

Significance
Androgens are primarily considered detrimental to women’s health. However, androgen-receptor KO mouse models have been used to establish that androgens are actually necessary for normal ovarian function and female fertility. Despite these observations, how androgens regulate female fertility is not known. Here we show that androgens promote follicular development via two mechanisms: (i) prevention of follicular atresia by inducing the expression of an antiapoptotic microRNA (miR), miR-125b; and (ii) promotion of follicle growth by increasing follicle-stimulating hormone receptor levels in a transcription-independent fashion. These data considerably change our understanding of androgen effects in female reproduction, and help explain the ovarian physiology seen in patients with too little or too much androgen.
Abstract
Although androgen excess is considered detrimental to women’s health and fertility, global and ovarian granulosa cell-specific androgen-receptor (AR) knockout mouse models have been used to show that androgen actions through ARs are actually necessary for normal ovarian function and female fertility. Here we describe two AR-mediated pathways in granulosa cells that regulate ovarian follicular development and therefore female fertility. First, we show that androgens attenuate follicular atresia through nuclear and extranuclear signaling pathways by enhancing expression of the microRNA (miR) miR-125b, which in turn suppresses proapoptotic protein expression. Second, we demonstrate that, independent of transcription, androgens enhance follicle-stimulating hormone (FSH) receptor expression, which then augments FSH-mediated follicle growth and development. Interestingly, we find that the scaffold molecule paxillin regulates both processes, making it a critical regulator of AR actions in the ovary. Finally, we report that low doses of exogenous androgens enhance gonadotropin-induced ovulation in mice, further demonstrating the critical role that androgens play in follicular development and fertility. These data may explain reported positive effects of androgens on ovulation rates in women with diminished ovarian reserve. Furthermore, this study demonstrates mechanisms that might contribute to the unregulated follicle growth seen in diseases of excess androgens such as polycystic ovary syndrome.
Other than the obligatory role of androgens as estrogen precursors in steroidogenesis (1), little is known about the direct involvement of androgens in the female ovary. For many decades, excess androgens in women have been considered detrimental to women’s health, as diseases such as polycystic ovary syndrome (PCOS) are associated with reduced fertility. In the past, these negative effects of androgens on female fertility were thought to occur primarily at the level of the hypothalamus and pituitary (2, 3), although important data across different species (4–7) suggested that androgens could also directly promote follicle growth (8, 9). Attitudes about androgen actions in female fertility changed with the development of global androgen-receptor knockout (ARKO) mice (10–12). The female ARKO mice had considerable reproductive defects, with decreased fertility, defective follicular development, reduced ovulation, and premature ovarian failure. In other words, the phenotype of these ARKO mice suggested that androgen signaling might actually be important for normal female reproductive health. Intriguingly, through the generation of granulosa cell (GC)-specific ARKO mice, we (13) and then others (14) demonstrated that essentially all the observed reproductive phenotypes in the complete AR-null mice are caused by androgen actions in GCs. These results highlighted that, with regard to fertility, androgen signaling in the ovary is at least as important as androgen signaling in the pituitary or hypothalamus. By using this GC-specific ARKO mouse model, we further demonstrated that androgens regulate follicular development and female fertility by attenuating follicular atresia while simultaneously promoting preantral follicle growth and development into antral follicles. However, these in vivo studies did not elucidate specific mechanisms used by androgens and ARs to mediate these processes.
Here we performed an in-depth analysis of androgen-induced signaling pathways that regulate ovarian folliculogenesis. Androgens signal via extranuclear (nongenomic) and nuclear (genomic) pathways. In fact, previous work by others and ourselves in androgen-sensitive prostate cancer cells showed that these two processes are tightly linked, with maximal AR-mediated nuclear transcription requiring extranuclear AR signaling (15). We reported that, through membrane-localized ARs, androgens trigger matrix metalloproteinase (MMP)-mediated transactivation of the epidermal growth factor receptor (EGFR), which in turn leads to cytoplasmic Akt and MAPK3/1 signaling. MAPK3/1 signaling is then required for normal nuclear AR transcriptional effects (16, 17). Interestingly, we have shown (16) that paxillin, a scaffold protein traditionally thought to regulate cytoskeletal remodeling and focal adhesion function, is an essential mediator of extranuclear and nuclear AR signaling. Paxillin first regulates Src-induced MAPK3/1 activation at the membrane. Upon activation, MAPK3/1 promotes serine phosphorylation of paxillin, resulting in the nuclear localization of phosphoserine (PS)-paxillin. Once in the nucleus, PS-paxillin then complexes with the AR to retain it in the nucleus, allowing AR-mediated transcription to occur. In fact, in the absence of paxillin, AR is no longer retained in the nucleus of prostate cancer cells or primary ovarian GCs, even in the presence of androgen (16). Thus, paxillin serves as a critical “liaison” between extranuclear kinase signaling and intranuclear transcription (17).
Here we describe two paxillin-dependent AR-induced regulatory mechanisms in GCs that use extranuclear and nuclear AR signaling. These pathways likely account for the majority of the observed androgen actions in follicular development. First, we show that ARs enhance the expression of the antiapoptotic microRNA (miR) miR-125b, which likely contributes to androgen-induced follicular survival. Second, we demonstrate that androgens increase follicle-stimulating hormone (FSH) receptor (FSHR) protein, but not mRNA, expression, thus sensitizing follicles to FSH actions and potentially contributing to androgen-mediated follicle growth. These positive effects of androgens on follicle development may play a critical role in gonadotropin-induced ovulation, and may serve as potential targets to enhance fertility in women with decreased follicle growth and ovulation as a result of diminished ovarian reserve.
Results
Nuclear and Extranuclear Actions of ARs Regulate Expression of miR-125b in GCs.
