

Significance
Oxidative stress is an important contributor to aging-associated diseases including cancer and neurodegeneration, and antioxidant stress responses are critical to limit manifestations of these diseases. We report that the tumor suppressor Homologous to the E6-AP Carboxyl Terminus domain and Ankyrin repeat containing E3 ubiquitin–protein ligase 1 (HACE1) promotes activity of the transcription factor, nuclear factor erythroid 2-related factor 2 (NRF2), a master regulator of the antioxidative stress response. In Huntington disease patients, HACE1 is lost in the brain region most affected by the disease, namely the striatum, and restoring HACE1 functions in striatal cells expressing mutant Huntingtin protein provides protection against oxidative stress. Therefore, the tumor suppressor HACE1 is a new regulator of NRF2 and an emerging player in neurodegeneration.
Keywords: ROS, glutathione, aging, transcription factor
Abstract
Oxidative stress plays a key role in late onset diseases including cancer and neurodegenerative diseases such as Huntington disease. Therefore, uncovering regulators of the antioxidant stress responses is important for understanding the course of these diseases. Indeed, the nuclear factor erythroid 2-related factor 2 (NRF2), a master regulator of the cellular antioxidative stress response, is deregulated in both cancer and neurodegeneration. Similar to NRF2, the tumor suppressor Homologous to the E6-AP Carboxyl Terminus (HECT) domain and Ankyrin repeat containing E3 ubiquitin–protein ligase 1 (HACE1) plays a protective role against stress-induced tumorigenesis in mice, but its roles in the antioxidative stress response or its involvement in neurodegeneration have not been investigated. To this end we examined Hace1 WT and KO mice and found that Hace1 KO animals exhibited increased oxidative stress in brain and that the antioxidative stress response was impaired. Moreover, HACE1 was found to be essential for optimal NRF2 activation in cells challenged with oxidative stress, as HACE1 depletion resulted in reduced NRF2 activity, stability, and protein synthesis, leading to lower tolerance against oxidative stress triggers. Strikingly, we found a reduction of HACE1 levels in the striatum of Huntington disease patients, implicating HACE1 in the pathology of Huntington disease. Moreover, ectopic expression of HACE1 in striatal neuronal progenitor cells provided protection against mutant Huntingtin-induced redox imbalance and hypersensitivity to oxidative stress, by augmenting NRF2 functions. These findings reveal that the tumor suppressor HACE1 plays a role in the NRF2 antioxidative stress response pathway and in neurodegeneration.
Oxidative stress contributes to the development of numerous late onset human diseases such as neurodegeneration and cancer (1, 2). Molecular mechanisms that sense and respond to increased reactive oxygen species (ROS) levels have evolved and are highly conserved. One such master regulator of the antioxidative stress response is the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) (3). In response to increased ROS, NRF2 induces expression of antioxidant proteins such as Heme Oxygenase (HO1) and NAD(P)H dehydrogenase (quinine; NQO1) and key enzymes in the glutathione (GSH) biosynthesis pathway such as GSH synthetase (GSS) (4), collectively known as phase II detoxifying enzymes (5). NRF2 activity is tightly regulated by cellular redox balance. Under normal conditions, NRF2 is bound to the oxidative stress sensor KEAP1 to promotes NRF2 ubiquitylation and proteosomal degradation (6), resulting in low basal NRF2 activity. Under oxidative stress, KEAP1 becomes oxidized, disrupting its interaction with NRF2 (7), thus leading to stabilization and nuclear translocation of NRF2, where it transcribes its specific target genes.
NRF2 is critical for maintaining redox homeostasis, as Nrf2 knockout (KO) mice are prone to urethane-induced tumors (8) and NRF2 hyperactivity or deficiency is observed in cancer and neurodegeneration, respectively (9). Although KEAP1 oxidation is the most well-established mechanism of NRF2 regulation, other factors also promote NRF2 accumulation. These include p21 and p62 that interfere with KEAP1 binding to NRF2 (10, 11), protein kinase C that phosphorylates NRF2 to promote its accumulation (12), Ras pathway activation that enhances NRF2 mRNA expression (13), and H2O2 that increases NRF2 mRNA translation (14).
Homologous to the E6-AP Carboxyl Terminus (HECT) domain and Ankyrin repeat containing E3 ubiquitin–protein ligase 1 (HACE1) is a tumor suppressor that is inactivated in human Wilms’ tumor and other cancers (15, 16). HACE1 is a HECT family E3 ligase (15–17) that binds to and ubiquitylates the GTP-bound active form of Rac1, a Rho family GTPase (17, 18). HACE1 is ubiquitously expressed with relatively higher expression in heart, placenta, kidney, and brain (15). We recently found that HACE1 specifically targets activated Rac1 when the latter is bound to Rac1-dependent NADPH oxidase complexes, and therefore reduces ROS generation by these complexes (19).
Of note, there are several reported similarities between Hace1 KO and Nrf2 KO mice. As in Nrf2 null mice (8), tumor incidence is dramatically enhanced in Hace1 KO mice when they age or are subjected to urethane treatment and ionizing radiation (16). Because these stresses are linked to enhanced oxidative stress, we hypothesized that HACE1 may play a role in the oxidative stress response. Here we report that HACE1 is an important component of the cellular ROS detoxification and antioxidative stress responses by facilitating optimal activation of NRF2. This has potential consequences for neurodegenerative diseases such as Huntington disease (HD), as HACE1 protects neuronal progenitor cells from mutant Huntingtin (mHTT) toxicity by augmenting the NRF2 response. Moreover, its expression is markedly reduced in HD striatum, implicating HACE1 inactivation in the pathogenesis of HD.
Results
HACE1 Mediates Resistance to Oxidative Stress.
To determine if HACE1 is involved in cellular detoxification of oxidant load, we challenged Hace1 WT and KO mouse embryonic fibroblasts (MEFs) with H2O2 or arsenite (AR) and measured their ability to survive acute oxidative stress. Hace1 KO MEFs were significantly more sensitive to both forms of oxidative stress compared with WT MEFs (Fig. 1A). Moreover, HACE1 knockdown (kd) using specific siRNAs in HEK293T cells markedly increases cell death following AR treatment (Fig. S1 A and B). Under homeostatic conditions, HACE1 deficiency in MEFs and HEK293T cells results in redox imbalance reflected by increased ROS (Fig. 1B and Fig. S1 C and D) and decreased GSH levels (Fig. 1C and Fig. S1E). Supplementing Hace1 WT and KO MEFs with GSH eliminated the ROS difference between these cells (Fig. 1B) and reduced sensitivity of Hace1 KO MEFs to H2O2 (Fig. S1F). Together these results suggest that HACE1 mitigates oxidative stress, at least in part by maintaining oxidant detoxification capacity.
Fig. 1.

