsense mutations include W402X and Q70X, the two most commonly reported mutations in IDUA [20,18]. Patients that are homozygous for a nonsense mutation or heterozygous for two different nonsense mutations are diagnosed with Hurler syndrome, due to a lack of any enzymatic activity. Patients who are heterozygous for one nonsense allele and a different mutation may have various phenotypes depending on the nature of the second allele [18]. Missense mutations have been found to be associated with mild and intermediate phenotypes in many patients. It is likely that missense mutations allow for some residual enzyme activity, thus patients with one allele with a missense mutation will likely retain some functional enzyme activity [21]. Yet the genotype-phenotype associations for missense mutations are not clear-cut and missense mutations have been reported in individuals with a wide variability of phenotypes, both severe and mild [18].
Splice site mutations similar to nonsense mutations, are often associated with the severe form of MPS I. These mutations greatly affect splicing which leads to unstable mRNA [19,22, 23, 24, 25]. Deletions and insertions that have been detected in patients with MPS I typically result in Hurler syndrome [18].
Recent structural analysis of the a-L-iduronidase enzyme molecule [26] does not implicate L238 in the key binding sites, and proposes that the buried hydrophobic core may be destabilized by the substitution of L238 with a hydrophilic Q moiety. Thus, L238Q likely alters protein structure in a manner that destabilizes the protein for premature degradation or very inefficient catalysis resulting in minimal residual enzyme activity. L238Q might be a good candidate for the proposed chaperone therapy.
The L238Q mutation appears to be specific for neurobehavioral differences in the patients with Hurler-Scheie syndrome in this study. L238Q/Nonsense patients are more severe than most other patients with Hurler-Scheie syndrome. There are no significant differences in phenotypic presentation in frequency and severity of organ system involvement between L238Q group and the Hurler-Scheie syndrome comparison group. Because these patients have so many physical problems and severity is difficult to define precisely, we were could not reliably quantify the relative severity of each symptom and each surgery. However, the summative score of numbers of major problems and surgeries, did not differ between the two groups.
The dramatic difference in IQ and attention span in L238Q patients indicates a relatively severe neurologic phenotype among this group of patient with Hurler-Scheie syndrome. On four of the six L238Q patients we have early data of 5 years or more ago indicating that at initial evaluation these children were within the average range and now are not. This could be attributed to hydrocephalus; but some evidence exists that the beginning of decline dates to before that diagnosis and the placement of ventriculo-peritoneal shunts. MRI analysis is currently underway for these patients and will be reported at a later time.
These patients developed psychiatric symptoms in adolescence after a period of cognitive loss. By history, all of the L238Q patients had at least one episode of depression, psychosis, or autism and prescription of psychotropic medications. Two were receiving psychotropic medications at the time of testing. The parent report measure also indicated withdrawal and depression among patients in this group. All patients had sleep problems; none of the comparison group did.
Because the psychiatric problems were various, no particular treatments were reported to be notably effective. Two of our patients had serious chronic problems; the others had significant depression and anxiety that remitted with combinations of psychological counseling and medication. Many patients with attenuated MPS I have stressful periods in adolescence due to social isolation and low self esteem. This was true for the control group as well as the subject group, but only in the subject group were psychiatric diagnoses made and psychotropic medications consistently needed.
It is possible that the increased incidence of spinal cord compression and hydrocephalus may have had an impact on the psychiatric status of these patients. However, we see spinal cord compression and hydrocephalus in many of our MPS VI and attenuated MPS II patients without similar neuropsychiatric consequence. We conclude that it is a phenomenon of more impact on the CNS compared to other mutations.
Limitations of this study may be due to family history factors and the confounding effects of sleep disturbance. Although we do not have detailed information about whether family history of psychiatric problems might be contributing to psychiatric problems, a family tendency to psychiatric problems could be magnified by having two siblings from each of two families. In addition, sleep disturbances might be confounding the psychiatric problems. In itself, the difference in incidence of sleep problems was remarkable.
An intrinsic weakness of this study is that some of the subjects in the comparison group did not have equivalent mutations; for example, two patients in the comparison group had two missense mutations. However, inspection of their data does not indicate that these two subjects had different findings than those who had other mutation types. The significant p values and the specificity of the findings help to validate the findings. Finding comparable mutations is challenging in such a rare disease.
