

Abstract
Minimal requirements for generating effective immunity include the delivery of antigenic (signal 1) and costimulatory (signal 2) signals to T lymphocytes. Recently, a class of 3rd signals, often delivered by antigen-presenting dendritic cells, has been shown greatly to enhance immune responses, especially against tumors. Among signal 3 factors, interleukin-12 is particularly effective and can be conditionally induced by agonists of Toll-like transmembrane receptors (TLRs). In this study, we assessed the therapeutic impact of adjuvant TLR agonist administration upon the capacity of DC-tumor electrofusion hybrids to eradicate established MCA-205 sarcomas in syngeneic mice. Paired, but not solitary combinations of polyinosine:polycytadilic acid (p[I:C], TLR3 agonist) and CpG DNA (ODN1826, TLR9 agonist) stimulated IL-12 secretion from DCs in vitro and synergized with vaccination to achieve potent tumor rejection. Therapeutic effects, however, required co-administration of paired TLR agonists and DC-tumor fusion hybrids. The administration of TLR agonists alone or with fusion vaccine induced transient splenomegaly, but without apparent toxicity. The therapeutic effects of this immunization regimen were significantly abrogated through the neutralization of IL-12p70, indicating that production of this 3rd signal was essential to the observed tumor regression. These results demonstrate the profound functional consequences of TLR cooperativity and further highlight the critical role of IL-12 in anti-tumor immunity.
Keywords: tumor immunity, vaccination, dendritic cells, rodent, cytokines
INTRODUCTION
Toll-like receptors (TLR) allow for the discrimination of self tissues from infectious non-self through molecular pattern recognition (1). It is this faculty of the innate immune system that sets in motion the adaptive arm of immunity, and ensures appropriate and vigorous responses against microbial infection. Vaccination against tumor, on the other hand, seeks to generate immunity against self-derived tissues that usually do not express classical pathogen-associated molecular patterns. Therefore, combining TLR agonists with anti-cancer vaccines can potentially induce the immune system to respond against tumor antigens with a quality and intensity usually reserved for microbial infection.
Recent studies have shown that stimulating dendritic cells (DCs) with select pairs of TLR agonists (e.g. ones that coordinately stimulate the My-D88- and TRIF-dependent signaling pathways) greatly enhances the synthesis of so-called DC1 polarization factors, including Delta Notch ligand, interleukin-23, and interleukin-12 p70 (IL-12) (2). IL-12 in particular has been shown to operate as a “third signal”, that in addition to antigen (signal 1) and co-stimulation (signal 2) greatly enhances aspects of T-dependent immune responses (3–7) which may enhance anti-tumor immunity. Mechanisms by which IL-12 achieve this enhancement include Th1-biasing (8), augmentation of CTL avidity for tumor targets (7), and a BCL-3-mediated anti-apoptotic effect which preserves high viability during Ag-driven T cell proliferation (6).
We therefore assessed the impact of parenteral TLR agonists as adjuvants for an extensively characterized DC-tumor electrofusion hybrid vaccine modality (9). Here, electrical fields are used to fuse and hybridize dendritic cells with tumor cells. The resulting heterokaryons retain the superior antigen-presenting capacity of the DC and acquire the entire antigenic complement of the tumor partner, creating an immunogen rich in signals 1 and 2. While this vaccine modality is exceptional for its capacity to bypass the constraints of exogenous processing to present endogenous tumor proteins in both MHC Class I and II-restricted, and highly co-stimulatory contexts, vaccination has consistently benefited from parenteral co-delivery of “third signals” for maximized therapeutic efficacy. Among tested parenteral third signals, IL-12 and OX40 ligating mAb have proved particularly effective (10, 11). Here we show that fusion hybrid vaccination plus a single TLR agonist induces no detectable therapeutic anti-tumor immunity, whereas vaccination plus paired TLR agonists show powerful therapeutic responses against established lung metastases derived from the MCA205 sarcoma. This therapeutic effect is apparently mediated through a mechanism requiring host production of IL-12. This is the first demonstration, to our knowledge, that dual administered TLR agonists can augment DC-based vaccination equivalently to recombinant Signal 3 factors, leading to greatly enhanced anti-tumor immunity in a generally non-toxic manner.
