

Significance
Tumor suppressor p53 has been known to have a broader role that extends to the regulation of energy metabolism. We investigated the role of islet p53 and found that genetic and pharmacological inhibition of p53 preserves insulin secretion and glucose tolerance in both streptozotocin-induced type 1 and db/db mouse models of type 2 diabetes. Glucolipotoxicy induces accumulation of p53 in the cytosol via oxidative stress and endoplasmic reticulum stress. Cytosolic p53 inhibits the autophagic clearance of damaged mitochondria by an inhibitory protein–protein interaction with Parkin, leading to the impairment of mitochondrial energetics and subsequent insulin secretion signals in islet β-cells.
Abstract
Mitochondrial compromise is a fundamental contributor to pancreatic β-cell failure in diabetes. Previous studies have demonstrated a broader role for tumor suppressor p53 that extends to the modulation of mitochondrial homeostasis. However, the role of islet p53 in glucose homeostasis has not yet been evaluated. Here we show that p53 deficiency protects against the development of diabetes in streptozotocin (STZ)-induced type 1 and db/db mouse models of type 2 diabetes. Glucolipotoxicity stimulates NADPH oxidase via receptor for advanced-glycation end products and Toll-like receptor 4. This oxidative stress induces the accumulation of p53 in the cytosolic compartment of pancreatic β-cells in concert with endoplasmic reticulum stress. Cytosolic p53 disturbs the process of mitophagy through an inhibitory interaction with Parkin and induces mitochondrial dysfunction. The occurrence of mitophagy is maintained in STZ-treated p53−/− mice that exhibit preserved glucose oxidation capacity and subsequent insulin secretion signaling, leading to better glucose tolerance. These protective effects are not observed when Parkin is deleted. Furthermore, pifithrin-α, a specific inhibitor of p53, ameliorates mitochondrial dysfunction and glucose intolerance in both STZ-treated and db/db mice. Thus, an intervention with cytosolic p53 for a mitophagy deficiency may be a therapeutic strategy for the prevention and treatment of diabetes.
It is crucial for organismal homeostasis to maintain glucose metabolism, in which insulin secreted from pancreatic β-cells in response to blood glucose levels stimulates glucose uptake into peripheral tissues. Diabetes is a chronic disorder of insulin insufficiency resulting in the disruption of glucose homeostasis and multiorgan complications. Type 1 diabetes results primarily from autoimmune β-cell destruction (1). In contrast, diverse environmental factors and genetic susceptibility combine to cause type 2 diabetes (2). Although these two types of diabetes have distinct pathogenic mechanisms, mitochondrial dysfunction is a common element of disease progression in both forms. Mitochondria are the principal organelles of energy production and cell death. Mitochondria also generate reactive oxygen species (ROS) as a by-product of oxidative phosphorylation. Impaired mitochondrial respiration capacity and ROS-mediated UCP2 expression lead to inadequate ATP production during glucose stimulation, which results in a defect in subsequent insulin secretion signaling (3). Moreover, mitochondria-dependent apoptosis is involved in the diminished β-cell mass observed in type 1 and type 2 diabetes (4).
Although p53 has a well-documented role as a tumor suppressor, recent detailed studies have identified broader functions for p53 that are independent of its effects in tumorigenesis. p53 had been shown to regulate the transcription of several genes involved in redox and glucose metabolism and autophagy (5, 6). It also possesses an extranuclear function in which cytosolic p53 inhibits the process of autophagy by a poorly known mechanism (7). We have previously demonstrated the contribution of p53 to mitochondrial integrity. p53 is required for mitochondrial aerobic respiration through the expression of SCO2 (8). Conversely, cytosolic p53 has been shown to disturb the clearance of damaged mitochondria by an inhibitory interaction with Parkin (9). Other recent studies have also revealed that p53 has a distinct cellular function according to its cellular concentration and distribution (10). Despite the intensive studies on p53 in glucose homeostasis, the role of islet p53 in β-cell function and diabetes remains poorly understood.