We have identified a missense mutation L238Q, when paired with a nonsense or other severe mutation, is associated with severe cognitive abnormality and possibly decline starting at a later age than is seen in Hurler syndrome, and with psychiatric problems emerging in adolescence. Understanding this particular genotype-phenotype may provide insight into the genesis of psychiatric illnesses more broadly. This information may be useful in predicting long-term outcomes early on for individual patients. We need to strongly consider methods of early, brain targeted treatment in MPS I patients with a L238Q and a nonsense mutation and with vigor even though they may not yet show cognitive decline at this early time. It is notable that all of the children with the L238Q group and one in the comparison group were eligible for and were referred for trial of intrathecal enzyme replacement therapy prior to this new insight into the L238Q mutations. In addition to intrathecal enzyme, other brain treatments such as hematopoietic cell transplant (HCT) and addition of chaperone therapy might be considered.
Highlights.
L238Q/Nonsense patients are more severe than other Hurler-Scheie patients.
Impaired IQ and attention were found patients with L238Q/Nonsense mutations.
Cognitive and psychological symptoms seem to worsen in L238Q patients with age.
Acknowledgments
We are thankful for the support provided by NIH U54-NS065768 and UL1TR000114 (NINDS, NIDDK. ORDR, and NCATS), National MPS Society, Genzyme-Sanofi and The Ryan Foundation for Orphan Disease Research as well as other investigators who contributed patient material and information.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pastores GM, Arn P, Beck M, Clarke JTR, Guffon N, Kaplan P, Muenzer J, Norato DYJ, Shapiro E, Thomas J, Viskochil D, Wraith JE. The MPS I registry: Design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Molecular Genetics and Metabolism. 2007;91:37–47. doi: 10.1016/j.ymgme.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Clarke LA, Heppner J. Mucopolyschharidosis Type I. 2002 Oct 31 (Updated 2011 Jul 21) In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews™ [Internet] Seattle (WA): University of Washington, Seattle; 1993–2013. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1162/ [Google Scholar]
- 3.Yogalingam G, Guo XH, Muller VJ, Brooks DA, Clements PR, Kakkis ED, Hopwood JJ. Identification and Molecular Characterization of α-L-Iduronidase Mutations Present in Mucopolysaccharidosis Type I Patients Undergoing Enzyme Replacement Therapy. Human Mutation. 2004;24:199–207. doi: 10.1002/humu.20081. [DOI] [PubMed] [Google Scholar]
- 4.Chandar SS, Mahalingam K. Mucopolysaccharidosis type I: Homology Modeling and docking analysis of the lysosomal enzyme, human α-L-iduronidase. African Journal of Pharmacy and Pharmacology. 2012;6(27):2027–2038. [Google Scholar]
- 5.Saito S, Ohno K, Maita N, Sakuraba H. Structural and clinical implications of amino acid substitutions in α-L-iduronidase: Insight into the basis of mucopolysaccharidosis type I. Molicular Genetics and Metabolism. 2013 doi: 10.1016/j.ymgme.2013.10.005. in press. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed A, Cooksley R, Rudser K, Whitley C, Shapiro E. Relationship of Genotype, Treatment and Age with Medical Phenotype in Mucopolysaccharidosis Type I. Molecular Genetics and Metabolism. 2013;108:S17–S18. [Google Scholar]
- 7.Chen HY, Dinmore A, Hildebrand D, Ledbetter M, McDonald J, Pike J, Wolley L, ASIW . Wechsler Abbreviated Scale of Intelligence Manual. The Psychological Corporation; San Antonio, TX: 1999. [Google Scholar]
- 8.Greenberg LM, Kindschi CL, Dupuy TR, Hughes SJ. Test of Variables of Attention continuous performance test. The TOVA Company; Los Alamitos, CA: 2007. [Google Scholar]