MATERIALS AND METHODS
Mice and tumors
Female C57BL/6N (B6) mice were purchased from the Biologic Testing Branch, Frederick Cancer Research and Development Center, National Cancer Institute (Frederick, MD). They were maintained in a specific pathogen-free environment and used for experiments at age 8–10 wk. All experiments using mice were approved by the Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic Lerner Research Institute and cared for according to institutional guidelines.
The 3-methylcholanthrene-induced MCA 205 fibrosarcoma (12), syngeneic to B6 mice, was maintained in vivo via serial s.c. transplantation in syngeneic mice and was used for experiments within the 10th transplant generation. Single cell suspensions were prepared from excised solid tumors via enzymatic digestion as described previously (13).
Dendritic cell production
Bone marrow DCs were prepared as previously described (14) by a modification of Inaba et al. (15). Briefly, cells harvested from femora and tibiae were depleted of B and T cells by negative selection with monoclonal antibody (mAb)-coated magnetic beads (Dynal Biotech, Olso, Norway). These cells (0.5 × 106/mL) were cultured in flasks in complete medium supplemented with 10 ng/mL GM-CSF and 10 ng/mL IL-4 (Peprotech Inc., Rocky Hill, NJ). Complete medium consisted of RPMI 1640 with 10% heat-inactivated fetal calf serum (FCS), l-glutamine, and antibiotics as previously described (Tanaka H Cell Immunol 2002). On day 6, nonadherent cells were harvested and further cultured (1.0 × 106/mL) with fresh medium for an additional 2 days.
Electrofusion
Procedures for electrofusion have been described previously (16–18). Briefly, tumor cells were irradiated (5000 cGy) and labeled with the green fluorescent dye, carboxyfluorescein diacetate succinimidyl ester (CFSE, Molecular Probes, Eugene, OR) before fusion. DCs and tumor cells at 1:1 ratio were then mixed and suspended in fusion medium. Fusion medium was composed of 5% glucose containing 0.1 mM Ca (CH3 COO2), 0.5 mM Mg CH3 COO2, and 0.3% bovine serum albumin (BSA). The pH of the fusion medium was adjusted to 7.2–7.4 with L -histidine. After brief centrifugation, the cells were resuspended in the same fusion medium without BSA at a concentration of 15 × 106 cells/ml. Electrofusion was carried out using a custom made concentric chamber, connected to the ECM 2001 pulse generator (BTX Instrument, Genetronics, San Diego, CA). Fusion was accomplished by dielectropheresis with an alternating current (ac) pulse of 220 V/cm for 10 s immediately followed by a direct current (dc) pulse of 1230 V/cm for 99 μs. The cell suspensions were then resuspended in CM and incubated overnight in a 37 °C, 5% CO2 incubator. The adherent cell fraction containing fusion hybrids was harvested and analyzed for fusion rates by staining with PE-labeled mAbs against characteristic DC markers such as CD80, CD86, and I-A. Double-positive cells represented heterokaryons of DCs and MCA 205 tumor cells.
Active immunotherapy
Pulmonary metastases were induced by i.v. inoculations of mice with 3 × 105 tumor cells suspended in 1.0 ml Hanks’ balanced salt solution (HBSS). Three days later, mice were vaccinated by the intranodal route as previously described (14). Routinely, 3x105 fusion cells in 10 μl HBSS were delivered to each of 2 superficial inguinal lymph nodes. Some mice were additionally supplied i.p. with 100 ug polyinosine:polycytadilic acid (p[I:C]), or 50 ug ODN-1826, or a combination of both in 0.5 ml HBSS on the day of vaccination and again three and seven days later. For comparison, vaccinated mice were given either IL-12 (a gift from Wyeth, Cambridge, MA), 0.2 μg in 0.5 ml HBSS, i.p. for 4 consecutive days to provide a third signal or adjuvant. Some of the vaccinated mice were also treated with the neutralizing anti-IL-12 p70 mAb (R2-9A5) to block the in vivo activity of this cytokine. R2-9A5 was administered i.p. (0.45 mg) for six consecutive days starting the first day of vaccination with fusion hybrids and adjuvant. .
All mice were sacrificed on days 21–23, and metastatic nodules on the surface of the lung were enumerated after counter-stain with India ink as previously described (16, 18).