In this study, we demonstrate the distinctive expression and pathogenic involvement of p53 in both type 1 and type 2 diabetes. Under diabetic conditions, the cellular distribution of p53 shifts to the cytosolic compartment with no alterations in expression levels in whole cells, in which oxidative stress and endoplasmic reticulum (ER) stress are collaboratively involved. Glucotoxicity and lipotoxicity provoke NADPH oxidase-dependent ROS generation via a receptor for advanced-glycation end products (RAGE) and Toll-like receptor 4 (TLR4), respectively. The cytosolic accumulation of p53 disturbs Parkin-mediated mitophagy and induces further mitochondrial compromise and subsequent defects in insulin secretion. The present study provides a mechanism for elucidating the pathogenesis of diabetes.
Results
Deletion of Islet p53 Preserves Glucose Oxidation and Insulin Secretion in Streptozotocin-Induced Type 1 Diabetes.
To assess the effects of islet p53 on diabetes, we first induced experimental type 1 diabetes with the multiple low dose of streptozotocin (STZ) treatment (11) in WT and p53−/− mice. Bone marrow (BM)-chimeric mice were generated by transplanting the BM cells of WT-GFP mice into WT or p53−/− mice to eliminate differences in the anti-inflammatory effect of p53 in the immune system. Eight-week-old mice underwent BM transplantation and five consecutive i.p. daily injections of 40 mg⋅kg−1 of STZ (Fig. S1A). A previous report showed that the p53 pathway was not involved in pancreatic β-cell death upon irradiation (12). We observed a few apoptotic cells in islets following irradiation in this study (Fig. S1B). Compared with WT mice that developed hyperglycemia 2 wk after the start of STZ injections, p53−/− mice exhibited markedly suppressed hyperglycemia with preserved serum insulin levels (Fig. 1 A and B). Furthermore, glucose tolerance tests revealed the significant amelioration of glucose metabolism in p53−/− mice (Fig. 1C). We observed no significant changes in insulin tolerance tests (Fig. 1D), whereas serum insulin levels were enhanced in p53−/− mice after glucose loading in vivo (Fig. 1E). These results suggested that disruption of islet p53 preserved insulin secretion and maintained glucose metabolism in STZ-induced type 1 diabetes.
Fig. 1.
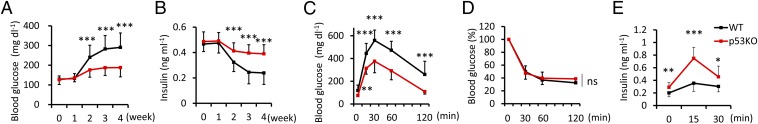
p53−/− mice were resistant to hyperglycemia in STZ-induced type 1 diabetes. (A) Blood glucose and (B) serum insulin concentrations from mice fed ad libitum were measured at the indicated time points after STZ treatment (n = 16–18). (C) Glucose and (D) insulin tolerance tests were performed 4 wk after STZ treatment (n = 12). (E) Serum insulin values were assessed before and at 15 and 30 min into the glucose tolerance test (n = 12). Data are shown as the means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
Serum insulin levels are governed by a combination of the insulin secretory capacity of individual β-cells and the number of β-cells. In this study, we first performed histological analysis of islets in STZ-treated mice. No significant change was observed in the islet mass, β-cell proliferation, and apoptosis between WT and p53−/− mice (Fig. 2A and Fig. S1 C and D). We then investigated β-cell function by measuring islet oxygen consumption rate (OCR) with the XF24 analyzer. The up-regulation of OCR at high glucose relative to basal glucose was stronger in STZ-treated p53−/− mice (Fig. 2B). Preserved capacity of mitochondrial glucose oxidation was associated with sustained insulin secretion.
Fig. 2.

