

Significance
Acetaminophen overdose is the most common cause of acute liver failure and the leading cause of chronic liver damage requiring liver transplantation in developed countries. There are limited options for early treatment. Acetaminophen liver toxicity leads to the formation of reactive oxygen and nitrogen species which cause an increase in intracellular Ca2+ and hepatocellular death. We show that acetaminophen-induced liver toxicity depends on Transient Receptor Potential Melanostatine 2 (TRPM2) cation channels in hepatocytes, which are activated in response to oxidative stress and are responsible for Ca2+ overload. Lack of TRPM2 channels in hepatocytes or their pharmacological inhibition protects liver from acetaminophen toxicity. This provides evidence that TRPM2 may present a potential therapeutic target for treatment of oxidative-stress related liver diseases.
Abstract
Acetaminophen (paracetamol) is the most frequently used analgesic and antipyretic drug available over the counter. At the same time, acetaminophen overdose is the most common cause of acute liver failure and the leading cause of chronic liver damage requiring liver transplantation in developed countries. Acetaminophen overdose causes a multitude of interrelated biochemical reactions in hepatocytes including the formation of reactive oxygen species, deregulation of Ca2+ homeostasis, covalent modification and oxidation of proteins, lipid peroxidation, and DNA fragmentation. Although an increase in intracellular Ca2+ concentration in hepatocytes is a known consequence of acetaminophen overdose, its importance in acetaminophen-induced liver toxicity is not well understood, primarily due to lack of knowledge about the source of the Ca2+ rise. Here we report that the channel responsible for Ca2+ entry in hepatocytes in acetaminophen overdose is the Transient Receptor Potential Melanostatine 2 (TRPM2) cation channel. We show by whole-cell patch clamping that treatment of hepatocytes with acetaminophen results in activation of a cation current similar to that activated by H2O2 or the intracellular application of ADP ribose. siRNA-mediated knockdown of TRPM2 in hepatocytes inhibits activation of the current by either acetaminophen or H2O2. In TRPM2 knockout mice, acetaminophen-induced liver damage, assessed by the blood concentration of liver enzymes and liver histology, is significantly diminished compared with wild-type mice. The presented data strongly suggest that TRPM2 channels are essential in the mechanism of acetaminophen-induced hepatocellular death.
Acetaminophen (N-acetyl-p-aminophenol), when used at prescribed doses, is a safe analgesic and antipyretic drug (1). Its overdose, however, can be life threatening, causing severe liver and kidney damage (2–5). In Western countries, acetaminophen-induced hepatotoxicity is a leading cause of acute liver failure requiring liver transplantation (6). Due to a widespread availability of acetaminophen and potentially lethal consequences of its overdose, the mechanisms of acetaminophen hepatotoxicity have been in focus of a large number of investigations (7). Despite significant progress, the exact pathways of acetaminophen hepatotoxicity that lead to hepatocellular death are still not completely understood. It is clear, however, that acetaminophen toxicity arises from its metabolic activation (8, 9).
In the liver, therapeutic doses of acetaminophen are metabolized by glucuronidation and sulfation into nontoxic compounds (1). Only a small amount of acetaminophen is converted by hepatic cytochrome P450 (CYP)-dependent mixed function oxidases to the reactive intermediate metabolite N-acetyl-parabenzo-quinoneimine (NAPQI). The NAPQI generated by a therapeutic dose of acetaminophen is rapidly metabolized to nontoxic products by conjugation with glutathione (GSH) (1, 10). With large doses of acetaminophen, however, hepatic GSH becomes depleted resulting in the accumulation of toxic amounts of NAPQI. Covalent binding of NAPQI to cellular proteins has previously been considered the main cause of liver cell death under these circumstances. Indeed it has been shown that covalent binding precedes hepatocellular death, and treatments that prevent covalent binding also prevent liver necrosis (11). More recently, however, it has been suggested that, by itself, the covalent binding of NAPQI is not sufficient to induce apoptosis or necrosis. The toxic signal produced by covalent binding undergoes further amplification through the formation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), deregulation of Ca2+ homeostasis, and increased intracellular Ca2+, causing oxidant stress in mitochondria and inducing the mitochondrial membrane permeability transition (12, 13). Although widely acknowledged, the role of Ca2+ in acetaminophen toxicity is poorly understood and has not been thoroughly investigated. Nonselective Ca2+ channels blockers chlorpromazine and verapamil have been shown to attenuate liver injury in mice (14, 15), however, the mechanism of their protective properties in acetaminophen overdose is not clear, and it is not known whether it involves any Ca2+-permeable channels on the plasma membrane of hepatocytes.
The only Ca2+-selective channel that has been clearly identified in hepatocytes so far is the Ca2+ release-activated Ca2+ channel activated by the depletion of intracellular Ca2+ stores downstream of phospholipase Cβ and phospholipase Cγ signaling (16, 17). In addition, a number of Ca2+-permeable nonselective cation channels with no clearly defined functions and mostly from the TRP family of channels have been shown to be present in hepatocytes and liver cells (18, 19). One of these channels, Transient Receptor Potential Melanostatine 2 (TRPM2), whose presence in the liver has only been demonstrated on an mRNA level (19), is activated in response to oxidative stress and, potentially, can be involved in acetaminophen-induced Ca2+ rise in hepatocytes.