In this study we report that androgens regulate follicular atresia by inducing the expression of miR-125b, an miR known to suppress proapoptotic protein expression (18). Results show that miR-125b is expressed in mouse GCs (Fig. 1A), human KGN GC tumor cells (Fig. 1B), and primary human GCs isolated during oocyte retrieval from women undergoing in vitro fertilization (Fig. S1D). Importantly, the androgens dihydrotestosterone (DHT) and testosterone, in contrast to other steroids (estradiol and R5020), induce expression of miR-125b (Fig. 1A), but not miR-125a, a related miR of the same miR family (Fig. S2B), in primary GCs.Notably, basal expression of miR-125b is high in KGN cells. Thus, DHT-induced induction of miR-125b expression is subdued but still significant. This may be because tumor cells such as KGN have suppressed apoptosis, perhaps maintained in part through increased expression of antiapoptotic miRs such as miR-125b.
Fig. 1.

(A and B) Androgens [DHT and testosterone (T)] but not estradiol (E2) or the progestin R5020 induce miR-125b expression in GCs after 18 h. Relative expression of miR-125b in (A) primary mouse GCs and (B) the human granulosa-like tumor cell line, KGN. Data are displayed as means ± SEM (n = 3) and normalized to GAPDH levels (*P ≤ 0.05 vs. media). (C and D) DHT stimulates AR association with the mouse miR-125b-2 promoter: (C) Schematics showing that the human and mouse 5′ UTRs of the miR-125b-2 promoter contain four (I, −316/−266; II, −616/−567; III, −816/−767; IV, −2,628/−2,579) and three (I, −2,400/−2,350; II, −2,533/−2,483; III, −2,733/−2,783) AREs, respectively. (D) ChIP assay in primary mouse GCs showing that ARs bind to the AREs in the miR-125b-2 promoter after 45 min with DHT (25 nM). Immunoprecipitation of AR was equal between media- and DHT-treated cells (Inset). IgG represents nonspecific antibody and “-C” represents nonspecific primers within the promoter. Values represent percentage input (means ± SEM, n = 3; *P ≤ 0.05 vs. media).
Mature miR-125b originates from two precursors: miR-125b-1 and miR-125b-2. In mice and humans, the 5′ UTRs of the miR-125b-2, but not miR-125b-1, promoters contain three and four androgen responsive elements (AREs; Fig. 1B), respectively, suggesting that the AR might interact directly with these AREs to induce miR-125b expression. In fact, ChIP assays in primary mouse GCs reveal that ARs associate with all three AREs in the 5′ UTR region of the mouse miR-125b-2 promoter in a DHT-dependent fashion (Fig. 1C); thus, androgens likely up-regulate miR-125b expression through AR-mediated transcription of miR-125b-2.
As mentioned, our previous work in prostate cancer cells showed that AR-mediated transcription in the nucleus requires extranuclear AR signaling through transactivation of the EGFR and subsequent MAPK3/1 signaling in the cytoplasm (16, 17, 19). Similarly, in primary mouse GCs, DHT promotes rapid extranuclear MAPK3/1 signaling (Fig. S1E). Addition of an AR antagonist (flutamide), an MMP inhibitor (Galardin), or an EGFR inhibitor (AG1478) inhibits DHT-induced MAPK3/1 activation (Fig. S1E), confirming that, as seen in prostate cancer cells, androgen-induced MAPK3/1 signaling occurs via transactivation of the EGFR through MMP-mediated release of membrane bound EGFR ligands in GCs. Furthermore, as in prostate cancer cells, androgen-triggered extranuclear MAPK3/1 signaling is critical for nuclear AR-mediated transcription in GCs, as the mitogen-activated protein kinase kinase (MEK) inhibitor U0126 completely abrogates DHT-induced up-regulation of miR-125b (Fig. 2A). Finally, consistent with our observation that paxillin is required for AR nuclear localization in GCs (16), siRNA-mediated knockdown of paxillin in primary mouse GCs inhibits DHT-induced MAPK3/1 signaling, whereas reexpression of paxillin restores MAPK3/1 signaling (Fig. 2B). Paxillin knockdown also blocks DHT-induced miR-125b expression, and EGF alone is not sufficient to promote miR-125b expression (Fig. 2A), confirming the need for paxillin-mediated nuclear and extranuclear AR crosstalk to regulate miR-125b expression in GCs.
Fig. 2.
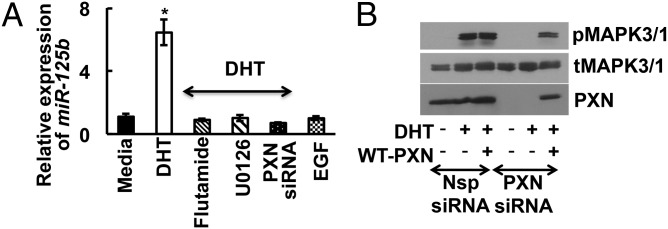
Extra- and intranuclear androgen signaling regulate miR-125b expression in GCs. (A) Relative expression of miR-125b in primary mouse GCs treated with vehicle (0.1% DMSO), flutamide (AR inhibitor; 100 nM), or U0126 (MEK inhibitor; 10 μM), and in siRNA-mediated paxillin (PXN) knockdown GCs stimulated with/without DHT (25 nM) for 30 min. EGF is a negative control. Data are displayed as means ± SEM (n = 3) and normalized to GAPDH levels (*P ≤ 0.05 vs. media). (B) Representative immunoblots of total MAPK3/1, phosphorylated MAPK3/1, and paxillin in nonspecific (Nsp) or paxillin-specific siRNAs treated mouse primary GCs. For rescue experiments, following siRNA treatment GCs were transfected with WT paxillin. Results are representative of three separate experiments.
Androgen-Induced miR-125b Suppresses Proapoptotic Protein Expression in GCs, Resulting in Follicular Survival.
We used TargetScan algorithm 6.2 (Whitehead Institute for Biomedical Research) to find that Bcl-2 homologous antagonist/killer (BAK1), Bcl-2 modifying factor (BMF), Bcl-2-associated X protein (BAX), and the tumor suppressor protein p53 (TP53), are potential targets of miR-125b. As these are antiapoptotic proteins, we tested whether androgens protect GCs from apoptosis. In fact, DHT significantly reduces apoptosis in primary mouse GCs, as determined by flow cytometry for Annexin V expression (Fig. 3A). Moreover, down-regulation of miR-125b in primary GCs using an miR-125b-2 inhibitor (Fig. S2A) increases expression of proapoptotic proteins (Fig. 3B) as well as the number of apoptotic cells (Fig. 3C). Notably, with reduction of miR-125b expression, DHT no longer protects cells from apoptosis (Fig. 3C), confirming that miR-125b at least in part mediates DHT-induced suppression of apoptosis.