Glucose oxidation capacity was preserved in STZ-treated p53−/− mice. (A) Histological analysis of insulin immunostaining of islets 4 wk after STZ treatment. (Scale bars, 500 μm.) The β-cell area relative to the pancreatic area was quantified (n = 4). (B) Representative results of the OCR of islets assessed using the Seahorse XF24 extracellular flux analyzer. The amplification rate of OCR at high (22.2 mM) glucose was calculated relative to basal (2.8 mM) glucose stimulation from five replicates per sample (n = 4). Data are shown as the means ± SD. *P < 0.05; ***P < 0.001.
Cytosolic p53 Inhibits Parkin-Mediated Mitophagy in β-Cells.
We examined the protein–protein interaction between p53 and Parkin in MIN6 β-cells. The endogenous Parkin–p53 complex was observed in both immunoprecipitates of endogenous Parkin and p53 and markedly in the cytosolic lysate of MIN6 β-cells under diabetic conditions composed of 30 mM glucose and 0.3 mM palmitate incubation for 7 d (Fig. 3A). We confirmed the contribution of p53 to mitophagy in MIN6 β-cells overexpressing GFP-LC3. Colocalization between LC3 and mitochondria was evaluated by blocking autophagy flux using bafilomycin-A1 (Baf-A1) to underscore mitophagy. This colocalization was reduced under diabetic conditions, but was preserved by siRNA targeting p53. siRNA targeting Parkin canceled the p53 knockdown-mediated restoration of colocalization (Fig. 3B and Fig. S2). We quantified mitophagy further by using the pMT-mKeima-Red that exhibits a bimodal excitation maximum at 430 and 590 nm in a neutral or acidic pH, respectively (13). The population of mitochondria fused to lysosomes can be determined by quantifying the 590/430-nm excitation ratio. Although diabetic conditions reduced the 590/430-nm ratio, siRNA targeting p53 enhanced the ratio to as high as the control medium, and this was not observed in Parkin knockdown MIN6 β-cells (Fig. S3A). These results demonstrated that p53 was involved in the significant decrease in Parkin-dependent mitophagy under diabetic conditions. Consequently, we examined the contribution of mitophagy to β-cell function. Insulin secretion from MIN6 β-cells under diabetic conditions in response to high glucose was blunted and was ameliorated by p53 knockdown. This improved glucose-stimulated insulin secretion (GSIS) was canceled by the knockdown of Parkin (Fig. 3C). Glucose metabolism has been shown to increase ATP concentrations as a signal to enhance insulin secretion (14). Increases in ATP content with increasing glucose levels were not marked under diabetic conditions. The knockdown of p53 enhanced ATP content in response to high glucose depending on Parkin (Fig. 3D). p53 knockdown-mediated improvement of GSIS was also negated by inhibiting the process of autophagy with Baf-A1 treatment or Atg5 knockdown as well as in preventing mitophagy with PINK1 knockdown (Fig. 3E and Fig. S3B), supporting evidence that preserved mitophagy was behind the protective effect of p53 ablation.
Fig. 3.

Cytosolic p53 inhibits Parkin-mediated mitophagy in β-cells. (A) The endogenous Parkin–p53 complex in β-cells. Cytosolic lysate of MIN6 β-cells exposed to 30 mM high glucose (HG) and 0.3 mM palmitate (PA) for 1 wk was immunoprecipitated with anti-p53, Parkin, and control IgG antibodies. (B) Representative images of GFP-LC3–overxpressing MIN6 β-cells exposed to HG and PA. Cells were transfected with siRNA targeting p53 and/or Parkin and were treated to underscore mitophagy with 100 nM Baf-A1, a specific inhibitor of H+-ATPases of the vacuolar type, for 6 h before the immunostaining of mitochondria with anti-TOM20 (red) (original magnification, ×1,000). (Scale bars, 10 μm.) (Right) Colocalization between LC3 and TOM20. A minimum of 50 GFP-positive cells were scored in three independent experiments. (C and D) MIN6 β-cells were stimulated with 2.8 or 22.2 mM glucose for 30 min. Insulin secretion (C) and ATP contents (D) were measured by ELISA and a luminometer, respectively, in duplicate (n = 4). (E) Glucose-stimulated insulin secretion was measured by ELISA in the context of mitophagy deficiency induced by the transfected with siRNA targeting Atg5 or PINK1. The experiments were performed in duplicate (n = 4). Data are shown as the means ± SD. *P < 0.05; **P < 0.01.