Results
Acetaminophen and H2O2 Activate Nonselective Cation Current in Hepatocytes.
To investigate the role of Ca2+-permeable channels in acetaminophen toxicity in the liver, first we examined the effects of acetaminophen on the free cytoplasmic Ca2+ concentration ([Ca2+]cyt) in rat hepatocytes. Hepatocytes were incubated with 10–15 mM acetaminophen for 60 min in a bath solution containing 1.3 mM Ca2+. Treated hepatocytes were then loaded with Fura-2 acetoxymethyl (AM) in a nominally Ca2+-free bath solution, and after washing, were transferred to the microscope stage. After introduction of 1.3 mM Ca2+ into the bath, [Ca2+]cyt increased to micromolar levels, indicating that acetaminophen activates Ca2+ entry across the plasma membrane through Ca2+-permeable channels. Control cells showed no change in [Ca2+]cyt (Fig. 1A). Ca2+ entry activated in hepatocytes by preincubation with acetaminophen was inhibited by 50 µM clotrimazole and 10μM N-(p-amylcinnamoyl)anthranilic acid (ACA) (Fig. 1A). Clotrimazole and ACA were previously shown to block heterologously expressed TRPM2 channels (20, 21).
Fig. 1.

Acetaminophen and H2O2 activate Ca2+ entry and a nonselective cation current in rat hepatocytes. (A) Ca2+ entry in hepatocytes treated with 10 mM acetaminophen for 60 min under the conditions indicated in A (average data from three separate cell preparations). Both clotrimazole (50 μM) and ACA (10 μM) were applied to the bath 5 min before the addition of Ca2+. (B) I-V plots of membrane currents measured in control hepatocytes, and hepatocytes treated with 10 mM acetaminophen for 60 min in control bath solution, after replacement of 140 mM NaCl in the bath solution with 140 mM NMDG Cl and after the addition of 50 μM clotrimazole to the bath (n = 22 for each trace). Error bars are omitted for clarity here and all other I-V plots. (C) Ca2+ entry in hepatocytes treated with 0.5 mM H2O2 (n = 3). (D) I-V plots of membrane currents measured in control hepatocytes, and hepatocytes treated with 0.5 mM H2O2 in control bath solution, after replacement of 140 mM NaCl in the bath solution with 140 mM NMDG Cl and after the addition of 50 μM clotrimazole to the bath (n = 7). (E) Activation of membrane conductance in hepatocytes by 10 mM H2O2 applied to the bath. Each data point represents amplitude of the current at −100 mV. (F) I-V plots of membrane currents measured before application of H2O2 (control), and after full development of the H2O2-activated current in control bath solution (H2O2) and after replacement of 140 mM NaCl with 140 mM NMDG Cl (n = 5).
To investigate the nature of the Ca2+-permeable channels responsible for acetaminophen-induced Ca2+ entry, we used whole-cell patch clamping. After isolation, hepatocytes were cultured for 24–48 h on glass coverslips and treated with or without acetaminophen for 60 min. The average density of baseline current at −100 mV in rat hepatocytes normally varies between 2 and 4 pA/pF and the current-voltage (I-V) plot shows some outward rectification due to Cl− conductance (22) (Fig. 1B). Hepatocytes preincubated with 10 mM acetaminophen for 60 min, however, showed significantly larger currents upon establishing whole-cell configuration, with the average current density of 8–10 pA/pF at −100 mV (Fig. 1B). The virtually linear I-V plot, near-zero reversal potential, and sensitivity of the inward current to the replacement of extracellular Na+ with the large cation N-methyl-d-glucamine (NMDG+) (Fig. 1B), suggested that treatment with acetaminophen resulted in the activation of nonselective cation channels. Similarly to acetaminophen-induced Ca2+ entry, this nonselective cation current was blocked by clotrimazole and ACA (Fig. 1B; only clotrimazole is shown, as ACA produced the same level of inhibition). When we increased the treatment time with acetaminophen, hepatocytes became increasingly damaged with extensive membrane blebbing so that they were not amenable to Fura-2 Ca2+ measurements and patch clamping. Direct application of acetaminophen to the bath in patch clamping or Ca2+ imaging experiments had no acute effect on membrane currents or [Ca2+]cyt.
Oxidant stress caused by ROS and RNS formed in the liver in acetaminophen overdose is considered a major mediator of hepatocellular death (23). To determine whether hepatocytes express ROS-sensitive Ca2+-permeable channels which can potentially be activated by ROS and RNS generated in hepatocytes treated with acetaminophen, we used Ca2+ imaging and patch clamping of isolated rat hepatocytes treated with H2O2. Fura-2 experiments indicated a robust rise of [Ca2+]cyt in response to incubation with 0.5 mM H2O2 for 25–30 min, whereas patch clamping showed activation of a nonselective cation current similar to that activated by acetaminophen (Fig. 1 C and D). Both Ca2+ entry and the nonselective cation current activated in hepatocytes by incubation with H2O2 were inhibited by 50 µM clotrimazole and 10 μM ACA (Fig. 1 C and D). As observed for 10–15 mM acetaminophen, 0.5 mM H2O2 produced some membrane blebbing in hepatocytes. Cells with damaged membranes were not amenable to patch clamping. Therefore, the amplitude of the cation current activated by incubation with 1 mM H2O2 was likely to be underestimated, as only visibly undamaged cells were used. Adding 1 mM H2O2 directly to the bath after achieving the whole-cell configuration with control pipette solution did not result in activation of a noticeable current within 10 min of recording or before the seal between the cell and the pipette was lost. However, application of 10 mM H2O2 to the bath in such experiments resulted in a relatively rapid development of a large cation current with properties similar to those of the currents activated by preincubation of hepatocytes with 0.5 mM H2O2 or 10 mM acetaminophen (Fig. 1 E and F).