Surface-staining FACS analysis
For direct immunofluorescence, PE-conjugated mAbs including CD11c, CD80, CD86, CD40, H-2Kb, I-Ab, ICAM-1 (CD54), and OX-40L mAbs (BD PharMingen, San Diego, CA) were used for analyses of fusion products. At least 10,000 cells from each sample were analyzed using the FACS Calibur (BD, Mountain View, CA).
Cytokine analysis
Culture supernatants from 24 hour stimulated DC cultures were subjected to ELISA analysis for IL-12 p70, via OPT-EIA kits (BD/Pharmingen) according to manufacturer’s recommended protocol. For intracellular FACS, lymph node cells harvested 7 days after vaccination were polyclonally expanded with anti-CD3 mAb and IL-2 as described previously (11). Cells were then harvested and co-cultured with tumor targets at a 2:1 ratio in the presence of brefeldin A. After 24 hours, cells were harvested, stained with anti-CD4 or anti-CD8, permeabilized and stained for intracellular IFN-γ (all stains from BD, Mountain View, CA), and subjected to FACS analysis.
RESULTS AND DISCUSSION
This laboratory has pioneered a unique tumor vaccination strategy wherein electric fields are used to fuse DC to tumor cells. (Figure 1A, upper two panels). These DC-tumor hybrids inherit the antigen-presenting phenotype of the DC (as evidenced by the continued expression of I-A and CD80 in the fused cells, which unfused tumor cells lack), and the full antigenic complement of the tumor cell. These DC-tumor fusion vaccines have been demonstrated much more effective than DCs plus either tumor lysates or irradiated tumor (14). The very high fusion efficiency of this process was evidenced when tumor cells were labeled with the fluorescent dye, CFSE prior to fusion with DC (Figure 1A, lower panel). The population of cells in the double-positive quadrant (up to 45%) of the two-dimensional dot-plots represented the fusion hybrids that possess the characteristic surface staining of the DC and the CFSE staining of the tumor fusion partner.
Figure 1.
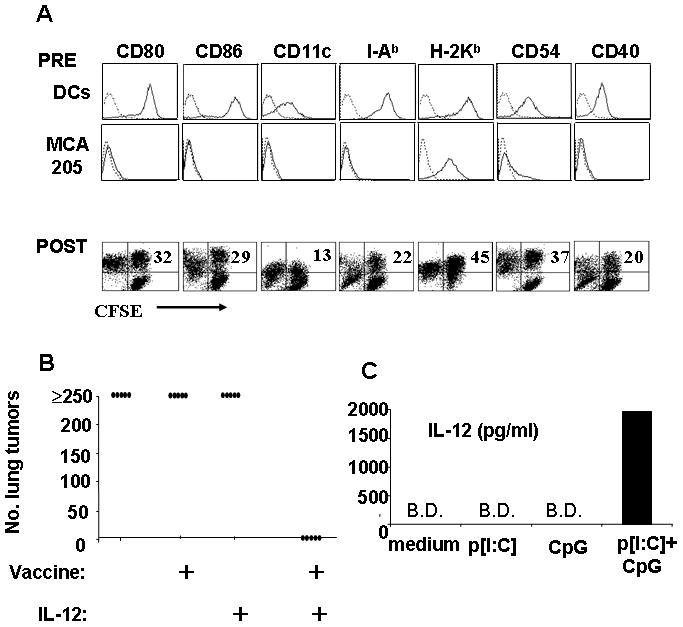
Electrofusion efficiently hybridizes MCA205 tumor cells and DCs to form anti-tumor vaccines that work in conjunction with IL-12 to suppress outgrowth of lung tumors. A, surface phenotype as determined by FACS of murine bone marrow-derived DCs and MCA205 tumor cells prior to fusion (“PRE”) and after fusion (POST). Tumor cells were labeled with CFSE prior to fusion. After 24 hours, adherent cells were stained with PE-conjugated mAbs as indicated. Numbers in upper right quadrants denote percentages of double-positive fusion hybrids. B, mice bearing 3-day MCA205 lung metastases were immunized i.n. with fusion hybrids or left unvaccinated. Some mice then received recombinant IL-12 adjuvant (0.2 μg/day over 4 consecutive days). On day 21–23 mice were sacrificed and lung surface metastases enumerated. C, murine bone marrow-derived DCs were cultured in medium with GM-CSF and IL-4 and then treated with combinations of p[I:C] (50 μg/ml) or ODN1826 (1μM) or both. Culture supernatants were removed 24h later and assayed for IL-12 p70 by ELISA (B.D.: below detection limit of 62 pg/ml).