To further confirm the role of Parkin-mediated mitophagy, we generated MIN6 β-cells stably overexpressing Parkin (Fig. 4A). Preserved mitophagy was observed in Parkin-overexpressing stable cell lines under diabetic conditions (Fig. 4B and Fig. S4). Parkin-mediated mitophagy was associated with preserved mitochondrial oxygen consumption in complexes II and IV, despite no significant differences in glucokinase and hexokinase activities (Fig. 4 C and D). The maintained respiration capacity provided higher elevation in ATP content and insulin secretion in response to high glucose (Fig. 4 E and F). Although Parkin has cellular functions other than the induction of mitophagy (15), the protective effect of Parkin overexpression was not observed in the context of compromised autophagosome formation (Fig. 4G).
Fig. 4.

Parkin-mediated mitophagy maintains insulin secretion under diabetic conditions. (A) Construction of the stable MIN6 β-cell lines overexpressing Parkin. Representative immunoblots are shown from three independent experiments. (B) Excitation ratio (590/430 nm) of pMT-mKeima-Red–overexpressing stable cell lines incubated with HG and PA. Baf-A1–treated cells were used as a negative control for the induction of mitophagy. The experiments were performed in eight replicates (n = 3). (C) The sequential state III OCR of isolated mitochondria from stable cell lines was assessed using the Seahorse XF24 analyzer. Representative results are shown, and the OCR of each complex was calculated from five replicates per sample (n = 3). (D) Glucokinase (GK) and hexokinase (HK) activities in stable cell lines. Results are shown from four independent experiments performed in duplicate. (E) ATP contents and (F and G) insulin secretion were measured by a luminometer and ELISA, respectively. The experiments were performed in duplicate (n = 4). Data are shown as the means ± SD. †P < 0.05 versus stable cell lines carrying control vectors under the same condition.
ER Stress and Oxidative Stress Increase Cytosolic p53 in Diabetic Islets.
We examined the expression of p53 in islets from STZ-induced type 1 and db/db mouse models of type 2 diabetes. Intriguingly, p53 expression was increased in the cytosol, but was decreased in the nucleus with no alteration in the whole-islet expression level in both STZ-treated mice and db/db mice (Fig. S5A). A similar result was observed in MIN6 β-cells under diabetic conditions (Fig. S5B). Consistent with these results, a detectable increase in p53 staining was observed in the cytosol (Fig. S5C). To investigate the mechanism underlying the altered subcellular localization of p53, we focused on ER stress and NADPH oxidase that were activated under diabetic conditions (Fig. S5 D–F). Previous studies showed that ER stress induced the cytosolic translocation and subsequent degradation of p53 via the glycogen synthase kinase (GSK)-3β pathway (16–18). We observed this redistribution in MIN6 β-cells following treatment with the ER stress inducer thapsigargin, which was canceled by the GSK-3 inhibitor LiCl (Fig. S5G). The export of nuclear p53 to the cytosolic compartment under diabetic conditions was also inhibited by LiCl treatment (Fig. S5H). The treatment with the antioxidant N-acetylcysteine (NAC) reduced the expression of p53 in both the nucleus and cytosol (Fig. S5I). Furthermore, altered p53 expression in MIN6 β-cells under diabetic conditions was canceled by the treatment with LiCl and NAC (Fig. S5J), indicating the involvement of ER stress and oxidative stress in altered p53 expression. Preventing the distinctive expression of p53 actually preserved GSIS under diabetic conditions (Fig. S5K).