Acetaminophen-Induced Nonselective Cation Current in Hepatocytes Is Mediated by TRPM2 Channels.
Although the data presented above demonstrates activation of a nonselective cation current across the plasma membrane and a rise in [Ca2+]cyt in hepatocytes in response to acetaminophen treatment, the identity of the channels responsible for this Ca2+ entry is not known. One possible candidate that is activated in response to oxidative stress is the TRPM2 (24–26). Evidence that acetaminophen-induced nonselective cation current is mediated by TRPM2 is as follows: (i) the current shows a linear I-V relationship; (ii) it is blocked by clotrimazole and ACA; and (iii) it is activated by H2O2 (19, 27).
The hallmark of TRPM2-mediated currents is activation by ADP-ribose (ADPR) and H2O2 (28–30). Several splice variants of TRPM2 channel have been reported (31). One of them, with a deletion in the C terminus (TRPM2ΔC), lacks the ADPR-binding motif and has been shown to be activated by H2O2, but not ADPR (31). To determine the main splice variants of TRPM2 expressed in hepatocytes we used RT-PCR and showed that rat hepatocytes express only long isoform of TRPM2 (LTRPM2) with the ADPR binding motif intact (Table S1 and Fig. S1). To establish if these channels are functional we used whole-cell patch clamping of hepatocytes cultured for 24 h after isolation and a pipette solution supplemented with ADPR. Addition of 1 mM ADPR resulted in activation of a large nonselective cation current (137 ± 27 pA/pF) 1–5 min after establishing the whole-cell configuration (Fig. 2 A and B). Lower concentrations of ADPR produced smaller currents (EC50 480 μM) with a longer time course of activation. The ADPR-activated current was inhibited by ACA and clotrimazole at the same concentrations as those that inhibited the currents activated by acetaminophen and H2O2 (Fig. 2 A and C).
Fig. 2.

TRPM2 current in rat hepatocytes. (A) Activation of TRPM2 current in a rat hepatocyte in response to intracellular perfusion with 1 mM ADPR. The current was recorded in response to 100-ms voltage ramps between −120 and 120 mV and applied every 2 s. Current amplitude at −100 mV is plotted against time. Application of 10 μM ACA to the bath resulted in a 90% block of the current. (B) The dose–response curve for ADPR. TRPM2 current amplitude at −100 mV is plotted against ADPR concentration in the pipette. The data points are fitted with a Hill equation with slope 1 and EC50 of 480 μM. (C and D) I-V plots of fully developed TRPM2 currents activated by ADPR in control bath solution, after replacement of 140 mM NaCl in the bath solution with 140 mM NMDG Cl, and after application of 50 µM clotrimazole or 100 µM chlorpromazine to the control bath solution, respectively (n = 3).
In addition to ACA and clotrimazole we investigated the effects of another broad ion channel and Ca2+–calmodulin inhibitor, chlorpromazine, on an acetaminophen- and ADPR-activated current in hepatocytes. It has previously been shown that chlorpromazine protects against acetaminophen toxicity in mouse liver (14, 32). ADPR- and acetaminophen-activated current in rat hepatocytes was fully blocked by 100 μM chlorpromazine (Fig. 2D and Fig. S2A), with an EC50 of ∼5 μM. The time course of inhibition of the current activated by ADPR was very similar to that of the current induced by acetaminophen (Fig. S2 A and B). To confirm that chlorpromazine blocks TRPM2 channels we used HEK293T cells transfected with TRPM2 cDNA (Fig. S2 C and D). TRPM2 current activated by either acetaminophen or ADPR in transfected HEK293T cells was blocked by chlorpromazine with similar time courses and concentrations as the currents activated in rat hepatocytes (Fig. S2 E and F).
Acetaminophen overdose has been shown to cause DNA fragmentation in hepatocytes and, as a consequence, activation of poly(ADP-ribose) polymerase (PARP), which generates polyADPR, the main precursor of cytoplasmic ADPR (33). Using antibodies against polyADPR and immunofluorescence we were able to demonstrate an increase in polyADPR production in hepatocytes treated for 45 min by either 10 mM acetaminophen or 1 mM H2O2 (Fig. 3A). Incubation with 10 mM acetaminophen overnight induced much stronger polyADPR production. To investigate whether generation of ADPR in hepatocytes in response to H2O2 and acetaminophen contributes to activation of Ca2+ entry we used PARP inhibitor 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinoline (DPQ) (34). Measurements of [Ca2+]cyt showed that DPQ strongly inhibited Ca2+ entry in rat hepatocytes, supporting the notion that Ca2+ rise induced by H2O2 and acetaminophen is mediated by ADPR-activated TRPM2 channels (Fig. 3 B and C).
