

Significance
Mitochondria, the powerhouses of the cell, possess their own DNA, which encodes proteins involved in cellular ATP production. During the aging process deleted versions of mtDNA accumulate inside cells and replace the wild type form. Currently no explanation exists that can explain the mechanism behind this accumulation, which is also compatible with experimental observations. We present here a new idea based on the distinctive connection between transcription and replication of mtDNA. Analysis of mtDNA deletion spectra strongly supports our hypothesis and computer simulations show the mechanism to be compatible with data from short- and long-lived species. We believe that this idea may provide a major breakthrough in understanding the complex links between mitochondrial mutations and their accumulation in aging.
Keywords: mitochondrial mutations, mathematical model
Abstract
The mitochondrial theory of aging is widely popular but confronted by several apparent inconsistencies. On the one hand, mitochondrial energy production is of central importance to the health and proper functioning of cells, and single-cell studies have shown that mtDNA deletion mutants accumulate in a clonal fashion in various mammalian species, displacing the wild-type mtDNAs. On the other hand, no explanation exists yet for the clonal expansion of mtDNA mutants that is compatible with experimental observations. We present here a new idea based on the distinctive connection between transcription and replication of metazoan mtDNA. Bioinformatic analysis of mtDNA deletion spectra strongly supports the predictions of this hypothesis and identifies specific candidates for proteins involved in transcriptional control of mtDNA replication. Computer simulations show the mechanism to be compatible with the available data from short- and long-lived mammalian species.
Mitochondria play central roles in health and disease and have long been implicated in aging (1–3). Point mutations and deletions of mtDNA have the capacity to impair mitochondrial ATP production with negative consequences for all aspects of cellular metabolism and viability. However, the exact role of acquired mitochondrial DNA (mtDNA) mutations remains unclear (4, 5). The possibility of a common underlying basis for mitochondrial diseases and intrinsic aging is intriguing (6). However, fundamental questions remain unanswered; resolving these will be necessary before the relevance of mtDNA mutations in aging and disease, including cancer, can be confirmed.
Single-cell studies have shown convincingly that tissue sections of old individuals display a mosaic pattern of healthy and affected cells, coexisting side by side. In the compromised cells of various mammalian species such as mice, rats, monkeys, and humans, mitochondrial mutants accumulate with age (7–13). Furthermore, within individual cells the mitochondrial population is frequently overtaken by a single mutant type via some form of “clonal expansion.”
The dynamics that lie behind such clonal expansion must be rooted in the biology of mtDNA replication and removal through turnover (autophagy). These processes have their origins in the genesis of the animal mitochondrion as a bacterially derived endosymbiont which, during the course of evolution, has undergone a complex coevolution with the host cell, when most of the original mitochondrial genome has transferred to the host cell nucleus. In this article, we examine core challenges in understanding the dynamics of clonal expansion of mtDNA mutations and present evidence in support of a novel hypothesis to explain the clonal expansion particularly of mtDNA deletions, which also has deep relevance for the role of mtDNA mutations in aging and disease. We infer that there is a feedback mechanism regulating mtDNA replication, which is compromised in those mtDNA deletions that have been reported to undergo clonal expansion in aged animal tissues. The significance of such a mechanism is that it not only explains much that is puzzling about the connection between mtDNA mutations and aging but also offers potential new targets for intervention.
Current ideas to explain expansion of mtDNA mutations fall into two groups: those that involve some sort of selection advantage and those that do not. The nonselection ideas comprise: (i) the “vicious cycle” hypothesis, which suggests that defective mitochondria generate more radicals, and thereby more mutations, through a self-amplifying feedback mechanism (14, 15), or (ii) the suggestion that expansion occurs simply by random drift (16, 17). The problem with the vicious cycle is that this is unlikely to lead to the expansion of a single mutant; rather, one should expect a plethora of different mtDNA mutants in a single cell, which is the opposite of what has been observed. As for random drift, although early computer simulations showed that such a process might conceivably work for long-lived species such as humans (16), recent studies reveal that in short-lived animals such as rodents, drift predicts an extremely high degree of mtDNA heterogeneity that is again incompatible with experimental results (18).