The main sources of ROS in the pancreatic islets are mitochondria and NADPH oxidase (19, 20), which may produce a feed-forward vicious cycle of ROS production (21). Under the high-glucose condition, the siRNA targeting RAGE, as well as p47phox, one of the NADPH oxidase components, partially canceled ROS production. However, siRNA targeting RAGE did not provide an additional reduction in ROS generation in p47phox knockdown MIN6 β-cells (Fig. S6A). Similarly, siRNA targeting TLR4 suppressed ROS production as much as siRNA targeting p47phox without any additive effects in palmitate-treated MIN6 β-cells (Fig. S6B). These results suggested that RAGE and TLR4 were involved in ROS generation through the activation of NADPH oxidase in glucotoxicity and lipotoxicity, respectively. These alterations in ROS production were reflected in the expression of p53 (Fig. S6 C and D).
p53 Deficiency Preserves Mitochondrial Integrity and Insulin Secretion in Diabetic Mice.
We examined the morphology of mitochondria in STZ-treated β-cells using electron microscopy. Islet β-cells in p53−/− mice had more mitochondria incorporated into autophagosomes and lower abnormal mitochondria. p53 deletion induced few transcriptional changes of mitochondrial quality-control factors, supporting the contribution of inhibitory interaction between cytosolic p53 and Parkin (Fig. S7). To directly test our hypothesis that Parkin-mediated mitophagy contributes to protection against diabetes by p53 deletion, we crossed p53−/− mice with Parkin−/− mice and treated mice with STZ after BM transplantation. Parkin deletion markedly reduced mitophagy and mitochondrial integrity that were preserved in p53−/− mice (Fig. 5A). Next we isolated islets from the four mouse groups and carried out static incubation experiments. Increases in ATP content with increasing glucose levels were suppressed in STZ-treated mice. p53 deletion enhanced ATP content and subsequent insulin secretion in response to high glucose, which was abolished by Parkin deletion (Fig. 5 B and C). Consistently, p53 deletion significantly improved hyperglycemia accompanied by preserved serum insulin levels, and these protective effects were not observed in p53−/−Parkin−/− mice (Fig. 5 D and E). These results suggested that p53 deficiency ameliorated the functional impairment of islets depending on Parkin, which improved glucose intolerance in STZ-induced type 1 diabetes. We conducted further experiments using pifithrin-α (PFT-α), a specific inhibitor of p53, to determine the effect of p53 deficiency. PFT-α was administered to mice transplanted with BM cells from p53−/− mice at a dose of 1.1 mg⋅kg−1 three times weekly for 4 wk starting on the first day of STZ treatment (Fig. S8). Under PFT-α treatment, WT mice exhibited glucose tolerance similar to that of p53−/− mice, whereas Parkin−/− mice had reduced glucose tolerance (Fig. 5F). Serum insulin levels after glucose loading were also not different between WT and p53−/− mice and were decreased in Parkin−/− mice (Fig. 5G). In the OCR of isolated islets, up-regulation at high glucose relative to basal glucose was restored in WT and p53−/− mice equivalently, but was attenuated in Parkin−/− mice (Fig. 5H). The protective effects of p53 deficiency against diabetes were also mostly achieved by PFT-α treatment, which was actually dependent on the function of Parkin as hypothesized.
Fig. 5.