Fig. 3.

(A) DHT protects GCs from apoptosis. Percentage of apoptotic cells measured by flow cytometry for Annexin V in primary mouse GCs treated with media or 25 nM DHT for 24 h. Data are means ± SEM (n = 5; *P ≤ 0.05 vs. media). (B and C) miR-125b suppresses proapoptotic proteins in GCs in vitro. Proapoptotic protein levels (B) and percentage of apoptotic cells (C) in primary mouse GCs transfected with mouse miR-125b-2 mirVana miR inhibitor or nonspecific control and stimulated with media or 25 nM DHT for 24 h. For C, data are means ± SEM (n = 3; *P ≤ 0.05 vs. nonspecific). (D–F) In vivo knockdown of miR-125b-2 increases follicular atresia. Relative expression of miR-125b (D) and proapoptotic protein levels and RPL19 control (E) in GCs and representative TUNEL-stained ovarian sections (F) from animals (n = 3) injected with LNA-containing oligonucleotides targeting miR-125b-2 into one ovarian bursa and vehicle control injected in the other corresponding ovary. For D, data are means ± SEM (n = 3) and normalized to GAPDH (*P ≤ 0.05 vs. control).
To determine the in vivo effects of miR-125b on ovarian apoptosis, we knocked down miR-125b expression in vivo (Fig. 3D) by injecting locked nucleic acid (LNA)-containing oligonucleotides targeting miR-125b-2 into one ovarian bursa whereas the other corresponding ovary received vehicle. Ovaries were collected 72 h postsurgery and analyzed for apoptosis. Levels of proapoptotic proteins BAK, BAX, BMF, and p53 in GCs (Fig. 3E), as well as follicular atresia (measured by TUNEL assay; Fig. 3F), are significantly elevated in miR-125b–knockdown ovaries compared with corresponding vehicle-treated ovaries. Morphological analysis (n = 3) reveals that 54.7% of follicles are atretic in LNA-oligonucleotide–injected vs. 33% in control ovaries (P ≤ 0.05).
Finally, in ovaries of GC-specific ARKO mice, our reported high rate of follicular atresia (13) corresponds with lower expression of miR-125b (Fig. 4A) as well as higher expression of proapoptotic proteins and caspase 3 activity compared with control littermates (Fig. 4 B and C). Thus, in vitro and in vivo results indicate that, in GCs, paxillin-mediated nuclear and extranuclear androgen signaling induces miR-125b, which then suppresses proapoptotic protein expression, leading to decreased follicular atresia and increased follicular survival.
Fig. 4.
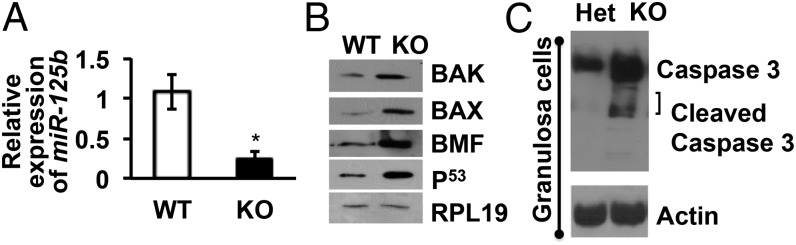
Expression of miR-125b and proapoptotic proteins are significantly low and high, respectively, in primary GCs isolated from GC-specific ARKO mice. (A) Relative expression of miR-125b as measured by quantitative real-time PCR. Data are displayed as means ± SEM (n = 3 mice per genotype) and normalized to GAPDH levels (*P ≤ 0.05 vs. WT). (B) Representative Western blots (from n = 3 mice per genotype) for proapoptotic proteins and RPL19 in GCs isolated from WT and GC-specific ARKO (KO) ovaries. (C) Representative Western blots (from n = 3 mice per genotype) detecting for caspase 3 activity and actin control in GCs isolated from ovaries of GC-specific ARKO (KO) mice and heterozygous (Het) littermates.
Androgens Increase FSHR Protein Levels in a Transcription-Independent (Nongenomic) Fashion.
Our GC-specific ARKO mouse model revealed that, whereas androgens suppress follicular atresia, they might also promote follicle growth and development. Previous work in animals (4–8, 20, 21) and humans (22, 23) suggested that androgens might augment FSH function in the ovary. To test whether androgens promote follicle growth by enhancing FSH actions, we examined androgen-induced FSHR mRNA and protein levels in primary mouse GCs. Neither DHT (Fig. 5A) nor testosterone (Fig S2D) alters FSHR mRNA levels, but both significantly increase FSHR protein expression (Fig. 5B and Fig. S2D), suggesting that androgens promote FSHR protein expression independent of transcription. Pulse-chase studies with cells exposed to the translation inhibitor cycloheximide (CHX) with or without DHT reveal no difference in FSHR protein degradation (Fig. 5C; quantified in Fig. S3A), indicating that DHT promotes FSHR protein expression by enhancing translation through extranuclear AR signaling. Accordingly, inhibition of the nongenomic AR-induced signaling by using flutamide, the MEK inhibitor U0126, or siRNA-mediated paxillin knockdown abrogates DHT-induced FSHR expression, whereas reexpression of paxillin in paxillin-knockdown GCs partially rescues DHT-induced FSHR expression (Fig. 5D; quantified in Fig. S3B).
Fig. 5.