Mice were immunized by direct injection of fusion vaccine into the superficial inguinal lymph nodes (14) to circumvent the need for lymph node trafficking. It was found that for vaccines to be effective against established tumors, an adjuvant was required, such as exogeneously-supplied recombinant interleukin 12 p70 (Figure 1B) or agonistic OX-40L monoclonal antibody (11). This property of adjuvant-dependency prompted us to examine the potential of alternative adjuvants for improving anti-tumor responses. Recently, the Toll-like family of transmembrane receptors (TLRs) have been identified as primary sensors for molecular patterns that distinguish self tissues from infectious non-self (19). We therefore sought to determine whether TLR agonist stimulation would serve an adjuvant function for DC-tumor fusion vaccination. Based on preliminary profiles of TLR expression in mouse-derived conventional DCs (not shown), we selected two TLR agonists, the double-stranded RNA analog, p[I:C] (TLR 3 agonist, TRIF pathway) and the synthetic CpG DNA analog ODN1826 (TLR9 agonist, MyD88 pathway). These TLR agonists were first used to stimulate cultured murine DCs in vitro to confirm synergistic induction of IL-12 p70 (Figure 1C). Supernatants from 24-hour stimulated cultures showed either low or no detectable IL-12 p70 in untreated DC, and in DC treated with single TLR agonists. On the other hand, combined treatment with p[I:C] and ODN1826 resulted in nanogram levels of IL-12 (Figure 1C).
We next evaluated this combination of TLR agonists as an adjuvant in conjunction with DC-tumor fusion hybrid vaccination in the murine MCA205 sarcoma model. Here, mice with 3-day established pulmonary metastases were vaccinated intranodally with fusion vaccine (day 0), and then supplied by the intra-peritoneal route with p[I:C] (100 ug/mouse), or ODN1826 (50 ug/mouse), or a combination of both on day 0, day 3 and day 7. On day 21, mice were sacrificed, and lung tumors enumerated. It was found, as historically demonstrated, that mice receiving no treatment displayed heavy tumor burdens, with over 250 metastatic nodules on the surface of the lung (Figure 2A). Likewise, immunization with fusion vaccine alone, or fusion vaccine plus single TLR agonist, had little or no significant effect on the outgrowth of tumor compared to untreated groups. However, mice receiving the fusion vaccine plus both TLR agonists proved to be either completely or mostly free of tumor (Figure 2A). Further experiments revealed that dual TLR agonist treatment alone did not impact tumor outgrowth, but instead had to be combined with vaccination, indicating that the agonists were serving an adjuvant function (Figure 2B). Results from a similar experiment are shown photographically in Figure 2C, with surface tumors visible as white nodules on a black counterstained background.
Figure 2.
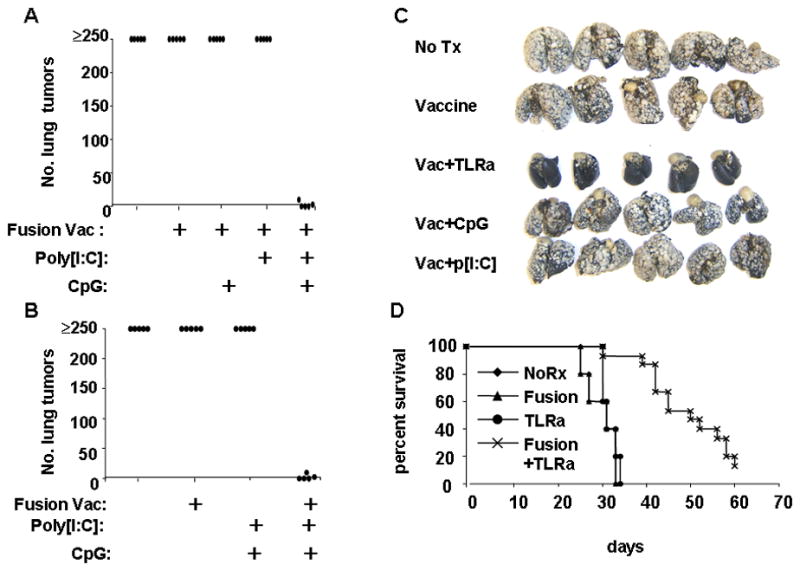
Therapeutic anti-tumor effect requires both vaccination and paired TLR agonist adjuvants. Mice were injected via the tail vein with MCA 205 tumor cells to establish 3-day lung metastases. Some groups of mice were then immunized intranodally with dendritic cell-tumor fusion vaccines. On the day of vaccination, and days 3 and 7 post-vaccination, combinations of p[I:C] (100 μg/mouse) and ODN 1826 (50 μg/mouse) were administered i.p. to both vaccinated and unvaccinated tumor-bearing mice. On days 21–23 mice were either sacrificed and lung surface metastatic nodules enumerated, or allowed to live until moribund or dead. A, Vaccination plus dual, but not single TLR agonists suppress lung metastases. B, Paired TLR agonists must be used in conjunction with vaccination for therapeutic effect. C, Photographic example of excised lungs counter-stained with India ink. D, Treated mice were then observed over a 60-day period with mortality as an endpoint. Panels A-C representative of 3 separate experiments.