Fig. 3.

The role of ADPR in activation of Ca2+ entry in hepatocytes. (A) Poly-ADPR–specific immunofluorescence in rat control hepatocytes (A, i) and hepatocytes treated with 10 mM acetaminophen (45 min in A, ii; 16 h in A, iv) or 1 mM H2O2 (45 min in A, iii). (B and C) Inhibition of H2O2 and acetaminophen-induced Ca2+ entry in hepatocytes by PARP inhibitor DPQ (10 μM). DPQ was added to the incubation medium 2 min before the addition of H2O2 or acetaminophen (n = 3).
To confirm that the current activated by ADPR, H2O2, and acetaminophen is mediated by TRPM2 channels we used siRNA-mediated knockdown of TRPM2 in rat hepatocytes. In cells transfected with siRNA against TRPM2, patch clamping showed that membrane currents activated by the application of intracellular ADPR, H2O2, or acetaminophen were each reduced by 65–70% (Fig. 4A). RT-PCR and Western blotting confirmed that by using siRNA-mediated knockdown we reduced TRPM2 expression in primary hepatocytes by about 60% (57 ± 5%, n = 3) within 48 h after transfection (Fig. 4B, Table S2, and Fig. S3). Measurements of [Ca2+]cyt using Fura-2AM revealed that siRNA-mediated knockdown of TRPM2 expression resulted in a significant reduction of Ca2+ entry in hepatocytes threated by H2O2 or acetaminophen (Fig. 4 C and D). These results confirm that the cation current activated by acetaminophen and H2O2 in hepatocytes is mediated by TRPM2.
Fig. 4.

The effect of TRPM2 knockdown on hepatocyte membrane currents and Ca2+ entry. (A) The effect of TRPM2 knockdown on the amplitude of membrane currents activated by intracellular ADPR, H2O2, and acetaminophen. The absolute amplitude of the current measured at −100 mV is shown (mean ± SEM). (B) Western blot of TRPM2 protein in hepatocytes treated with control siRNA (lane 2) and anti-TRPM2 siRNA (lane 3). (C) The effect of TRPM2 knockdown on Ca2+ entry in rat hepatocytes preincubated with 0.5 mM H2O2 for 30 min. (D) The effect of TRPM2 knockdown on Ca2+ entry in rat hepatocytes preincubated with 10 mM acetaminophen for 60 min.
To further test whether H2O2 and acetaminophen-activated Ca2+ entry is mediated by TRPM2 channels, we conducted measurements of [Ca2+]cyt using Fura-2AM and hepatocytes isolated from TRPM2 knockout (KO) and (wild-type) WT mice, and the inhibitors of TRPM2 channel, ACA and clotrimazole (Fig. 5 A–C). RT-PCR and Western blot analysis confirmed that WT mouse hepatocytes express the same isoform of TRPM2, LTRPM2, as rat hepatocytes and that TRPM2 protein is absent in TRPM2 KO mouse hepatocytes (Table S3 and Figs. S4 and S5). In response to H2O2 or acetaminophen treatment, hepatocytes isolated from TRPM2 WT mice showed Ca2+ entry similar to that of rat hepatocytes, which was blocked by ACA and clotrimazole (Fig. 5 A and C; compare with Fig. 1 A and C). In contrast, hepatocytes isolated from TRPMKO mice had a significantly smaller Ca2+ entry activated by H2O2 and virtually no Ca2+ entry in response to the treatment with acetaminophen (Fig. 5 B and C). A relatively small Ca2+ rise in TRPM2 KO mouse hepatocytes induced by H2O2 was blocked by clotrimazole, but not ACA, suggesting the presence of a minor H2O2-activated Ca2+ entry pathway not mediated by TRPM2 channels. Results of Ca2+ imaging were supported by patch clamping of hepatocytes isolated from TRPM2 WT and KO mice. Treatment of hepatocytes with H2O2 or acetaminophen resulted in activation of a significantly larger nonselective cation current in TRPM2 WT hepatocytes compared with hepatocytes from KO animals (Fig. S6 A–D).
Fig. 5.
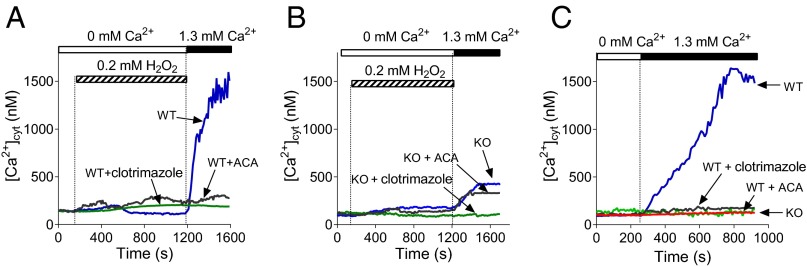
H2O2- and acetaminophen-activated Ca2+ entry is attenuated in TRPM2 KO mouse hepatocytes. (A and B) Ca2+ entry in TRPM2 WT (A) and KO (B) mouse hepatocytes activated by the addition of H2O2 to the bath solution (n = 3). (C) Ca2+ entry in TRPM2 WT and KO mice hepatocytes treated with 10 mM acetaminophen for 60 min (n = 3). Both clotrimazole (50 μM) and ACA (10 μM) were applied to the bath 5 min before the addition of Ca2+.