Androgens increase FSHR protein, but not mRNA, levels in a transcription-independent fashion. (A) Relative expression of FSHR mRNA levels by quantitative PCR in primary mouse GCs stimulated with or without DHT for 24 h. Results are normalized to GAPDH. (B) Quantitative densitometric analysis of immunoblots from three separate experiments (means ± SEM; *P ≤ 0.05) showing relative increase in FSHR protein levels in mouse primary GCs upon DHT stimulation. Representative Western blot for FSHR and RPL19 expression (Inset). (C) Androgens do not suppress FSHR protein degradation. Time course of FSHR protein degradation in presence of the translational inhibitor cycloheximide (CHX) (1 μM) in primary mouse GCs treated with or without DHT. (D) Extranuclear androgen signaling regulates DHT-induced increase in FSHR protein levels. Representative Western blots of FSHR and RPL19 protein levels (n = 3 with similar results) in primary mouse GCs treated with flutamide (100 nM), U0126 (10 μM) or in siRNA-mediated paxillin (PXN) knockdown primary mouse GCs following DHT for 30 min. For rescue experiments, paxillin-knockdown GCs were transfected with WT paxillin (PXN). Quantitative analysis for C and D is shown in Fig. S3.
Androgen Treatment Enhances Follicular Sensitivity Toward FSH Actions.
As androgens increase FSHR expression in primary mouse GCs, we postulated that this might enhance FSH-mediated follicle growth. Preantral follicles were collected and treated with FSH (10 ng/mL) plus increasing amounts of DHT. Although FSH alone promotes follicular growth, addition of DHT in a dose-dependent manner significantly increases follicular growth (Fig. 6A), suggesting that androgens indeed enhance FSH-mediated follicular growth.
Fig. 6.

(A) Androgens enhance FSH-mediated in vitro follicle growth in a dose-dependent manner. Increase in diameter of follicles cultured 4 d with 10 ng/mL FSH and with or without different concentrations of DHT. The difference in follicular diameter from day 0 and day 4 of culture is presented as a measure of follicle growth (*P ≤ 0.05 vs. no DHT, n = 15 follicles per treatment). (B) Androgens enhance FSH-mediated preantral to antral follicle transition in a whole ovary culture. Percentage of antral follicles in ovaries cultured with different concentrations of FSH in presence or absence of DHT (25 nM) for 5 d. Data are presented as percent of antral follicles relative to total follicles counted (*P ≤ 0.05 vs. no DHT, n = 3 ovaries per treatment). (C) Proposed model for androgen actions in follicular development. Based on work in prostate cancer cells (16, 17), androgens signal via classical ARs and extranuclear paxillin to promote MAPK3/1 signaling. MAPK3/1 then promotes increased FSHR protein expression that enhances the sensitivity of preantral follicles to FSH-mediated growth. In addition, extranuclear AR-mediated activation of MAPK3/1 leads to phosphorylation of paxillin on serines (P, paxillin), allowing paxillin to enter the nucleus and mediate AR-induced transcription of miR-125b. This miR suppresses apoptosis, thus attenuating preantral follicle atresia. The resultant androgen-mediated increased follicle growth plus decreased follicular atresia promotes follicle development and subsequent ovulation.
To determine whether androgens similarly enhance FSH-mediated transition from preantral to antral follicles in a whole ovary, intact mouse ovaries were cultured (Fig. 6B) with different concentrations of FSH in the presence or absence of DHT (25 nM). After culture and treatment, ovaries were fixed, sectioned, and stained for morphological analysis. DHT alone has no effect on antral follicle formation relative to untreated ovaries, whereas FSH alone increases follicle progression to the antral stage in a dose-dependent manner (Fig. 6B). Importantly, addition of DHT significantly enhances FSH-induced antral follicle formation (Fig. 6B). Thus, although androgens are neither sufficient to promote follicle progression nor necessary for FSH-induced follicle progression, they increase the sensitivity of preantral follicles toward FSH, thus optimizing transition from preantral to antral follicles.
Preovulatory Androgen Priming Enhances Oocyte Ovulation.
As androgens enhance gonadotropin-induced follicle growth and development, we determined whether androgens augment gonadotropin-mediated ovulation. We injected mice for 3 d with DHT (0, 0.25, or 25 mg) before superovulation with pregnant mare's serum gonadotropin (PMSG) and human chorionic gonadotropin. To avoid overwhelming DHT effects with excess stimulation, 2 IU of PMSG and 2 IU of hCG were administered. Ovulated oocytes were then isolated from the oviducts of the animals and counted. DHT pretreatment has no effect on the number of ovulated oocytes in prepubertal (4 wk old) animals (Table 1), consistent with our previous data showing that GC-specific ARKO ovaries of 4-wk-old animals are normal. In contrast, pretreatment with 0.25 mg of DHT significantly increases the number of ovulated oocytes in 8- to 12-wk-old (13.6 ± 0.8 vs. 19 ± 0.6) and 24- to 28-wk-old (12.2 ± 0.5 vs. 16.6 ± 1.2) animals (Table 1), consistent with our model that androgens enhance gonadotropin-mediated follicle progression. Intriguingly, animals pretreated with higher amounts of DHT (25 mg) trend toward fewer superovulated oocytes compared with controls (Table 1).
Table 1.
Preovulatory androgen treatment enhances ovulation
| Time point | DHT (mg) |
||
| 0 | 0.25 | 25 | |
| Prepubertal | 14 ± 0.6 | 13.5 ± 0.6 | 14.1 ± 0.8 |
| 8–12 wk | 13.6 ± 0.8 | 19.0 ± 0.6* | 11.2 ± 1.0 |
| 24–28 wk | 12.2 ± 0.5 | 16.6 ± 1.2* | 11.0 ± 1.0 |
Mice were injected i.p. for 3 d with DHT as indicated before superovulation (i.p. injection of 2 U PMSG followed by 2 U hCG 48 h later). At 18 h after the hCG injection, ovulated oocytes were counted. *P ≤ 0.05 vs. no DHT (n = 10 mice per condition).
Discussion
Although global (11, 12) and GC-specific (13) ARKO mouse models, as well as in vitro studies (4–6, 8, 9, 20, 21, 24, 25), established the importance of androgens and ARs in follicular development, the present study provides insight into the molecular mechanisms of AR actions in the ovary. Previous work (26, 27) established that the balance between follicular atresia and follicle growth is essential for normal ovarian function, and we find that androgens play a key role in this regulation. We show that the physiological effects of androgens in GCs involve paxillin-mediated synergistic action between nuclear and extranuclear signaling of ARs that induces expression of the antiapoptotic miR-125b and up-regulates expression of the proproliferative FSHR.