Thus, it seems that a selection advantage is necessary to achieve clonal expansion of mtDNA mutations with the observed low-mtDNA heterogeneity in short-lived species. Here again there have been several alternative suggestions. The survival-of-the-slowest hypothesis identifies such a selection advantage by proposing that defective mitochondria are degraded less frequently than wild-type organelles (19). However, the proposed mechanism is incompatible with mitochondrial dynamics (20) and furthermore it has been shown that dysfunctional mitochondria are preferentially degraded (21–23), instead of being spared. It has also been proposed that reactive oxygen species (ROS) signal a deficiency of respiratory chain complexes back to the local mtDNA, which replicates in response (24). In this scenario mitochondrial mutants that produce more ROS would be selectively amplified. Although feasible, this idea does not readily explain the predominance of mtDNA deletions over point mutations and it predicts a central involvement of ROS in the aging process, which has been questioned by several recent reports (25–27). Most straightforwardly, the reduced genome size of deletion mutants has been suggested to provide a direct selection advantage through faster replication (28, 29). Because deletions inside the major arc lead to a juxtaposition of the heavy and light strand origins, the effects on replication time are even more severe than the pure size difference would suggest. As a consequence, a mutant with a complete deletion of the major arc should replicate 4.3 times faster than wild type and not 2.6 times as may be expected (30). This idea has been questioned, however, because the time required for the replication of the mtDNA is only 1–2 h (31, 32), whereas the half-life of mtDNA is on the order of 1–3 wk (33–35). It is therefore difficult to see how mtDNA replication could be a rate-limiting step for mitochondrial growth. This problem has been confirmed by simulations, which show that the large discrepancy between half-life and replication time diminishes the resulting selection advantage so much that it leads to unrealistically slow accumulation times (36). Thus, this mechanism might work for extremely long-lived species, but cannot be a general explanation for the accumulation of deletion mutants in species with a shorter lifespan.
The intriguing challenge is therefore that no current hypothesis satisfactorily explains the clearly demonstrable clonal expansion of mitochondrial deletion mutants in both long- and short-lived species. We here present evidence to resolve this enigma.
Results
Transcription Primes Replication.
Metazoan mitochondrial DNA replicates via the strand-displacement model with two unidirectional origins of replication, OH for the heavy strand and OL for the replication of the light strand (31, 37) [although evidence also exists hinting at a bidirectional mechanism (38, 39)]. All known DNA polymerases require an RNA primer with a free 3′-OH group to begin DNA synthesis. This is also true for mitochondrial DNA polymerase-γ. The primer for the heavy strand is provided by processing mRNA transcripts of the L-strand promoter (37). This promoter normally produces a huge poly-cistronic mRNA, transcribing all of the light strand genes. Similarly, another large poly-cistronic mRNA is generated from the H-strand promoter, transcribing all heavy strand genes (40).
Based on this tight linkage of transcription and replication we assume, following Shadel and Clayton (37), that initiation of replication is positively correlated with the transcription rate. We further propose the existence of a product inhibition loop that down-regulates the transcription rate when sufficient proteins are available. Such a mechanism, however, presents a specific threat. If a deletion removes the genes coding for protein products involved in this feedback, transcription will fail to be down-regulated in these mutants and consequently such mutants will have a higher initiation rate of replication than wild-type mtDNA. We suggest that this is the basis for the selection advantage that drives the observed clonal expansion of deletion mutants. This requires that an individual mtDNA molecule should be sensitive to the amount of protein produced from this specific mtDNA. Such a feat might seem implausible if mtDNA and proteins mix freely during the mitochondrial life cycle, which includes frequent fission and fusion. However, it is precisely because there must be some coupling between genotype and phenotype, to maintain mitochondrial integrity by selection, that we recently suggested how the link between mtDNA molecules and their gene products might be maintained during fusion and fission processes (20). The hypothesis proposes a physical connection between mitochondrial nucleoids and respiratory chain complexes, for which there exists considerable evidence. Furthermore, recent evidence suggests that most mammalian mitochondria contain only a single copy of mtDNA per nucleoid, which facilitates the preservation of a clear genotype–phenotype association (41).
Identification of Candidate Feedback Proteins.
If this proposed product-inhibition mechanism is correct, examination of published data on clonally amplified mitochondrial deletions causing a COX-deficient phenotype should be revealing. Suitable data sets are available for rats (7), rhesus monkeys (10), and humans (42, 43), where PCR has been used to amplify mtDNA deletions in single cells of skeletal muscle fibers and neurons.