Ablation of TRPM2 Channels Protects Against Acetaminophen Toxicity in the Liver.
Treatment of isolated rat and mouse hepatocytes in culture with acetaminophen causes progressive cell death through oncotic necrosis, thus mimicking the effects of acetaminophen on the intact liver (35). In the next experiment we investigated whether inhibition of TRPM2 channels by ACA affords protection to hepatocytes against high doses of acetaminophen. Indeed, ACA (1 μM) reduced cellular death by ∼60% in hepatocytes treated with 10 mM acetaminophen for 16 h compared with hepatocytes treated with acetaminophen alone (n = 3; P < 0.01) (Fig. S6E).
To establish whether activation of TRPM2 channels plays a role in acetaminophen-induced liver damage in vivo we used TRPM2 KO mice (36). I.p. injection of acetaminophen (500 mg/kg) to WT mice resulted in a large increase in the blood concentrations of the liver enzymes, alanine transaminase (ALT) and aspartate transaminase (AST) compared with vehicle-injected animals (Fig. 6A), suggesting severe liver damage 24 h postinjection (cf. ref. 37). In TRPM2 Het mice this dose of acetaminophen also caused a large elevation of liver enzymes. However, in TRPM2 KO animals the blood levels of ALT and AST were ∼5–7 times lower than in WT and Het mice (Fig. 6A). Hematoxalin/eosin (H&E) staining of liver sections of acetaminophen-treated TRPM2 WT and Het mice revealed widespread hepatocellular damage (Fig. 6B). This was characterized by areas of necrosis, infiltration by lymphocytes, and hemorrhage, and was prominent in zones 2 and 3 (hepatocytes around the hepatic vein) (Fig. 6B). Liver damage was substantially reduced in TRPM2 KO mice treated with acetaminophen compared with the treated WT and Het mice (Fig. 6B). The area of necrotic damage was much smaller in TRPM2 KO mice and was localized to zone 3. In some sections of livers from acetaminophen-treated TRPM2 WT and Het mice, hemorrhagic necrosis and extravasations of blood were detected along with necrotic damage. Because the prominent and consistent effect of acetaminophen was necrotic damage, the area of necrotic tissue was quantified. The results indicate that the area of necrosis in the livers of TRPM 2KO mice treated with acetaminophen was substantially smaller than in the livers of acetaminophen-treated TRPM2 WT and Het mice (Fig. S6F).
Fig. 6.

TRPM2 KO mice are substantially protected against acetaminophen-induced damage. (A) Blood concentrations of the liver enzymes ALT and AST in TRPM2 KO, TRPM2 Het, and WT mice pretreated with acetaminophen or vehicle for 24 h. The results are the means ± SEM of the number of mice indicated. The degree of significance, determined using the one way ANOVA test was P < 0.007 for comparison of TRPM2 KO with each of TRPM2 WT and TRPM2 Het. (B) Representative bright field images of H&E-stained liver sections at 20× magnification. The light colored areas in the WT acetaminophen and KO acetaminophen images represent areas of necrosis.
Discussion
In this study we show that hepatocytes express long isoform of TRPM2 channels. These channels function as a cation entry pathway across the hepatocyte plasma membrane and similarly to TRPM2 channels heterologously expressed in HEK293 cells are activated by ADPR included in the patch pipette and H2O2 added to the bath solution (24, 38). The important finding of this study is that TRPM2 channels mediate a substantial increase in [Ca2+]cyt in hepatocytes treated with toxic concentrations of acetaminophen. Furthermore, we show that Ca2+ entry through TRPM2 channels plays a significant role in acetaminophen-induced hepatocellular death both in vitro and in vivo.
The mechanism of acetaminophen hepatotoxicity has been under intensive investigation for several decades (7). It has been established that acetaminophen overdose causes a multitude of interrelated cellular events (7), but the relative importance of each of these events in hepatocellular death is not well understood. Briefly, the main steps that lead to acetaminophen hepatotoxicity can be summarized as follows. Saturation of glucuronidation and sulfation pathways by excessive levels of acetaminophen leads to the increased acetaminophen metabolism by several isoforms of CYP (CYP2E1, CYP1A2, CYP3A4, and CYP2D6) into the reactive metabolite NAPQI (8, 39). NAPQI saturates and depletes intracellular GSH and covalently binds to proteins (8, 9). Lack of GSH causes accumulation of ROS and RNS and oxidative stress. Increased oxidative stress, together with covalent binding, causes mitochondrial dysfunction, DNA fragmentation, and deregulation of Ca2+ homeostasis (7, 10). Oxidants and increased [Ca2+]cyt promote mitochondrial permeability transition, which, in turn, initiates further oxidative stress, loss of mitochondrial potential, and cessation of ATP synthesis (24). Finally, loss of ATP triggers necrosis of hepatocytes.