First, we use primary GCs, GC-specific AR-null mice and in vivo ovary knockdown experiments to show that androgens promote miR-125b expression, which then suppresses expression of proapoptotic protein markers and subsequent apoptosis and follicle atresia. Notably studies report (18, 28) that miR-125b is similarly up-regulated by androgens in prostate cancer, and may play a crucial role in cancer cell proliferation and metastasis (29). With respect to miRs in female reproductive tissue, most work has focused on miR profiling rather than mechanism, and miR-125b is reported to be one of the most abundant miRs in the ovary among different species (30, 31). In fact, little is known about the expression, regulation, and function of miRs in the ovary (32). Exceptions include miR-17-5p and let-7b, which regulate corpus luteum development and function (33); miR-21, an LH-induced miR (34) that may play an antiapoptotic role during ovulation (35); miR-224, a TGF-β1–induced miR that targets Smad4 (36); and miR-378, which regulates estradiol synthesis by targeting aromatase (37). Our study not only demonstrates miR-125b as an important regulator of follicular atresia, but also provides detailed insight as to the hormonal regulation of miR-125b by androgens in the ovary. Importantly, few transcriptional targets of androgens have been identified in the ovary (10–12, 38–40); our results add to this short list by establishing miR-125b as a direct target of AR in GCs.
Second, we perform an in-depth analysis of the androgen signaling pathways that regulate miR-125b expression in GCs, demonstrating the importance of extranuclear androgen signaling in ovarian follicles. We show that, in GCs, androgens use a conserved nongenomic pathway (16, 17) to rapidly activate kinase signaling via transactivation of the EGF receptor. Furthermore, we show that extranuclear AR signaling is required for AR-mediated transcriptional effects on miR-125b, confirming and adding to earlier data (16) demonstrating that extranuclear androgen signaling is essential for AR nuclear localization and subsequent genomic androgen signaling (16).
Third, we show that paxillin is a critical regulator of extranuclear, and therefore nuclear, AR signaling in primary mouse GCs. These results mirrors previous work in prostate cancer cells (17) showing that paxillin is required for AR-induced Erk activation, after which Erk-dependent serine phosphorylation of paxillin leads to nuclear localization of PS-paxillin. Once in the nucleus, PS-paxillin then regulates AR nuclear localization and AR-mediated transcription (16). This demonstration of a conserved function of paxillin in vastly different cell types validates the importance of paxillin as a general liaison between extranuclear and nuclear AR signaling.
Fourth, we examine the molecular mechanisms behind observations (6, 41, 42) that androgens can up-regulate FSHR expression. We use mouse primary GCs, whole ovary culture, and primary follicle culture experiments to show that androgens promote FSHR protein translation in an exclusively nongenomic fashion, by using the same paxillin-dependent extranuclear pathway described earlier (Fig. 6C). By increasing FSHR expression, we show that androgens then enhance sensitivity to FSH-induced follicle growth and progression to the antral stage. Detailed mechanisms explaining how AR and paxillin selectively regulate FSHR protein are still not known; however, previous studies (43, 44) suggest that paxillin might up-regulate protein expression by complexing with polyadenylate-binding protein 1 (PABP1) to assist in translation. Notably, some studies (24, 45) report that androgens may induce FSHR mRNA expression. However, these studies were performed in prepubertal animals (45) or in human luteinized GCs isolated during oocyte retrieval for in vitro fertilization (24). Furthermore, concentrations of androgens used to stimulate these cells were quite high. Thus, androgenic effects on follicle development may be concentration- and age-dependent, consistent with reports of age-dependent differential effects of hormones and growth factors (46, 47). For our studies, we did not use prepubertal animals because prepubertal GC-specific ARKO animals still had normal follicular development (13). Furthermore, to avoid unwanted effects caused by pretreatment with pharmacologic gonadotropin concentrations, we used freshly isolated primary GCs from unprimed mice. Although androgen-induced increases in FSHR levels may be one mechanism by which androgens facilitate follicular growth, it is likely that androgens also modulate post-FSHR signaling, such as FSH-induced induction of cAMP (25, 48, 49).
In summary, our studies show that androgens regulate two critical stages of follicular development: follicular atresia and preantral follicle growth. In GCs, androgens through extranuclear AR signaling and nuclear AR actions synergistically induce the expression of miR-125b. The latter targets proapoptotic proteins, thus suppressing follicular atresia (Fig. 6C). In addition, androgens increase FSHR protein levels in a transcription-independent (nongenomic) fashion that increases the sensitivity of the follicles toward FSH, promoting preantral follicle growth and progression to antral follicles. Importantly, both these AR actions are regulated by paxillin (Fig. 6C). As we show that preovulatory androgens can promote follicle development and increase ovulation rates, whereas androgen excess is well established as one of the major causes of PCOS, we propose that there exists a critical balance between the essentiality of androgens in normal follicular development and the detrimental effects of androgens in the setting of androgen excess. In fact, the concept that androgens can promote follicle development is supported by the reported positive effects of androgen treatment in animals (7) as well as in patients with diminished ovarian reserve (22, 23, 50), in whom androgen priming is now being used in clinical settings to enhance ovulation (51, 52).
Materials and Methods
Animals and Cell Culture.
Mouse studies were performed in accordance with the guidelines for the care and use of laboratory animals and were approved by the University Committee on Animal Resources at the University of Rochester. Unless otherwise mentioned, mouse experiments were performed in 8- to 12-wk-old C57BL/6J mice (Jackson Laboratories). Collection and culture of mouse GCs were performed as described previously (13, 16). KGN cells (RIKEN BioResource Center) were cultured in DMEM:F-12 medium containing 10% (vol/vol) FBS and 1% penicillin and streptomycin. Cells were serum starved for 4 h, followed by 25 nM of DHT stimulation for 18 h unless mentioned otherwise. GCs were also stimulated with 100 nM testosterone (Steraloids), 50 nM estradiol (Sigma), or 10 nM R5020 (Steraloids) for 18 h. In studies examining MAPK3/1 activation, GCs were stimulated with 25 nM DHT for 30 min. The concentration of DHT was based on dose–response experiments (Fig. S1 A–C). For experiments involving GC-specific ARKO animals, RNA and protein samples were obtained from GCs of GC-specific ARKO animals generated previously by crossing AR-flox and MisRII-Cre mice (13).
miR-125b Isolation and Detection.