We next sought to determine whether a single course of fusion vaccination plus dual TLR agonist treatment provided survival benefit (Figure 2D). It was found that mice receiving either no treatment, vaccination alone, or paired TLR agonist alone all succumb to growing metastases with a median survival time of around 30 days. However, mice receiving both vaccination plus paired TLR agonists displayed statistically prolonged survival, with median survival time extended to about 45 days.
Finally, we investigated mechanisms by which dual TLR agonist adjuvant enhanced vaccine therapeutic efficacy. Our original hypothesis was that induced secretion of IL-12 would be critical in this regard. We therefore designed experiments that would both directly test the necessity of IL-12 for tumor control as well as detect quantifiable changes in T cell activity consistent with the known effects of IL-12. To achieve this, we first prepared lymph node cells from tumor-bearing mice that had received either no treatment, paired TLR adjuvant only, fusion vaccine only, or combined vaccine plus adjuvant. These cells were polyclonally expanded in vitro and then co-cultured with either MCA205 tumors or, as a specificity control, MCA207 tumor cells, with IFN-γ production by CD4+ or CD8+ cells measured 24 hours later via intracellular FACS (Figure 3A). We observed little evidence of IFN-γ-secreting CD4+ or CD8+ cells in untreated, or adjuvant-only groups. However, specific IFN-γ secreting cells were seen from mice receiving fusion vaccine, with numbers and specificity increasing when dual TLR agonist adjuvant was combined with vaccination. This is an expected finding if IL-12 is influencing vaccine efficacy, since IL-12 enhances IFN-γ secretion by polarizing T cells toward the TH1 phenotype (8).
Figure 3.
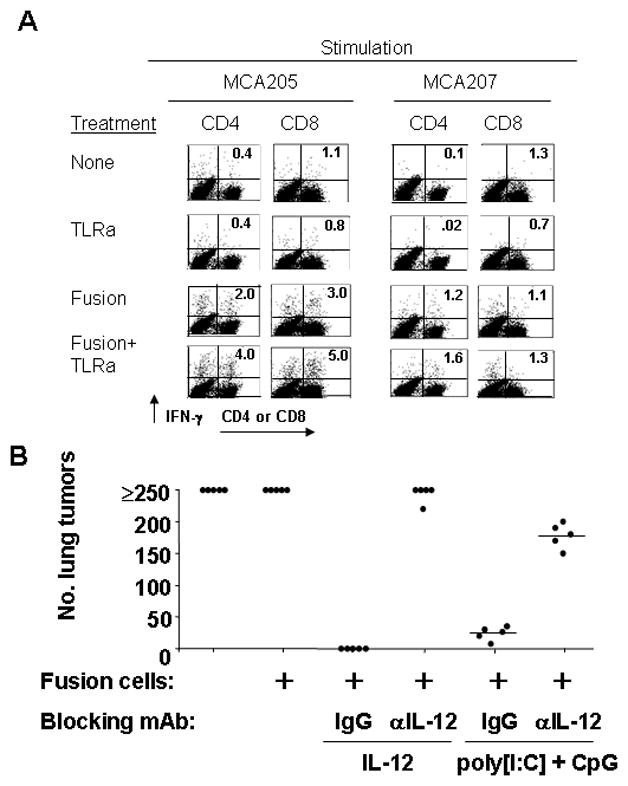
Vaccination plus paired TLR agonists enhance tumor-specific IFN-γ-secreting T cells and promote therapeutic effect through a mechanism mediated by IL-12. A, mice with 3-day MCA 205 lung metastases were immunized or not with DC-tumor fusion vaccines. Some mice then received paired TLR agonist adjuvant (p[I:C], 100 μg/ml, ODN1826, 50 μg/ml) as described previously. On day 7 mice were sacrificed and lymph node cells cultured for 5 days in the presence of anti-CD3 mAb (Day 1) and 4 IU/ml IL-2 (Day 3). Cells were then harvested and co-cultured at a 2:1 ratio with MCA 205 or MCA207 targets and IFN-γ production by CD4+ and CD8+ cells determined by intracellular FACS. B, mice bearing 3-day MCA 205 pulmonary metastases were likewise immunized intranodally with fusion vaccine. Some groups received either 0.45 mg IL-12-neutralizing monoclonal antibody (R2-9A5) or a rat IgG control for six consecutive days. Select groups also received adjuvants consisting of either recombinant murine IL-12 (0.2 μg/day over 4 consecutive days), or a suboptimal dose schedule of p[I:C] and ODN1826 ( 100 and 50 μg/mouse, respectively on days 0 and 3). Mice were sacrificed on says 21–23 and surface lung metastases enumerated.