Two cellular processes in this sequence are likely to result in an increase in cytoplasmic concentration of ADPR, the main ligand of the Ca2+-permeable TRPM2 channels (29, 40). Opening of the mitochondrial permeability transition pore in the inner membrane releases ADPR from mitochondria, whereas activation of PARP by DNA damage results in generation of ADPR precursor, polyADPR (40, 41). The importance of the events that produce cytoplasmic ADPR is emphasized by the findings that the inhibitors of mitochondrial permeability transition and the PARP inhibitors protect the liver against acetaminophen overdose (33). The results of this study show that the increase in polyADPR production in hepatocytes treated with 10 mM acetaminophen can be detected by immunofluorescence within 45 min of the start of the treatment, and progressively increases with time, which creates favorable conditions for activation of TRPM2 channels. Inhibition of acetaminophen-induced Ca2+ entry in hepatocytes by PARP inhibitor DPQ shown here suggest a mechanism for the known protective effects of PARP inhibitors against acetaminophen toxicity (33).
Deregulation of Ca2+ homeostasis in acetaminophen hepatotoxicity has been demonstrated in earlier studies (14, 32), however, it has been suggested that intracellular Ca2+ rise, mainly due to inhibition of Ca2+-Mg2+ ATPase, accompanies rather than causes hepatocellular injury (42). Ca2+ channel antagonists, verapamil and chlorpromazine, have been shown to protect the liver against acetaminophen toxicity, but whether they actually block any channels in hepatocytes has never been investigated. We have shown here that chlorpromazine blocked TRPM2 currents activated in rat hepatocytes by ADPR, H2O2, or acetaminophen. It also blocked TRPM2 channels heterologously expressed in HEK293T cells and activated by acetaminophen or ADPR. This suggests that the protective properties of chlorpromazine may be due to its inhibition of TRPM2 channels, although chlorpromazine may have other relevant targets in hepatocytes (14). More definitive data about the role of TRPM2 mediated Ca2+ entry in acetaminophen toxicity come from TRPM2 KO mice experiments. Lack of TRPM2 channels results in significantly improved blood levels of the liver enzymes ALT and AZT and significantly reduced liver damage 24 h post-acetaminophen injection, suggesting that activation of TRPM2 channels contribute to hepatocellular death. The deleterious effects of TRPM2 activation may not be just due to Ca2+ entry, but also to a very high Na+ and K+ conductance through these channels. Accumulation of Na+ and loss of K+ leads to a loss of the plasma membrane potential and activation of Na+/K+ ATPase which contributes to the reduction of cellular ATP levels and promote cell necrosis.
These findings add considerably to our current understanding of the mechanism of acetaminophen liver toxicity. Currently, the only clinically available treatment for acetaminophen overdose is N-acetyl-cysteine, a GSH precursor, which has to be administered within 15–16 h after acetaminophen ingestion to be effective (43). If this time window is lost, the efficacy of N-acetyl-cysteine in preventing liver damage is significantly reduced, and liver failure is the likely outcome (43). The TRPM2 channel offers an alternative therapeutic target which may allow treatment over a wider window of time. Moreover, inhibitors of TRPM2 offer the potential to treat other ROS-mediated liver diseases such as nonalcoholic liver disease, hepatitis, and hepatocellular carcinoma.
Materials and Methods
Animals.
Hooded Wistar (HW) rats and TRPM2 KO and WT mice were housed and bred in the controlled environment with a 12-h light-dark cycle. Animals had access to food and water ad libitum. TRPM2 KO mice were obtained from Yasuo Mori’s laboratory (Kyoto University, Kyoto, Japan). All animal studies were approved by the Animal Ethics Committees of the University of Adelaide and Flinders University of South Australia.
Hepatocyte Isolation and Culture.
Hepatocytes were isolated from HW rats by liver perfusion with collagenase using the protocol described previously (44). The isolated hepatocytes were cultured on glass coverslips at 37 °C in 5% CO2 in air (vol/vol) in DMEM containing penicillin (100 U/mL), streptomycin (100 µg/mL), and 10% FBS (vol/vol) for 16–72 h before experiments. Isolated mouse hepatocytes were prepared by retrograde perfusion of the liver through the inferior vena cava and cut-open portal vein. The rest of the protocol was similar to that used to prepare isolated rat hepatocytes (44).
Calcium Imaging.
Hepatocytes cultured on glass coverslips for 24 h were loaded with Fura2-AM (5 µM) for 30 min, washed, and incubated in Krebs–Ringer–Hepes solution for 10 min in a CO2 incubator at 37 °C. The fluorescence of Fura-2 was measured using a Nikon TE300 Eclipse microscope equipped with a Sutter DG-4/OF wavelength switcher, Omega XF04 filter set for Fura-2, Photonic Science ISIS-3 intensifiedCCD camera, and Universal Interface Card MetaFluor software. Fluorescence images were obtained every 20 s using a 20× objective. Fluorescence ratio values (340:380 nm) were transformed to [Ca2+]cyt using the equation derived by Grynkiewicz et al. (45).
Patch-Clamp Recording.