Fig. 1 shows the positions of the deletions in relation to schematic maps of the mitochondrial genomes for the three species. These reveal that all (rat, human neurons) or almost all (rhesus, human muscle) deletions overlap the NADH dehydrogenase subunit (ND)4 gene. For all studies that looked at muscle fibers the deletions overlap the two genes ND4 and ND5, two subunits of complex I. In the case of human neurons the deletions overlap ND3 and ND4.
Fig. 1.

Locations of mitochondrial deletions found in rat (7) (Upper Left), rhesus monkey (10) (Upper Right), and humans (42, 43) (Lower). The highlighted area indicates a stretch of mtDNA that is overlapped by all 30 deletions in rat, 38 of 39 deletions in rhesus monkey, all 89 deletions in human neurons, and 46 of 48 deletions in human muscle. The mtDNA maps are based on GenBank entries X14848 (rat), AY612638 (rhesus monkey), and 251831106 (human).
It is tempting to speculate, therefore, that the gene ND4 (and possibly ND3/5) is involved in the feedback mechanism we propose. However, it has been suggested that mitochondrial deletions are a consequence of slippage replication (44) involving repeats or the hybridization of longer stretches of mtDNA (45–47). Thus, it could be that the overlapping deletion areas are simply a consequence of appropriately placed DNA sequences. To exclude this possibility we used the software package UNAFold 3.8 (48) to calculate the free energies ∆G of the mitochondrial deletions based on an assumed hybridization length of 100 bp (Materials and Methods as well as SI Text). Next we calculated the free energies of all possible 100-bp hybridizations that result by stepping through the large arc of the mtDNA [like Guo et al. (47)]. From the resulting 5,565 possible deletions in the Homo sapiens mtDNA, we omitted all those whose free energy was above the mean of the free energy calculated for the 48 deletions that were found in muscle fibers (−16.91 kcal/mol). From the remaining 1,070 deletions, 544 contained the shared deletion area, whereas 526 did not. After this correction for ∆G, we calculated the probability that the presence of the shared deletion area in 46 of the 48 observed deletions was caused by pure chance to be
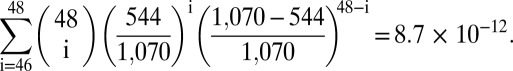 |
Equivalent calculations for the human data set based on neurons resulted in a probability of 2.1 × 10−38, the rhesus data gave 4.7 × 10−17, and the deletions in rats yielded a probability of 5 × 10−14. Thus, the existence of the shared deletion area in rat, rhesus monkey, and humans is statistically highly significant and not explicable by free-energy considerations. We therefore suggest that the subunit ND4 and possibly subunits ND3 and ND5 of complex I of the electron transport chain are involved in the proposed transcriptional feedback mechanism.
Kinetics of Clonal Expansion.
To examine whether the kinetics of the proposed clonal-expansion mechanism are compatible with observations, we ran computer simulations based on a system of ordinary differential equations (ODE) that describe the time course of wild-type and mutant mtDNA molecules as well as the amount of ATP. The first terms in Eqs. 1 and 2 describe the synthesis of mtDNAs, whereas the second terms describe their degradation. Synthesis is proportional to the synthesis parameters swt and smt, and negatively regulated by the ATP level. If the ATP is equal to the constant c, the synthesis rate drops to 50% of its maximal value. Degradation is controlled by the half-life parameter, halfL, and assumed to be identical for wild type and mutant.
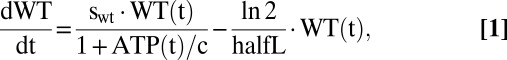 |
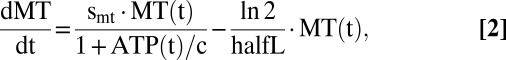 |
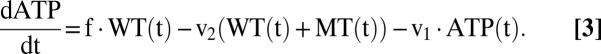 |
The equation for ATP comprises three terms, the first describing the production by wild-type mtDNA and the other two describing its consumption. The second term reflects our assumption that the mere existence of mtDNA molecules imposes some energetic costs: because mRNA and proteins are synthesized from it, new membrane has to be synthesized to accommodate the proteins, and all these components also have to be degraded again. ATP is also consumed by many other processes in the cell, which is represented as an aggregate by the last term. In total the model contains seven parameters that are summarized in Table 1 with their standard values.
Table 1.