Total RNA was isolated by using mirVANA miRNA Isolation Kit (Ambion/Life Technologies). Quantitative RT-PCR was performed using TaqMan MicroRNA reverse transcription kit and mouse/human miR-125b TaqMan MicroRNA assays (Applied Biosystems). GAPDH was used as an endogenous control and relative expression of miR-125b was calculated by using the Δ/Δ Ct method.
ChIP Assay.
GCs were stimulated with DHT for 45 min, and ChIP was performed as described (16). Quantitative PCR was performed using EXPRESS SYBR GreenER qPCR SuperMixes (Invitrogen) with primers designed for ARE I, ARE II, ARE III, and control regions (Table S1).
Paxillin Knockdown and Rescue Experiments.
siRNA-mediated paxillin knockdown and rescue experiments were performed as described previously (16) using nontargeting siRNA pool or mouse paxillin siRNA ON-TARGET plus SMARTpool according to the manufacturer’s instructions (Themo Fisher Scientific).
miR-125b Inhibition.
In vitro, primary mouse GCs were transfected with mouse miR-125b-2 mirVana miR inhibitor (mmu-miR-125b-2–3p) or with nonspecific control (Ambion). After 48 h, GCs were serum-starved for 4 h and then stimulated with DHT for 18 h. Specificity of knockdown was determined by measuring miR-125b and miR-125a expression levels (Fig. S2 A and B). In vivo, ovarian bursal injections were performed as described previously (33, 35). miRCURY LNA miR-125b-2 inhibitor (Exiqon) or vehicle control were injected into ovarian bursa. The LNA miR-125b-2 inhibitor (0.5 nM) was mixed with FuGENE 6 (Promega) before bursal injection. Saline solution plus FuGENE 6 was used as vehicle control. Each animal (n = 6) served as its own control, with one ovary receiving the blocking oligonucleotide and the other vehicle control. Animals were euthanized 72 h after injection and ovaries processed for TUNEL assay as well as GC isolation to detect miR-125b and proapoptotic protein expression.
Western Blot Analysis.
Western blots were performed as described previously (13, 16, 53). Primary antibodies used were as follows: anti-rabbit BAX, anti-rabbit BMF (G81), anti-rabbit BAK (D2D3), anti-rabbit p53 and anti-rabbit Caspase-3 (Cell Signaling Technology), and anti-rabbit FSH receptor anti-RPL19 (Abcam).
TUNEL Assay.
TUNEL assay was performed by using an ApopTag Plus Peroxidase In Situ Apoptosis detection kit (EMD Millipore) as described previously (13).
In Vitro Follicle Culture.
In vitro follicle culture was performed as described previously (13).
In Vitro Organ Culture.
Mouse whole ovaries (n = 3 ovaries per treatment) were cultured as described previously (54) with 0, 1, 10, and 100 ng/mL of recombinant FSH (National Hormone and Peptide Program, Harbor–University of California, Los Angeles, Medical Center) in the presence or absence of DHT. Ovaries were cultured for 5 d at 37 °C under 5% CO2 in air with media replaced every 48 h. Ovaries were then processed for sectioning (5-μm sections taken at 30-μm intervals) and stained with H&E for morphological analysis by using previously published criteria (13).
Oocyte Collection and Counting.
Hormonal stimulation for superovulation as well as oocyte collection and counting were performed as described previously (13).
Flow Cytometry.
Flow cytometry was performed in the University of Rochester Medical Center flow cytometry core facility by using Alexa Fluor 488 Annexin V/dead cell apoptosis kit (Invitrogen).
Statistical Analysis.
Data are displayed as means ± SEM of replicate samples. Statistical analysis was performed using Prism version 6 (GraphPad). ANOVA was used to detect differences between treatments. P ≤ 0.05 was considered significant.
Supplementary Material
Acknowledgments
This study was supported by a Foundation for Reproductive Medicine Grant (to A.S. and S.R.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318978111/-/DCSupplemental.