Membrane currents were measured at room temperature (23 °C) using standard patch clamping in a whole-cell mode and a computer-based EPC-9 patch-clamp amplifier run by PULSE software (HEKA) (22). To monitor the development of membrane currents, voltage ramps between −120 and +120 mV were applied every 2 s following the achievement of whole-cell configuration. The holding potential was −40 mV. The data were analyzed using PULSEFIT software (HEKA). For current measurements in rat hepatocytes, patch pipettes were pulled from borosilicate glass and fire polished to a resistance between 1.5 and 2.5 MΩ. Mouse hepatocytes were generally less amenable for patch clamping, therefore to increase the probability of forming a gigaseal, smaller patch pipettes with a resistance between 3 and 5 MΩ were used. Series resistance was 50–70% compensated.
Statistical Analysis.
Data are presented as means ± SEM. Statistical significance was assessed using ANOVA followed by the Bonferroni post hoc test or using unpaired two-tailed t test with Welch’s correction.
Chemicals, solutions, and methods for RT-PCR, cell transfections, Western blot analysis, cell viability assay, immunofluorescence, in vivo acetaminophen toxicity, blood liver enzymes assay, and histopathology are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Prof. Yasuo Mori (Kyoto University) for kindly providing TRPM2 KO mice, Ms. Jin Hua (Flinders University) for performing RT-PCR on TRPM2 mouse hepatocytes, and the late Dr. John Phillips for advice on the preparation of mouse hepatocytes.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322657111/-/DCSupplemental.
References
- 1.Thomas SHL. Paracetamol (acetaminophen) poisoning. Pharmacol Ther. 1993;60(1):91–120. doi: 10.1016/0163-7258(93)90023-7. [DOI] [PubMed] [Google Scholar]
- 2.Davidson DG, Eastham WN. Acute liver necrosis following overdose of paracetamol. BMJ. 1966;2(5512):497–499. doi: 10.1136/bmj.2.5512.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchell JR, et al. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther. 1973;187(1):185–194. [PubMed] [Google Scholar]
- 4.Edwards OM, Edwards P, Huskisson EC, Taylor RT. Paracetamol and renal damage. BMJ. 1971;2(5753):87–89. doi: 10.1136/bmj.2.5753.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyer TD, Rouff SL. Acetaminophen-induced hepatic necrosis and renal failure. JAMA. 1971;218(3):440–441. [PubMed] [Google Scholar]
- 6.Simpson KJ, et al. The utilization of liver transplantation in the management of acute liver failure: Comparison between acetaminophen and non-acetaminophen etiologies. Liver Transpl. 2009;15(6):600–609. doi: 10.1002/lt.21681. [DOI] [PubMed] [Google Scholar]
- 7.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handbook Exp Pharm. 2010;(196):369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273(28):17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 9.Srivastava A, et al. Role of reactive metabolites in drug-induced hepatotoxicity. Handbook Exp.Pharm. 2010;(196):165–194. doi: 10.1007/978-3-642-00663-0_7. [DOI] [PubMed] [Google Scholar]
- 10.Bessems JGM, Vermeulen NPE. Paracetamol (acetaminophen)-induced toxicity: Molecular and biochemical mechanisms, analogues and protective approaches. Crit Rev Toxicol. 2001;31(1):55–138. doi: 10.1080/20014091111677. [DOI] [PubMed] [Google Scholar]
- 11.Cohen SD, Khairallah EA. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab Rev. 1997;29(1-2):59–77. doi: 10.3109/03602539709037573. [DOI] [PubMed] [Google Scholar]
- 12.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89(1):31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 13.Muriel P. Role of free radicals in liver diseases. Hepatol Int. 2009;3(4):526–536. doi: 10.1007/s12072-009-9158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray SD, Balasubramanian G, Bagchi D, Reddy CS. Ca(2+)-calmodulin antagonist chlorpromazine and poly(ADP-ribose) polymerase modulators 4-aminobenzamide and nicotinamide influence hepatic expression of BCL-XL and P53 and protect against acetaminophen-induced programmed and unprogrammed cell death in mice. Free Radic Biol Med. 2001;31(3):277–291. doi: 10.1016/s0891-5849(01)00562-7. [DOI] [PubMed] [Google Scholar]
- 15.Tsutsui S, et al. D-galactosamine induced hepatocyte apoptosis is inhibited in vivo and in cell culture by a calcium calmodulin antagonist, chlorpromazine, and a calcium channel blocker, verapamil. Exp Anim. 2003;52(1):43–52. doi: 10.1538/expanim.52.43. [DOI] [PubMed] [Google Scholar]
- 16.Litjens T, Harland ML, Roberts ML, Barritt GJ, Rychkov GY. Fast Ca(2+)-dependent inactivation of the store-operated Ca2+ current (ISOC) in liver cells: A role for calmodulin. J Physiol. 2004;558(Pt 1):85–97. doi: 10.1113/jphysiol.2004.065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barritt GJ, Litjens TL, Castro J, Aromataris E, Rychkov GY. Store-operated Ca2+ channels and microdomains of Ca2+ in liver cells. Clin Exp Pharmacol Physiol. 2009;36(1):77–83. doi: 10.1111/j.1440-1681.2008.05095.x. [DOI] [PubMed] [Google Scholar]