Parameters and standard values used for the simulations
| Name | Value | Description |
| halfL | 240 h | Half-life of mtDNA in hours. Based on data by Gross et al. (36), Huemer et al. (34), and Korr et al. (35), we chose a half-life of 10 d as standard value. |
| swt | 1 h−1 | Parameter controlling the synthesis rate of wild-type mtDNA based on a replication time of 1 h (31, 32). |
| smt | 1.5 h−1 | Parameter controlling the synthesis rate of mutant mtDNA. |
| c | 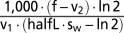 |
Carry capacity of ATP. Calculated such that without mutants the steady state of wild-type mtDNA is equal to 1,000. |
| f | 0.1 h−1 | Parameter describing how much ATP is produced by wild-type mtDNAs. Free parameter. |
| v1 | 0.2 h−1 | Parameter controlling how much ATP is consumed by cellular processes. Free parameter. |
| v2 | 0.01 h−1 | Parameter controlling how much ATP is consumed as maintenance cost per existing mtDNA. Free parameter. |
Fig. 2 shows the time course of the system using standard parameters, which means a half-life of 10 d and a 50% increased replication rate for mutant mtDNAs that have lost the transcriptional feedback. Furthermore, for all our simulations we adjusted the parameter “c” such that in the absence of any mutants the steady-state level of wild-type mtDNAs would be 1,000 (this is possible because the equation system can be solved analytically for steady-state conditions). The simulation was initiated with 1,000 wild-type and a single molecule of mutant mtDNA.
Fig. 2.

Typical time course of a single-cell simulation with the standard parameters given in Table 1. The simulation was started with 1,000 wild-type mtDNAs and 1 mutant mtDNA. The mutant accumulates exponentially until the system collapses after approximately 250 d.
Because of the constant turnover, and driven by its selection advantage, the amount of mutant increases exponentially with time. This imposes increasing energetic costs and, although the system tries to compensate this by an increase of the total number of mtDNA molecules, it leads to the gradual decline of the ATP steady-state level. Eventually, the unlimited accumulation of mutant mtDNA causes the system to collapse through ATP exhaustion.
Using the standard set of parameters, the system breaks down after approximately 250 d, with the amount of wild-type mtDNA changing only slowly until the very end of the simulation. At the moment of collapse the mutant is present in roughly ninefold excess over the wild type. This resembles very closely what has been observed experimentally by Herbst et al. (11).
Two parameters influence the timespan until collapse: the half-life of mtDNA, and the ratio of replication rates, sm/sw. A long half-life prolongs the time, because fewer mtDNAs are degraded and, other things being equal, the total synthesis rate of new mtDNAs will be diminished, which slows down the accumulation rate of the mutant.
The simulations show that (i) the proposed mechanism can explain the accumulation of mutant mtDNA within a time frame that is compatible with the lifespan of short-lived mammals and (ii) the levels of mutant and wild-type mtDNA after breakdown are compatible with experimental observations.
Predictions for Aged Tissue.
Although a system of ODEs can give a deterministic description of the competition between the wild type and a mutant within a single cell, it is not sufficient to understand what consequences the hypothesis will have at the tissue level for a given mutation rate. In muscle tissue of old organisms, single cells can be found that display a COX-negative phenotype, a commonly used marker for abnormalities of the electron transport chain (7). As in earlier studies (16, 18), we assumed for our simulations that COX-negative cells contain at least 60% mutant mtDNA, and we developed a Java program that performs stochastic simulations based on the reactions described by the ODEs. In contrast with the single-cell simulations, the system was initiated containing exclusively wild-type molecules and allowed mutations to arise by chance. We assumed that mitochondrial deletions appear with a probability Pmut during replication, caused by slippage replication at perfect and imperfect direct repeats (44, 47). The simulation was performed for a certain number of virtual years during which degradation and replication were calculated on an hourly basis. To get statistical insight into the results we performed 1,000 repetitions of each single simulation and calculated average values.
Clonally expanded deletions have been observed in species with a 30-fold difference in life expectancy (rodents vs. humans) and a critical test for each hypothesis is hence to demonstrate its applicability across such a range of time scales. We therefore performed simulations for 3, 10, 40, and 80 y and adjusted the mutation rate such that at the end of the simulation, 10 ± 0.5% of the cells belonged to the COX-negative phenotype. The results are shown in Fig. 3A, together with corresponding simulations from earlier studies modeling random drift (18) and the effects of a smaller genome size (36). In principle, all ideas can generate the required level of COX-negative cells after the various simulation times if the mutation rate is adjusted accordingly.
Fig. 3.