Parkin-mediated mitophagy links p53 deficiency and protection against diabetes. (A) Representative electron micrographs of STZ-treated islets. Arrows indicate abnormal mitochondria defined by marked swelling associated with an increased number of disarrayed or disappeared cristae and by the reduced electron density of the matrix (original magnification ×5,000). (Scale bars, 2 μm.) Magnified photograph represents an autophagic vacuole containing mitochondria (red box) (original magnification ×12,000). (Scale bar, 500 nm.) (Right) Abnormal mitochondria and mitochondria incorporated into the autophagic vacuole were quantified from at least 10 β-cells (n = 3). (B and C) Batches of 10 pancreatic islets isolated from STZ-treated WT, p53−/−, Parkin−/−, and p53−/−Parkin−/− mice were stimulated with 2.8 or 22.2 mM glucose for 30 min. ATP contents (B) and insulin secretion (C) were measured by a luminometer and ELISA, respectively. The amplification rates of ATP contents and insulin secretion at high glucose were calculated relative to basal glucose stimulation. Results are shown from duplicate experiments per sample (n = 6). (D) Blood glucose levels and (E) serum insulin levels from mice fed ad libitum (n = 11–20). (F) Glucose tolerance tests were performed after 4 wk of PFT-α or the carrier DMSO treatment (n = 12). (G) Serum insulin values were assessed before and at 15 and 30 min into the glucose tolerance test (n = 12). (H) The OCR of islets was assessed using the Seahorse XF24 analyzer. The amplification rate of OCR at high glucose was calculated relative to basal glucose stimulation from five replicates per sample (n = 4). Data are shown as the means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
We also examined the protective effects of PFT-α against diabetes in type 2 diabetes. Six-week-old male db/db mice were treated with 1.1 mg⋅kg−1 PFT-α three times weekly for 4 wk. Hyperglycemia was improved with higher serum insulin levels in db/db mice treated with PFT-α (Fig. 6 A and B). Glucose tolerance tests also revealed that PFT-α significantly ameliorated glucose metabolism (Fig. 6C). Similar to the experiment on STZ-induced type 1 diabetes, insulin tolerance remained unchanged, whereas an increase in insulin secretion contributed to improved glucose homeostasis (Fig. 6 D and E). Histological analysis showed no significant changes in islet mass, cell proliferation, or apoptosis in the PFT-α treatment (Fig. S9 A–C). Morphological analysis of mitochondria in β-cells using electron microscopy revealed that PFT-α increased mitochondria incorporated into autophagosomes, which inversely correlated with the presence of abnormal mitochondria (Fig. 6F). In the OCR of isolated islets, PFT-α restored up-regulation at high glucose relative to basal glucose (Fig. 6G). Improved glucose oxidation resulted in a higher ATP content and GSIS (Fig. 6 H and I). These results indicated that p53 inhibition with PFT-α treatment promoted mitophagy and prevented mitochondrial compromise and subsequent β-cell failure in db/db mice.
Fig. 6.

PFT-α preserves β-cell integrity in db/db mice. (A) Six-week-old mice were injected with PFT-α or the carrier DMSO three times per week for 4 wk. Blood glucose levels were measured at the indicated time points (n = 14). (B) Serum insulin levels from mice fed ad libitum were measured after 4 wk of PFT-α or the carrier DMSO treatment (n = 14). (C) Glucose and (D) insulin tolerance tests were performed after 4 wk of treatment (n = 8). (E) Serum insulin values were assessed before and at 15 and 30 min into the glucose tolerance test (n = 8). (F) Representative electron micrographs of islets in db/db mice treated with PFT-α or the carrier DMSO. Arrows indicate abnormal mitochondria (original magnification ×5,000). (Scale bars, 2 μm.) Magnified photograph represents an autophagic vacuole containing mitochondria (red box) (original magnification ×12,000). (Scale bar, 500 nm.) (Lower) Abnormal mitochondria and mitochondria incorporated into the autophagic vacuole were quantified from at least 10 β-cells (n = 3). (G) Batches of five pancreatic db/db mice islets were stimulated with 2.8 or 22.2 mM glucose. The OCR of islets was assessed using the Seahorse XF24 analyzer. The amplification rate of OCR at high glucose was calculated relative to basal glucose stimulation from five replicates per sample (n = 4). (H) ATP contents and (I) insulin secretion were measured by a luminometer and ELISA, respectively. The amplification rates of ATP contents and insulin secretion at high glucose were calculated relative to basal glucose stimulation. Results are shown from duplicate experiments per sample (n = 4). Data are shown as the means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