References
- 1.Hillier SG, Whitelaw PF, Smyth CD. Follicular oestrogen synthesis: The ‘two-cell, two-gonadotrophin’ model revisited. Mol Cell Endocrinol. 1994;100(1-2):51–54. doi: 10.1016/0303-7207(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan SD, Moenter SM. GABAergic integration of progesterone and androgen feedback to gonadotropin-releasing hormone neurons. Biol Reprod. 2005;72(1):33–41. doi: 10.1095/biolreprod.104.033126. [DOI] [PubMed] [Google Scholar]
- 3.Blank SK, McCartney CR, Helm KD, Marshall JC. Neuroendocrine effects of androgens in adult polycystic ovary syndrome and female puberty. Semin Reprod Med. 2007;25(5):352–359. doi: 10.1055/s-2007-984741. [DOI] [PubMed] [Google Scholar]
- 4.Vendola K, et al. Androgens promote oocyte insulin-like growth factor I expression and initiation of follicle development in the primate ovary. Biol Reprod. 1999;61(2):353–357. doi: 10.1095/biolreprod61.2.353. [DOI] [PubMed] [Google Scholar]
- 5.Vendola KA, Zhou J, Adesanya OO, Weil SJ, Bondy CA. Androgens stimulate early stages of follicular growth in the primate ovary. J Clin Invest. 1998;101(12):2622–2629. doi: 10.1172/JCI2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weil S, Vendola K, Zhou J, Bondy CA. Androgen and follicle-stimulating hormone interactions in primate ovarian follicle development. J Clin Endocrinol Metab. 1999;84(8):2951–2956. doi: 10.1210/jcem.84.8.5929. [DOI] [PubMed] [Google Scholar]
- 7.Cárdenas H, Herrick JR, Pope WF. Increased ovulation rate in gilts treated with dihydrotestosterone. Reproduction. 2002;123(4):527–533. doi: 10.1530/rep.0.1230527. [DOI] [PubMed] [Google Scholar]
- 8.Harlow CR, Shaw HJ, Hillier SG, Hodges JK. Factors influencing follicle-stimulating hormone-responsive steroidogenesis in marmoset granulosa cells: Effects of androgens and the stage of follicular maturity. Endocrinology. 1988;122(6):2780–2787. doi: 10.1210/endo-122-6-2780. [DOI] [PubMed] [Google Scholar]
- 9.Lenie S, Smitz J. Functional AR signaling is evident in an in vitro mouse follicle culture bioassay that encompasses most stages of folliculogenesis. Biol Reprod. 2009;80(4):685–695. doi: 10.1095/biolreprod.107.067280. [DOI] [PubMed] [Google Scholar]
- 10.Walters KA, et al. Female mice haploinsufficient for an inactivated androgen receptor (AR) exhibit age-dependent defects that resemble the AR null phenotype of dysfunctional late follicle development, ovulation, and fertility. Endocrinology. 2007;148(8):3674–3684. doi: 10.1210/en.2007-0248. [DOI] [PubMed] [Google Scholar]
- 11.Shiina H, et al. Premature ovarian failure in androgen receptor-deficient mice. Proc Natl Acad Sci USA. 2006;103(1):224–229. doi: 10.1073/pnas.0506736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu YC, et al. Subfertility and defective folliculogenesis in female mice lacking androgen receptor. Proc Natl Acad Sci USA. 2004;101(31):11209–11214. doi: 10.1073/pnas.0404372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sen A, Hammes SR. Granulosa cell-specific androgen receptors are critical regulators of ovarian development and function. Mol Endocrinol. 2010;24(7):1393–1403. doi: 10.1210/me.2010-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walters KA, et al. Targeted loss of androgen receptor signaling in murine granulosa cells of preantral and antral follicles causes female subfertility. Biol Reprod. 2012;87(6):151. doi: 10.1095/biolreprod.112.102012. [DOI] [PubMed] [Google Scholar]
- 15.Hammes SR, Levin ER. Extranuclear steroid receptors: Nature and actions. Endocr Rev. 2007;28(7):726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- 16.Sen A, et al. Paxillin mediates extranuclear and intranuclear signaling in prostate cancer proliferation. J Clin Invest. 2012;122(7):2469–2481. doi: 10.1172/JCI62044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen A, et al. Paxillin regulates androgen- and epidermal growth factor-induced MAPK signaling and cell proliferation in prostate cancer cells. J Biol Chem. 2010;285(37):28787–28795. doi: 10.1074/jbc.M110.134064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi XB, et al. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci USA. 2007;104(50):19983–19988. doi: 10.1073/pnas.0706641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Migliaccio A, et al. Crosstalk between EGFR and extranuclear steroid receptors. Ann N Y Acad Sci. 2006;1089:194–200. doi: 10.1196/annals.1386.006. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, et al. Effect of adrenal and ovarian androgens on type 4 follicles unresponsive to FSH in immature mice. Endocrinology. 2001;142(11):4930–4936. doi: 10.1210/endo.142.11.8482. [DOI] [PubMed] [Google Scholar]
- 21.Hamel M, Vanselow J, Nicola ES, Price CA. Androstenedione increases cytochrome P450 aromatase messenger ribonucleic acid transcripts in nonluteinizing bovine granulosa cells. Mol Reprod Dev. 2005;70(2):175–183. doi: 10.1002/mrd.20194. [DOI] [PubMed] [Google Scholar]
- 22.Fábregues F, et al. Transdermal testosterone may improve ovarian response to gonadotrophins in low-responder IVF patients: A randomized, clinical trial. Hum Reprod. 2009;24(2):349–359. doi: 10.1093/humrep/den428. [DOI] [PubMed] [Google Scholar]
- 23.Gleicher N, Weghofer A, Barad DH. The role of androgens in follicle maturation and ovulation induction: Friend or foe of infertility treatment? Reprod Biol Endocrinol. 2011;9(1):116. doi: 10.1186/1477-7827-9-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Velasco JA, et al. FSH receptor in vitro modulation by testosterone and hCG in human luteinized granulosa cells. Eur J Obstet Gynecol Reprod Biol. 2012;165(2):259–264. doi: 10.1016/j.ejogrb.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 25.Hillier SG, de Zwart FA. Androgen/antiandrogen modulation of cyclic AMP-induced steroidogenesis during granulosa cell differentiation in tissue culture. Mol Cell Endocrinol. 1982;28(3):347–361. doi: 10.1016/0303-7207(82)90132-0. [DOI] [PubMed] [Google Scholar]
- 26.Craig J, et al. Gonadotropin and intra-ovarian signals regulating follicle development and atresia: The delicate balance between life and death. Front Biosci. 2007;12:3628–3639. doi: 10.2741/2339. [DOI] [PubMed] [Google Scholar]
- 27.Matsuda F, Inoue N, Manabe N, Ohkura S. Follicular growth and atresia in mammalian ovaries: Regulation by survival and death of granulosa cells. J Reprod Dev. 2012;58(1):44–50. doi: 10.1262/jrd.2011-012. [DOI] [PubMed] [Google Scholar]