Comparison of different mechanisms that might explain the accumulation of mitochondrial mutant mtDNAs. “Random drift” indicates data taken from Kowald and Kirkwood (18), “reduced genome size” specifies simulation results from Kowald et al. (36), and “replication via transcription” labels the simulation outcomes of the mechanism presented in the current work. (A) Mutation rates required to achieve 10% COX-negative cells at the end of the indicated lifespan and (B) degree of resulting heteroplasmy given by the number of different mtDNA mutants per COX negative.
Another critical test relates to the resulting degree of heteroplasmy. In all experimental systems, ranging from rat to man, it was almost always found that a single mutant accumulated in the affected cell, i.e., the observed degree of heteroplasmy among different mutant types is extremely low. Earlier studies have shown that this is the main problem of the random drift as well as the smaller genome hypothesis, which predict a high degree of heteroplasmy for short-lived species (18, 36). Fig. 3B shows that this is not the case for the transcription-based mechanism. The low mutation rate, together with the selection advantage, results in a very low level of heteroplasmy even for short-lived species. For rats our hypothesis results on average in only 1.26 different types of mtDNA mutants per COX-negative cell, whereas the alternative mechanisms generate around 30 different types.
Thus, the tissue-based simulations show that a transcription-based replication advantage of mitochondrial deletion mutants predicts, for a wide range of animal lifespans, a degree of heteroplasmy that is fully compatible with experimental findings.
Discussion
The accumulation of mitochondrial deletion mutants in single cells of aged organisms is a hallmark of aging that has been observed in several mammalian species. Although these species display a 30-fold difference in life expectancy, they exhibit similar proportions of COX-negative cells at the end of their respective lifespans and the population of mutant mtDNAs is in each case clonal, i.e., it shows a very low level of heterogeneity. Clearly, a better understanding of how this occurs will help to judge its relevance for the aging process and to design strategies for its prevention or reversal. However, the current hypotheses are incompatible with the data.
We propose here a new mechanism based on the intimate connection of transcription and replication found in mitochondria of metazoans. We suggest that the transcription rate is controlled by a negative feedback loop that decreases transcription if there is a surplus of product. We further suggest that a higher transcription rate leads to a higher initiation rate of replication, because the RNA primer necessary for replication is generated from messenger transcripts (37, 40). This creates the potential vulnerability that if deletion events remove the genes for the proteins that are involved in this product inhibition, then mtDNA molecules harboring such deletions will have a higher effective replication rate than wild-type mtDNAs.
We used single-cell simulations to show that a few months are sufficient for a single mutant to outgrow the wild-type population giving rise to a COX-negative phenotype (Fig. 2). The simulation results agree with findings of Herbst et al. (11), who showed that the phenotype is caused by a runaway accumulation of mutant mtDNAs and not by the disappearance of wild-type mtDNAs. Next we performed stochastic simulations of many cells to investigate the consequences of our proposal at the tissue level. In contrast with other ideas, such as random drift or a reduced genome size, the transcription-based mechanism leads to an extremely low level of heterogeneity, for lifespans ranging from 3 to 80 y (Fig. 3).
Analysis of single-cell data for rat, rhesus monkey, and human (7, 10, 42, 43) found, in all three species, that a shared area of mtDNA was overlapped by all or almost all deletions. In the three studies using muscle cells this area affects the genes ND4 and ND5, whereas in the work investigating substantia nigra neurons the genes ND3, ND4L, and ND4 were affected. The probability that the deletions overlap this area by coincidence is vanishingly small, even after correcting for possible effects of DNA hybridization energies. Furthermore, data from mice are also consistent with our idea. Although single-cell data based on COX-negative cells are clearly most suitable for our analysis, there exists a tissue-based study in mice which also shows that deletions overlapped the genes ND4 and ND5 (49) (see also SI Text). The most parsimonious explanation is, therefore, that the subunit ND4 of complex I of the respiratory chain is part of the proposed transcriptional feedback system. However, if the feedback is modified in a tissue-specific manner (muscle vs. neurons), numerous combinations are possible involving ND3, ND4, or ND5. The proposed mechanism would not work for tRNAs, because they can diffuse throughout the mitochondrial matrix and therefore do not provide a feedback of the local protein requirements.