We demonstrated that cytosolic p53 inhibited Parkin-mediated autophagic degradation of damaged mitochondria and promoted mitochondrial compromise, leading to impaired insulin secretion in the β-cells of diabetic mice. Oxidative stress up-regulated p53 and ER stress induced its translocation from the nucleus to the cytosol, which resulted in the cytosolic accumulation of p53. Glucotoxicity and lipotoxicity activated NAPDH oxidase via RAGE and TLR4, respectively, and increased p53 expression and ROS-generating damaged mitochondria. In this study, the recent development of a high-throughput method for measuring islet oxygen consumption (22) revealed a poor increase in glucose oxidation in response to high glucose in diabetic islets. Moreover, the direct measurement of oxygen consumption of mitochondria isolated from MIN6 β-cells demonstrated the reduced respiration capacity under diabetic conditions without any marked changes in glucokinase activity. Cytosolic p53-mediated mitophagy deficiency is involved in an impaired electron transport system and subsequent ATP insufficiency. Based on the proposed mechanism (Fig. S9D), PFT-α treatment, as well as the genetic deletion of p53, ameliorated insulin secretion and glucose intolerance in both type 1 and type 2 diabetes.
Mitochondria are remarkably dynamic organelles that migrate, divide, and fuse. Membrane fusion and fission allow mitochondrial content mixing within a cell to maintain integrity, and severely damaged mitochondria are selectively removed by macroautophagy, termed “mitophagy.” Given the importance of mitochondria in insulin secretion, mitochondrial quality control has been assumed to contribute to β-cell integrity and glucose homeostasis. Evidence is accumulating for the relationship between mitochondrial dynamics and diabetes. Mitochondrial morphology had been shown to shift from an interconnected tubular form to a disconnected shorter appearance according to the diabetes status in β-cells. The tubular form obtained by the dominant negative expression of fission-promoting DRP1 is resistant to apoptosis in β-cells (23). In contrast, the fragmented pattern produced by the genetic deletion of Opa1 induces mitochondrial dysfunction and subsequent hyperglycemia (24). These results indicate the association between the postfission state in mitochondria and pancreatic β-cell failure. Intriguingly, impaired autophagy caused by the genetic deletion of Atg7 in β-cells also leads to hyperglycemia as a result of mitochondrial compromise and defective GSIS (25), which suggests that mitophagy may be indispensable for mitochondrial integrity and β-cell physiology. Recent detailed investigations identified the molecular mechanism behind the process of mitophagy, whereas genetic deletions of these molecules were not reported to contribute to physiological glucose homeostasis. One of the reasons for this may be that mitochondria are also nonselectively removed by macroautophagy. However, this study demonstrated that loss of Parkin-mediated mitophagy induced further β-cell failure under pathological stress conditions including STZ exposure and leptin receptor defects.
Identifying functions for p53 that are independent of its tumor suppressor effects is an intriguing and growing area of research. Recent studies on p53 produced some confusing and apparently contradictory results for glucose homeostasis. Peripheral tissue p53 was shown to play a controversial role in insulin resistance via its antioxidant and inflammatory effects (26, 27). As an indirect effect on pancreatic β-cells, immune cell p53 protected β-cells against STZ toxicity by inhibiting islet inflammation (28). A broader role for p53 has extended to the regulation of glycolysis (29), mitochondrial function (8, 30), and autophagy (10), suggesting the involvement of β-cell p53 in insulin secretion. This study shows that p53 has a previously undescribed pathogenic effect on pancreatic β-cells in which p53 promotes mitochondrial dysfunction by inhibiting Parkin-mediated mitophagy.