- 18.Rychkov GY, Barritt GJ. Expression and function of TRP channels in liver cells. Adv Exp Med Biol. 2011;704:667–686. doi: 10.1007/978-94-007-0265-3_35. [DOI] [PubMed] [Google Scholar]
- 19.Fonfria E, et al. Tissue distribution profiles of the human TRPM cation channel family. J Recept Signal Transduct Res. 2006;26(3):159–178. doi: 10.1080/10799890600637506. [DOI] [PubMed] [Google Scholar]
- 20.Harteneck C, Frenzel H, Kraft R. N-(p-amylcinnamoyl)anthranilic acid (ACA): A phospholipase A(2) inhibitor and TRP channel blocker. Cardiovasc Drug Rev. 2007;25(1):61–75. doi: 10.1111/j.1527-3466.2007.00005.x. [DOI] [PubMed] [Google Scholar]
- 21.Hill K, McNulty S, Randall AD. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch Pharmacol. 2004;370(4):227–237. doi: 10.1007/s00210-004-0981-y. [DOI] [PubMed] [Google Scholar]
- 22.Aromataris EC, Roberts ML, Barritt GJ, Rychkov GY. Glucagon activates Ca2+ and Cl- channels in rat hepatocytes. J Physiol. 2006;573(Pt 3):611–625. doi: 10.1113/jphysiol.2006.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Letelier ME, López-Valladares M, Peredo-Silva L, Rojas-Sepúlveda D, Aracena P. Microsomal oxidative damage promoted by acetaminophen metabolism. Toxicol In Vitro. 2011;25(7):1310–1313. doi: 10.1016/j.tiv.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 24.Ishii M, et al. Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat beta-cell line RIN-5F. Cell Calcium. 2006;39(6):487–494. doi: 10.1016/j.ceca.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Kaneko S, et al. A critical role of TRPM2 in neuronal cell death hy hydrogen peroxide. J Pharm Sci. 2006;101(1):66–76. doi: 10.1254/jphs.fp0060128. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, et al. 2013. Loss of TRPM2 function protects against irradiation-induced salivary gland dysfunction. Nature Comm 4:1515.
- 27.Nagamine K, et al. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54(1):124–131. doi: 10.1006/geno.1998.5551. [DOI] [PubMed] [Google Scholar]
- 28.Hara Y, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9(1):163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 29.Perraud AL, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411(6837):595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 30.Perraud AL, Schmitz C, Scharenberg AM. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium. 2003;33(5-6):519–531. doi: 10.1016/s0143-4160(03)00057-5. [DOI] [PubMed] [Google Scholar]
- 31.Wehage E, et al. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277(26):23150–23156. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- 32.Ray SD, Kamendulis LM, Gurule MW, Yorkin RD, Corcoran GB. Ca2+ antagonists inhibit DNA fragmentation and toxic cell death induced by acetaminophen. FASEB J. 1993;7(5):453–463. doi: 10.1096/fasebj.7.5.8462787. [DOI] [PubMed] [Google Scholar]
- 33.Cover C, et al. Pathophysiological role of poly(ADP-ribose) polymerase (PARP) activation during acetaminophen-induced liver cell necrosis in mice. Toxicol Sci. 2005;84(1):201–208. doi: 10.1093/toxsci/kfi065. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi K, et al. Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res. 1999;829(1-2):46–54. doi: 10.1016/s0006-8993(99)01335-9. [DOI] [PubMed] [Google Scholar]
- 35.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40(5):1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto S, et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14(7):738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kheradpezhouh E, et al. Curcumin protects rats against acetaminophen-induced hepatorenal damages and shows synergistic activity with N-acetyl cysteine. Eur J Pharmacol. 2010;628(1-3):274–281. doi: 10.1016/j.ejphar.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 38.Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18(1):61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 39.Chen C, Krausz KW, Idle JR, Gonzalez FJ. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem. 2008;283(8):4543–4559. doi: 10.1074/jbc.M706299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fonfria E, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143(1):186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buelow B, Song Y, Scharenberg AM. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem. 2008;283(36):24571–24583. doi: 10.1074/jbc.M802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsokos-Kuhn JO, Hughes H, Smith CV, Mitchell JR. Alkylation of the liver plasma membrane and inhibition of the Ca2+ ATPase by acetaminophen. Biochem Pharmacol. 1988;37(11):2125–2131. doi: 10.1016/0006-2952(88)90570-9. [DOI] [PubMed] [Google Scholar]
- 43.Chun LJ, Tong MJ, Busuttil RW, Hiatt JR. Acetaminophen hepatotoxicity and acute liver failure. J Clin Gastroenterol. 2009;43(4):342–349. doi: 10.1097/MCG.0b013e31818a3854. [DOI] [PubMed] [Google Scholar]
- 44.Berry MN, Edwards AM, Barritt GJ. In: Laboratory Techniques in Biochemistry and Molecular Biology. Burdon RH, van Knippenberg PH, editors. Vol 21. Amsterdam: Elsevier; 1991. pp. 15–81. [Google Scholar]
- 45.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.