The RNA primer for the heavy strand origin of replication OH is created from the transcript of the L promoter (40). However, because ND6 is the only protein encoded on the light strand, one might argue that only ND6 has the potential to act as feedback protein. However, the subunits of the respiratory chain complexes are needed in stoichiometric ratios and thus it is likely that the transcription rates of the L and H2 promoters are regulated in a coordinated fashion. In birds and amphibians, for instance, this is automatically assured through the existence of a bidirectional promoter (50, 51). We therefore propose that the feedback protein(s) influence the regulation of both mitochondrial promoters. This idea also requires that mtDNA molecules can sense the amount of protein that has been synthesized from individual molecules, a capacity that is supported by strong arguments in favor of a physical link between mtDNAs and the synthesized proteins (20).
The proposed selection mechanism for mitochondrial mutants immediately leads to several interesting predictions. All deletions that damage the genes for the feedback proteins destroy transcriptional control and thus gain a selection advantage. However, the size of the deletion is not likely to matter. Thus, we would predict that there is no correlation between the deletion size and the resulting selection advantage. Fukui and Moraes (52) generated transgenic mice with a large spectrum of deletion mutants using an inducible restriction enzyme targeted to mitochondria. They used PCR to quantify the ratio of short (3.8 kb) to long (10 kb) deletions and found a preference of long deletions. However, because it is unknown if all sizes are generated in equal quantities, these results need verification. Investigating the correlation between deletion size and the length of the COX-negative segment of skeletal muscle fibers might be a better approach that does not require genetic manipulations. In rare cases where the deletion removes only a part of the feedback protein it might be that the feedback is not completely destroyed, but in general we would not expect to see a correlation between deletion size and the length of the COX-negative area.
A further prediction of the proposed mechanism is that not only deletions but also certain point mutations, if they occur in the feedback proteins, could affect transcriptional control and cause amplification. Payne et al. (53) studied mtDNA mutations in COX-negative muscle fibers of patients treated with antiretroviral drugs, which are known to inhibit polymerase-γ. Most of the fibers contained large-scale mtDNA deletions which spanned ND4, but several instead harbored point mutations, which were located all over the mitochondrial genome. Whereas such dispersed point mutations are not predicted by the specific mechanism we propose, our mechanism does not preclude that there may also, in long-lived humans, be scope for clonal expansion by random drift processes, especially if these are exacerbated by the action of the antiviral drugs. The authors themselves suggest that the observed increased frequency of COX-deficient fibers is caused via an accelerated mtDNA turnover. A reduced mtDNA half-life would indeed speed up the accumulation of mtDNA deletions via transcriptional priming, but also accumulation of point mutations via random drift.
Our proposed mechanism depends on the tight connection between transcription and priming of replication seen in metazoan mitochondria. Plant mtDNA, however, is much larger, reaching more than 11 mega base pairs in Silene conica (54), and replication is primed by a specialized primase (55). Because this decouples the connection between transcription and replication, deletions can no longer influence the initiation of replication and hence there should be no accumulation of deletion mutants in plants.
In summary, we present here a specific mechanism that can provide the biochemical basis for the observed clonal amplification of mitochondrial deletion mutants in animals with a wide range of lifespans and that also explains the existence of the overlapping deletion area found in the data sets of rats, rhesus monkeys, and humans. Hopefully this new idea will motivate further experiments and lead to better understanding of the role of mitochondrial mutations in aging and disease.
Materials and Methods
Calculating Hybridization Energies.
To calculate the free energy ∆G for various DNA hybridizations we used the hybrid-min program of the software package UNAFold 3.8 (48) under default parameters regarding temperature and salt concentrations. We performed several tests to access the influence of different conditions and found that whereas the absolute values of free energy change considerably, the relative ranking (which is important for our conclusions) is hardly affected at all. Further details are provided in SI Text.
Computer Simulations of mtDNA Competition.
Deterministic simulations of the competition between wild-type and mutant mtDNA were performed by solving the system of differential equations represented by Eqs. 1–3. For this purpose we used the software tool Mathematica (56). To compute the effects of the competition between wild-type and mutant mtDNA at the tissue level, we developed a Java program that iterates through the mitochondrial lifecycle in steps of 1 h, stochastically performing degradation, replication, mutation, and ATP production according to the rules laid out by the system of ODEs. The program can be obtained from the authors.
Supplementary Material
Acknowledgments
We thank D.M. Turnbull for constructive comments and helpful discussion. This work was funded by a Glenn Foundation award to T.B.L.K. The research was also supported by the National Institute for Health Research Newcastle Biomedical Research Centre based at Newcastle upon Tyne Hospitals National Health Service Foundation Trust and Newcastle University.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.W.G. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314970111/-/DCSupplemental.