Finally, we more directly tested the role of IL-12 through in vivo neutralization. To accomplish this, either IL-12-neutralizing or normal rat IgG was given i.p. to fusion vaccine-treated, tumor-bearing mice that received either recombinant IL-12 or paired TLR agonists (two rather than the optimal three doses) as adjuvant. When mice were sacrificed, and lung metastases enumerated, it was apparent that IL-12-neutralizing, but not control antibody inhibited the adjuvant properties of paired TLR agonists (Figure 3B). This inhibition was quite strong, but nonetheless incomplete, suggesting either that induction of other factors, possibly Delta Notch Ligand or IL-23 (known to be up-regulated by paired TLR agonists) (2) contribute to the adjuvant effect, or that IL-12 exerts some of its effects via close cell-cell interactions (such as across an immunosynapse between a DC and T cell), and therefore cannot be readily neutralized to completion. Nonetheless, these experiments show that IL-12 plays significant role in the adjuvant properties of paired TLR agonists for therapeutic vaccination against tumors.
Agonists for TLRs have found use as adjuvants or immune response modifiers in a variety of settings, both experimental and clinical for infections and malignancy, but relatively limited adjuvant impacts have been observed with single TLR agonists. To our knowledge, this is the first example of in vivo, low toxicity synergy between paired TLR agonists (that collectively signal through both MyD88 and TRIF pathways) enhancing DC-based vaccines through apparent in vivo induction of IL-12.
For its part, IL-12 has steadily gained prominence as a major factor in controlling malignancies. We have demonstrated that appropriately stimulated DCs can achieve two surges of IL12p70 production ex vivo, but strategies are required to ensure that the timing of surges is optimal to maximize T cell sensitization. Towards this end, we have explored the feasibility of treating extracorporealized monocyte-derived DCs with the combination of lipopolysaccharide and IFN-γ just prior to their intranodal administration to patients. We recently employed this strategy to treat early breast cancer (20). Intranodal vaccination with HER-2/neu peptide antigen-pulsed DCs that secreted large amounts of IL-12 elicited powerful immunity as well as apparent reduction of HER-2/neu expression in tumors from over half the vaccinated subjects (20).
Remarkably, however, the present murine studies demonstrate that DC1-polarization, or at least therapeutically significant host IL12p70 production, can also be safely triggered by parenteral administration of appropriately paired TLR agonists, potentially reducing the need to expose DCs to these stimuli prior to their administration. We observed no detectable toxicities at the studied TLR agonist doses (save for transient spleenic enlargement), despite the high therapeutic activity. The demonstration of TLR agonist synergy in vivo for a DC-based tumor vaccination regimen may have profound implications for the formulation and use of small synthetic TLR ligands as adjuvants or immune response modifiers. These studies supply further evidence that IL-12 is a critical factor for tumor control and argue that methods to elicit powerful 3rd signals such as IL-12 should be a high priority for future cancer vaccination strategies.