- 28.Sun D, et al. Regulation of several androgen-induced genes through the repression of the miR-99a/let-7c/miR-125b-2 miRNA cluster in prostate cancer cells. Oncogene. 2013 doi: 10.1038/onc.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun YM, Lin KY, Chen YQ. Diverse functions of miR-125 family in different cell contexts. J Hematol Oncol. 2013;6:6. doi: 10.1186/1756-8722-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donadeu FX, Schauer SN, Sontakke SD. Involvement of miRNAs in ovarian follicular and luteal development. J Endocrinol. 2012;215(3):323–334. doi: 10.1530/JOE-12-0252. [DOI] [PubMed] [Google Scholar]
- 31.Hossain MM, et al. Altered expression of miRNAs in a dihydrotestosterone-induced rat PCOS model. J Ovarian Res. 2013;6(1):36. doi: 10.1186/1757-2215-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christenson LK. MicroRNA control of ovarian function. Anim Reprod. 2010;7(3):129–133. [PMC free article] [PubMed] [Google Scholar]
- 33.Otsuka M, et al. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J Clin Invest. 2008;118(5):1944–1954. doi: 10.1172/JCI33680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiedler SD, Carletti MZ, Hong X, Christenson LK. Hormonal regulation of MicroRNA expression in periovulatory mouse mural granulosa cells. Biol Reprod. 2008;79(6):1030–1037. doi: 10.1095/biolreprod.108.069690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carletti MZ, Fiedler SD, Christenson LK. MicroRNA 21 blocks apoptosis in mouse periovulatory granulosa cells. Biol Reprod. 2010;83(2):286–295. doi: 10.1095/biolreprod.109.081448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao G, et al. MicroRNA-224 is involved in transforming growth factor-beta-mediated mouse granulosa cell proliferation and granulosa cell function by targeting Smad4. Mol Endocrinol. 2010;24(3):540–551. doi: 10.1210/me.2009-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu S, Linher-Melville K, Yang BB, Wu D, Li J. Micro-RNA378 (miR-378) regulates ovarian estradiol production by targeting aromatase. Endocrinology. 2011;152(10):3941–3951. doi: 10.1210/en.2011-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen A, Hammes SR. Androgens: They don’t just make a man out of you. Expert Review of Obstetrics and Gynecology. 2011;6(1):19–22. [Google Scholar]
- 39.Wu YG, Bennett J, Talla D, Stocco C. Testosterone, not 5{alpha}-dihydrotestosterone, stimulates LRH-1 leading to FSH-independent expression of Cyp19 and P450scc in granulosa cells. Mol Endocrinol. 2011;25(4):656–68. doi: 10.1210/me.2010-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walters KA, Allan CM, Handelsman DJ. Androgen actions and the ovary. Biol Reprod. 2008;78(3):380–389. doi: 10.1095/biolreprod.107.064089. [DOI] [PubMed] [Google Scholar]
- 41.Hillier SG, Tetsuka M. Role of androgens in follicle maturation and atresia. Baillieres Clin Obstet Gynaecol. 1997;11(2):249–260. doi: 10.1016/s0950-3552(97)80036-3. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen ME, et al. In human granulosa cells from small antral follicles, androgen receptor mRNA and androgen levels in follicular fluid correlate with FSH receptor mRNA. Mol Hum Reprod. 2011;17(1):63–70. doi: 10.1093/molehr/gaq073. [DOI] [PubMed] [Google Scholar]
- 43.Woods AJ, Kantidakis T, Sabe H, Critchley DR, Norman JC. Interaction of paxillin with poly(A)-binding protein 1 and its role in focal adhesion turnover and cell migration. Mol Cell Biol. 2005;25(9):3763–3773. doi: 10.1128/MCB.25.9.3763-3773.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hervy M, Hoffman L, Beckerle MC. From the membrane to the nucleus and back again: Bifunctional focal adhesion proteins. Curr Opin Cell Biol. 2006;18(5):524–532. doi: 10.1016/j.ceb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 45.Xue K, Liu JY, Murphy BD, Tsang BK. Orphan nuclear receptor NR4A1 is a negative regulator of DHT-induced rat preantral follicular growth. Mol Endocrinol. 2012;26(12):2004–2015. doi: 10.1210/me.2012-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, et al. A comparative study on transforming growth factor-beta and activin A for preantral follicles from adult, immature, and diethylstilbestrol-primed immature mice. Endocrinology. 1999;140(6):2480–2485. doi: 10.1210/endo.140.6.6827. [DOI] [PubMed] [Google Scholar]
- 47.Lebedeva IY, Lebedev VA, Grossmann R, Parvizi N. Age-dependent role of steroids in the regulation of growth of the hen follicular wall. Reprod Biol Endocrinol. 2010;8:15. doi: 10.1186/1477-7827-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Hefnawy T, Zeleznik AJ. Synergism between FSH and activin in the regulation of proliferating cell nuclear antigen (PCNA) and cyclin D2 expression in rat granulosa cells. Endocrinology. 2001;142(10):4357–4362. doi: 10.1210/endo.142.10.8438. [DOI] [PubMed] [Google Scholar]
- 49.Hillier SG, De Zwart FA. Evidence that granulosa cell aromatase induction/activation by follicle-stimulating hormone is an androgen receptor-regulated process in-vitro. Endocrinology. 1981;109(4):1303–1305. doi: 10.1210/endo-109-4-1303. [DOI] [PubMed] [Google Scholar]
- 50.Gleicher N, et al. Starting and resulting testosterone levels after androgen supplementation determine at all ages in vitro fertilization (IVF) pregnancy rates in women with diminished ovarian reserve (DOR) J Assist Reprod Genet. 2013;30(1):49–62. doi: 10.1007/s10815-012-9890-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gleicher N, Barad DH. Dehydroepiandrosterone (DHEA) supplementation in diminished ovarian reserve (DOR) Reprod Biol Endocrinol. 2011;9:67. doi: 10.1186/1477-7827-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bosdou JK, et al. The use of androgens or androgen-modulating agents in poor responders undergoing in vitro fertilization: A systematic review and meta-analysis. Hum Reprod Update. 2012;18(2):127–145. doi: 10.1093/humupd/dmr051. [DOI] [PubMed] [Google Scholar]
- 53.Evaul K, Hammes SR. Cross-talk between G protein-coupled and epidermal growth factor receptors regulates gonadotropin-mediated steroidogenesis in Leydig cells. J Biol Chem. 2008;283(41):27525–27533. doi: 10.1074/jbc.M803867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nilsson E, Rogers N, Skinner MK. Actions of anti-Mullerian hormone on the ovarian transcriptome to inhibit primordial to primary follicle transition. Reproduction. 2007;134(2):209–221. doi: 10.1530/REP-07-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.