References
- 1.Harman D. The biologic clock: The mitochondria? J Am Geriatr Soc. 1972;20(4):145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 2.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1(8639):642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 3.Miquel J, Economos AC, Fleming J, Johnson JE., Jr Mitochondrial role in cell aging. Exp Gerontol. 1980;15(6):575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 4.Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagouge M, Larsson NG. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med. 2013;273(6):529–543. doi: 10.1111/joim.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinnery PF, Samuels DC, Elson J, Turnbull DM. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: Is there a common mechanism? Lancet. 2002;360(9342):1323–1325. doi: 10.1016/S0140-6736(02)11310-9. [DOI] [PubMed] [Google Scholar]
- 7.Cao Z, Wanagat J, McKiernan SH, Aiken JM. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: Analysis by laser-capture microdissection. Nucleic Acids Res. 2001;29(21):4502–4508. doi: 10.1093/nar/29.21.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: Implications for the central nervous system and muscle. Ann Neurol. 1998;43(2):217–223. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 9.Khrapko K, et al. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27(11):2434–2441. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gokey NG, et al. Molecular analyses of mtDNA deletion mutations in microdissected skeletal muscle fibers from aged rhesus monkeys. Aging Cell. 2004;3(5):319–326. doi: 10.1111/j.1474-9728.2004.00122.x. [DOI] [PubMed] [Google Scholar]
- 11.Herbst A, et al. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: Evidence for a causal role in muscle fiber loss. J Gerontol A Biol Sci Med Sci. 2007;62(3):235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor RW, et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 2003;112(9):1351–1360. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greaves LC, Barron MJ, Campbell-Shiel G, Kirkwood TB, Turnbull DM. Differences in the accumulation of mitochondrial defects with age in mice and humans. Mech Ageing Dev. 2011;132(11-12):588–591. doi: 10.1016/j.mad.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 14.Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: Implications for carcinogenesis and aging? Free Radic Biol Med. 1990;8(6):523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 15.Arnheim N, Cortopassi G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat Res. 1992;275(3-6):157–167. doi: 10.1016/0921-8734(92)90020-p. [DOI] [PubMed] [Google Scholar]
- 16.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68(3):802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chinnery PF, Samuels DC. Relaxed replication of mtDNA: A model with implications for the expression of disease. Am J Hum Genet. 1999;64(4):1158–1165. doi: 10.1086/302311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowald A, Kirkwood TB. Mitochondrial mutations and aging: Random drift is insufficient to explain the accumulation of mitochondrial deletion mutants in short-lived animals. Aging Cell. 2013;12(4):728–731. doi: 10.1111/acel.12098. [DOI] [PubMed] [Google Scholar]
- 19.de Grey ADNJ. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays. 1997;19(2):161–166. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- 20.Kowald A, Kirkwood TB. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc Natl Acad Sci USA. 2011;108(25):10237–10242. doi: 10.1073/pnas.1101604108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal. 2011;14(10):1919–1928. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA. 2010;107(26):11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lane N. Mitonuclear match: Optimizing fitness and fertility over generations drives ageing within generations. Bioessays. 2011;33(11):860–869. doi: 10.1002/bies.201100051. [DOI] [PubMed] [Google Scholar]
- 25.Pérez VI, et al. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790(10):1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapointe J, Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci. 2010;67(1):1–8. doi: 10.1007/s00018-009-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Speakman JR, Selman C. The free-radical damage theory: Accumulating evidence against a simple link of oxidative stress to ageing and lifespan. Bioessays. 2011;33(4):255–259. doi: 10.1002/bies.201000132. [DOI] [PubMed] [Google Scholar]
- 28.Wallace DC. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science. 1992;256(5057):628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 29.Lee CM, Lopez ME, Weindruch R, Aiken JM. Association of age-related mitochondrial abnormalities with skeletal muscle fiber atrophy. Free Radic Biol Med. 1998;25(8):964–972. doi: 10.1016/s0891-5849(98)00185-3. [DOI] [PubMed] [Google Scholar]