Materials and Methods
Detailed methods can be found in SI Materials and Methods. The murine MIN6 β-cell line was kindly provided by J. Miyazaki (Osaka University, Osaka) and cultured in DMEM containing 10 mM glucose, 0.1 mM 2-mercaptoethanol, 100 units/mL penicillin, 0.05 mg/mL streptomycin, and 10% FBS (HyClone) in humidified 5% CO2 at 37 °C (31). Cells were incubated with 30 mM glucose and 0.3 mM palmitate for 1 wk for mimicking diabetes.
Supplementary Material
Acknowledgments
We thank Dr. T. Yoshimori and Dr. J. Miyazaki for providing materials mentioned in the text. This study was supported in part by Grants-in-Aid from the Ministry of Education, Science and Culture of Japan.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318951111/-/DCSupplemental.
References
- 1.Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: Translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol. 2013;13(4):243–256. doi: 10.1038/nri3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muoio DM, Newgard CB. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(3):193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 3.Zhang CY, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105(6):745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 4.Szabadkai G, Duchen MR. Mitochondria mediated cell death in diabetes. Apoptosis. 2009;14(12):1405–1423. doi: 10.1007/s10495-009-0363-5. [DOI] [PubMed] [Google Scholar]
- 5.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9(10):691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107(16):7461–7466. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tasdemir E, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matoba S, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 9.Hoshino A, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 10.Maiuri MC, et al. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22(2):181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: New model of diabetes mellitus. Science. 1976;193(4251):415–417. doi: 10.1126/science.180605. [DOI] [PubMed] [Google Scholar]
- 12.Nam SY, Lee MK, Sabapathy K. The tumour-suppressor p53 is not required for pancreatic beta cell death during diabetes and upon irradiation. J Physiol. 2008;586(2):407–417. doi: 10.1113/jphysiol.2007.142612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18(8):1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 14.Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest. 2011;121(6):2118–2125. doi: 10.1172/JCI45680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim KY, et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest. 2011;121(9):3701–3712. doi: 10.1172/JCI44736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qu L, et al. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004;18(3):261–277. doi: 10.1101/gad.1165804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pluquet O, Qu LK, Baltzis D, Koromilas AE. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Mol Cell Biol. 2005;25(21):9392–9405. doi: 10.1128/MCB.25.21.9392-9405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee MJ, Kee KH, Suh CH, Lim SC, Oh SH. Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress- and calpain-mediated mitochondrial cell death pathways. Toxicology. 2009;264(3):205–214. doi: 10.1016/j.tox.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Syed I, et al. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: Role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes. 2011;60(11):2843–2852. doi: 10.2337/db11-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor-Fishwick DA. NOX, NOX who is there? The contribution of NADPH oxidase one to beta cell dysfunction. Front Endocrinol (Lausanne) 2013;4:40. doi: 10.3389/fendo.2013.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51(7):1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wikstrom JD, et al. A novel high-throughput assay for islet respiration reveals uncoupling of rodent and human islets. PLoS ONE. 2012;7(5):e33023. doi: 10.1371/journal.pone.0033023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng L, et al. Involvement of dynamin-related protein 1 in free fatty acid-induced INS-1-derived cell apoptosis. PLoS ONE. 2012;7(11):e49258. doi: 10.1371/journal.pone.0049258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell. 2011;22(13):2235–2245. doi: 10.1091/mbc.E10-12-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung HS, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8(4):318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Armata HL, et al. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol Cell Biol. 2010;30(24):5787–5794. doi: 10.1128/MCB.00347-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minamino T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15(9):1082–1087. doi: 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- 28.Zheng SJ, Lamhamedi-Cherradi SE, Wang P, Xu L, Chen YH. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes. 2005;54(5):1423–1428. doi: 10.2337/diabetes.54.5.1423. [DOI] [PubMed] [Google Scholar]
- 29.Bensaad K, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 30.Sahin E, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyazaki J, et al. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: Special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127(1):126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.