Acknowledgments
This work was supported by NIH grant CA100163 (G.K.K). CA089511 (P.A.C), CA103946 (S.S.), CA129815 (P.A.C)
Reference List
- 1.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–98. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 2.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–76. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–51. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curtsinger JM, Lins DC, Johnson CM, Mescher MF. Signal 3 tolerant CD8 T cells degranulate in response to antigen but lack granzyme B to mediate cytolysis. J Immunol. 2005;175:4392–99. doi: 10.4049/jimmunol.175.7.4392. [DOI] [PubMed] [Google Scholar]
- 5.Valenzuela J, Schmidt C, Mescher M. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J Immunol. 2002;169:6842–49. doi: 10.4049/jimmunol.169.12.6842. [DOI] [PubMed] [Google Scholar]
- 6.Valenzuela JO, Hammerbeck CD, Mescher MF. Cutting edge: Bcl-3 up-regulation by signal 3 cytokine (IL-12) prolongs survival of antigen-activated CD8 T cells. J Immunol. 2005;174:600–4. doi: 10.4049/jimmunol.174.2.600. [DOI] [PubMed] [Google Scholar]
- 7.Xu S, Koski GK, Faries M, et al. Rapid high efficiency sensitization of CD8+ T cells to tumor antigens by dendritic cells leads to enhanced functional avidity and direct tumor recognition through an IL-12-dependent mechanism. J Immunol. 2003;171:2251–61. doi: 10.4049/jimmunol.171.5.2251. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–49. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 9.Shu S, Cohen P. Tumor-Dendritic Cell Fusion Technology and Immunotherapy Strategies. J Immunother. 2001;24:99–00. [PubMed] [Google Scholar]
- 10.Kjaergaard J, Wang LX, Kuriyama H, Shu S, Plautz GE. Active immunotherapy for advanced intracranial murine tumors by using dendritic cell-tumor cell fusion vaccines. J Neurosurg. 2005;103:156–64. doi: 10.3171/jns.2005.103.1.0156. [DOI] [PubMed] [Google Scholar]
- 11.Kuriyama H, Watanabe S, Kjaergaard J, et al. Mechanism of third signals provided by IL-12 and OX-40R ligation in eliciting therapeutic immunity following dendritic-tumor fusion vaccination. Cell Immunol. 2006;43:30–40. doi: 10.1016/j.cellimm.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Shu SY, Rosenberg SA. Adoptive immunotherapy of newly induced murine sarcomas. Cancer Res. 1985;45:1657–62. [PubMed] [Google Scholar]
- 13.Plautz GE, Touhalisky JE, Shu S. Treatment of murine gliomas by adoptive transfer of ex vivo activated tumor-draining lymph node cells. Cell Immunol. 1997;178:101–7. doi: 10.1006/cimm.1997.1140. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu K, Kuriyama H, Kjaergaard J, Lee W, Tanaka H, Shu S. Comparative analysis of antigen loading strategies of dendritic cells for tumor immunotherapy. J Immunother. 2004;27:265–72. doi: 10.1097/00002371-200407000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–2. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi T, Tanaka H, Tanaka J, et al. Immunogenicity and therapeutic efficacy of dendritic-tumor hybrid cells generated by electrofusion. Clin Immunol. 104:14–20. doi: 10.1006/clim.2002.5224. [DOI] [PubMed] [Google Scholar]
- 17.Kjaergaard J, Shimizu K, Shu S. Electrofusion of syngeneic dendritic cells and tumor generates potent therapeutic vaccine. Cell Immunol. 2003;225:65–74. doi: 10.1016/j.cellimm.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka H, Shimizu K, Hayashi T, Shu S. Therapeutic immune response induced by electrofusion of dendritic and tumor cells. Cell Immunol. 2002;220:1–12. doi: 10.1016/s0008-8749(03)00009-1. [DOI] [PubMed] [Google Scholar]
- 19.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol. 2005;560:11–18. doi: 10.1007/0-387-24180-9_2. [DOI] [PubMed] [Google Scholar]
- 20.Czerniecki BJ, Koski GK, Koldovsky U, et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007;67:1842–52. doi: 10.1158/0008-5472.CAN-06-4038. [DOI] [PubMed] [Google Scholar]