- 30.Kowald A, Jendrach M, Pohl S, Bereiter-Hahn J, Hammerstein P. On the relevance of mitochondrial fusions for the accumulation of mitochondrial deletion mutants: A modelling study. Aging Cell. 2005;4(5):273–283. doi: 10.1111/j.1474-9726.2005.00169.x. [DOI] [PubMed] [Google Scholar]
- 31.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28(4):693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 32.Berk AJ, Clayton DA. Mechanism of mitochondrial DNA replication in mouse L-cells: Asynchronous replication of strands, segregation of circular daughter molecules, aspects of topology and turnover of an initiation sequence. J Mol Biol. 1974;86(4):801–824. doi: 10.1016/0022-2836(74)90355-6. [DOI] [PubMed] [Google Scholar]
- 33.Huemer RP, Lee KD, Reeves AE, Bickert C. Mitochondrial studies in senescent mice. II. Specific activity, buoyant density, and turnover of mitochondrial DNA. Exp Gerontol. 1971;6(5):327–334. doi: 10.1016/0531-5565(71)90001-5. [DOI] [PubMed] [Google Scholar]
- 34.Korr H, Kurz C, Seidler TO, Sommer D, Schmitz C. Mitochondrial DNA synthesis studied autoradiographically in various cell types in vivo. Braz J Med Biol Res. 1998;31(2):289–298. doi: 10.1590/s0100-879x1998000200012. [DOI] [PubMed] [Google Scholar]
- 35.Gross NJ, Getz GS, Rabinowitz M. Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J Biol Chem. 1969;244(6):1552–1562. [PubMed] [Google Scholar]
- 36.Kowald A, Dawson M, Kirkwood TB. Mitochondrial mutations and ageing: Can mitochondrial deletion mutants accumulate via a size based replication advantage? J Theor Biol. 2014;340:111–118. doi: 10.1016/j.jtbi.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 38.Holt IJ, Lorimer HE, Jacobs HT. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100(5):515–524. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- 39.Yang MY, et al. Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell. 2002;111(4):495–505. doi: 10.1016/s0092-8674(02)01075-9. [DOI] [PubMed] [Google Scholar]
- 40.Fernández-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2003;88(1):41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- 41.Kukat C, et al. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA. 2011;108(33):13534–13539. doi: 10.1073/pnas.1109263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bua E, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79(3):469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reeve AK, et al. Nature of mitochondrial DNA deletions in substantia nigra neurons. Am J Hum Genet. 2008;82(1):228–235. doi: 10.1016/j.ajhg.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shoffner JM, et al. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc Natl Acad Sci USA. 1989;86(20):7952–7956. doi: 10.1073/pnas.86.20.7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khaidakov M, Siegel ER, Shmookler Reis RJ. Direct repeats in mitochondrial DNA and mammalian lifespan. Mech Ageing Dev. 2006;127(10):808–812. doi: 10.1016/j.mad.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 46.Lakshmanan LN, Gruber J, Halliwell B, Gunawan R. Role of direct repeat and stem-loop motifs in mtDNA deletions: Cause or coincidence? PLoS ONE. 2012;7(4):e35271. doi: 10.1371/journal.pone.0035271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo X, et al. Repeats, longevity and the sources of mtDNA deletions: Evidence from ‘deletional spectra’. Trends Genet. 2010;26(8):340–343. doi: 10.1016/j.tig.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Markham NR, Zuker M. UNAFold: Software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 49.Chen H, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.L’Abbé D, Duhaime JF, Lang BF, Morais R. The transcription of DNA in chicken mitochondria initiates from one major bidirectional promoter. J Biol Chem. 1991;266(17):10844–10850. [PubMed] [Google Scholar]
- 51.Bogenhagen DF, Yoza BK, Cairns SS. Identification of initiation sites for transcription of Xenopus laevis mitochondrial DNA. J Biol Chem. 1986;261(18):8488–8494. [PubMed] [Google Scholar]
- 52.Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2009;18(6):1028–1036. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Payne BA, et al. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat Genet. 2011;43(8):806–810. doi: 10.1038/ng.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sloan DB, et al. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012;10(1):e1001241. doi: 10.1371/journal.pbio.1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shutt TE, Gray MW. Twinkle, the mitochondrial replicative DNA helicase, is widespread in the eukaryotic radiation and may also be the mitochondrial DNA primase in most eukaryotes. J Mol Evol. 2006;62(5):588–599. doi: 10.1007/s00239-005-0162-8. [DOI] [PubMed] [Google Scholar]
- 56.Wolfram Research . Mathematica. Champaign, IL: Wolfram Research; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.