- 9.Smerbeck AM, Parrish J, Yeh EA, Hoogs M, Krupp LB, Weinstock-Guttman B, Benedict RH. Regression-Based Pediatric Norms for the Brief Visuospatial Memory Test –Revised and the Symbol Digit Modalities Test. The Clinical Neuropsychologist. 2011;25(3):402–412. doi: 10.1080/13854046.2011.554445. [DOI] [PubMed] [Google Scholar]
- 10.Cohen M. Children’s memory scale (CMS) The Psychological Corporation; San Antonio TX: 1997. [Google Scholar]
- 11.Wechsler D. WMS-III: Wechsler memory scale administration and scoring manual. Psychological Corporation; San Antonio, TX: 1997. [Google Scholar]
- 12.Benton AL, Hamsher K, Varney N, Spreen O. Contributions to neuropsychological assessment. New York: Oxford University Press; 1983. [Google Scholar]
- 13.Sparrow SS, Cicchetti DV, Balla DA. Vineland adaptive behavior scales. 2. Psychological Corporation; San Antonio, TX: 2005. [Google Scholar]
- 14.Reynolds CR, Kamphaus RW. Behavior assessment system for children. 2. Circle Pines, MN: American Guidance Service; 2004. [Google Scholar]
- 15.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2012. URL http://www.R-project.org/ [Google Scholar]
- 16.Neufeld EF, Muenzer J. The Metabolic and Moleculer Basis of Inherited Disease. 6. McGraw-Hill; New York: 1989. The Mucopolysaccharidosis. [Google Scholar]
- 17.Hopwood JJ, Morris CP. The Mucopolysaccharidosis: diagnosis, molecular genetics and treatment. Molecular Biology and Medicine. 1990;7(5):381–404. [PubMed] [Google Scholar]
- 18.Terlato NJ, PharmD, Cox GF., MD, PhD Can mucopolysaccharidosis type I disease severity be predicted based on a patient’s genotype? Acomprehensive review of the literature. Genetics in Medicine. 2003;5:286–294. doi: 10.1097/01.GIM.0000078027.83236.49. [DOI] [PubMed] [Google Scholar]
- 19.Beesley CE, Meaney CA, Greenland G, Adams V, Vellodi A, Young EP, Winchester BG. Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Human Genetics. 2001;109:503–511. doi: 10.1007/s004390100606. [DOI] [PubMed] [Google Scholar]
- 20.Scott HS, Litjens T, Hopwood JJ, Morris CP. A Common Mutation for Type I Mucopolysaccharidosis Associated With a Severe Hurler Syndrome Phenotype. Human Genetics. 1992;109:103–108. doi: 10.1002/humu.1380010204. [DOI] [PubMed] [Google Scholar]
- 21.Scott HS, Bunge S, Gal A, Clarke LA, Morris CP, Hopwood JJ. Molecular genetics of Muccpolysaccharidosis type I: Diagnostic, Clinical, and Biological Implications. Human Mutation. 1995;6:288–302. doi: 10.1002/humu.1380060403. [DOI] [PubMed] [Google Scholar]
- 22.Teng YN, Wang TR, Hwu WL, Lin SP, Lee-Chen G. Identification and characterization of -3c–g acceptor splice site mutation in human α-L-iduronidase associated with mucopolysaccharidosis type IH/S. Clinical Genetics. 2000;57:131–136. doi: 10.1034/j.1399-0004.2000.570207.x. [DOI] [PubMed] [Google Scholar]
- 23.Clarke LA, Nelson PV, Warrington CL, Morris CP, Hopwood JJ, Scott HS. Mutation Analysis of 19 North American Mucopolysaccharidosis Type 1 Patients: Identification of Two Additional Frequent Mutations. Human Mutation. 1994;3:275–282. doi: 10.1002/humu.1380030316. [DOI] [PubMed] [Google Scholar]
- 24.Scott HS, Litjens T, Nelson PV, Thompson PR, Brooks DA, Hopwood JJ, Morris CP. Identification of Mutations in the α-L-Iduronidase Gene (IDUA) That Cause Hurler and Scheie Syndromes. American Journal of Human Genetics. 1993;53:973–986. [PMC free article] [PubMed] [Google Scholar]
- 25.Bach G, Moskowitz SM, Tieu PT, Matynia A, Neufeld EF. Molecular Analysis of Hurler Syndrome in Druze and Muslim Arab Patients in Israel: Multiple Allelic Mutation of the IDUA Gene in a Small Geographic Area. American Journal of Human Genetics. 1993;53:330–338. [PMC free article] [PubMed] [Google Scholar]
- 26.Bie H, Yin J, He X, Kermode AR, Goddard-Borger ED, Withers SG, James MNG. Insights into mucopolysaccharidosis I from the structure and action of α-L-iduronidase. Nature Chemical Biology. 2013;9:739–745. doi: 10.1038/nchembio.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]