HACE1 loss reduces antioxidative stress responses. (A) Hace1 WT or KO MEFs were treated with H2O2 (400 μM) or AR (10 μM) for 8 h, and cell death was determined by annexin V and propidium iodide (PI) staining. (Left) Typical FACS plots. (Right) Cell death relative to control. n = 3; *P < 0.05. (B) ROS levels in Hace1 WT and KO MEFs treated with GSH (1 μM; 16 h) or untreated controls were determined using 2′,7′–dichlorofluorescein diacetate (DCFDA) and FACS. (Left) Typical histograms. (Right) Relative fluorescence levels. n = 3; *P < 0.05; NS, nonsignificant. (C) GSH levels were measured in lysates obtained from Hace1 WT or KO MEFs. Values are relative to Hace1 WT. n = 3; *P < 0.05.
HACE1 Is Essential for Optimal NRF2 Activation.
To better characterize how HACE1 impacts cellular antioxidant levels, we investigated whether HACE1 might regulate NRF2 directly. We first measured NRF2 transcriptional activity using a vector encoding luciferase driven by the NQO1 promoter (20). NRF2 activity was significantly reduced in Hace1 KO compared with WT MEFs under resting and oxidative stress conditions (Fig. 2A). In addition, expression levels of the NRF2 target genes Hmox1 and Nqo1 were significantly reduced in Hace1 KO MEFs compared with WT MEFs under oxidative stress (Fig. 2B), suggesting that HACE1 is required for optimal NRF2 activation in response to oxidative stress.
Fig. 2.

HACE1 promotes NRF2 activity, stability, and synthesis. (A) Hace1 WT or KO MEFs cotransfected with antioxidative stress response element-luciferase (ARE-LUC) and pRenilla vectors were treated with H2O2 (200 μM; 6 h), as indicated. NRF2 activity was determined by measuring luciferase activity. n = 3; *P < 0.01; **P < 0.005. (B) Hace1 WT or KO MEFs were treated with H2O2 (200 μM) for the indicated times. Hmox1 and Nqo1 mRNA levels were determined using qRT-PCR. n = 3; *P < 0.01. (C) Hace1 WT or KO MEFs were treated with H2O2 (200 μM) for the indicated times. NRF2 protein levels were determined using Western blot. NRF2 levels were normalized to HSC70. n = 3; *P < 0.05. (D) Hace1 WT or KO MEFs were treated with H2O2 (200 μM) for the indicated time points. NRF2 localization was determined using immunofluorescence. Typical images are shown on the left. Cells exhibiting nuclear or cytosolic localization were scored. n = 200 from three independent experiments; *P < 0.01. (E) Hace1 WT or KO MEFs transfected with NRF2-expressing or control vector were treated with H2O2 (600 μM) or AR (10 μM) for 8 h. Cell death was measured by annexin V and PI staining and presented relative to WT cells. n = 3; *P < 0.01. (F) HEK293T cells were transfected as in E. Cells were pulsed with AHA (250 μM; 1 h) and MG132 to block protein degradation with or without AR (1 μM) as indicated. HACE1 kd in total cell lysates was confirmed using Western blot. Cell lysates were subjected to a Click-it reaction with biotin alkyne followed by streptavidin pulldown, and newly synthetized proteins were detected by Western blot.
To determine how HACE1 affects NRF2 activity, we first assessed NRF2 protein accumulation in Hace1 WT and KO MEFs treated with H2O2, as NRF2 protein stability is a major homeostatic mechanism (21). Although NRF2 levels were induced by H2O2 in both cell lines at the early time point (0.5 h), there was an additional increase in NRF2 protein levels at the latter time point (3 h) in WT cells that was attenuated in Hace1 KO MEFs (Fig. 2C). This was confirmed using subcellular fractionation, whereby NRF2 levels increased in nuclear fractions of H2O2 treated Hace1 WT and KO MEFs at 0.5 h, but were further elevated at 3 h only in WT MEFs (Fig. S2A). Immunofluorescence showed enhanced NRF2 nuclear localization in both WT and Hace1 KO cells at 0.5 h after H2O2 treatment (Fig. 2D). Nuclear localization was retained in Hace1 WT MEFs at the 3 h time point, whereas, in contrast, it was markedly reduced in Hace1 KO MEFs at this time point (Fig. 2D). To confirm this in a second cell line, we performed HACE1 kd in HEK293T cells, which led to reduced total NRF2 levels under AR treatment (Fig. S2 B and C). Moreover, HACE1 kd reduced nuclear NRF2 accumulation in HEK293T cells upon AR treatment (Fig. S2D). These data suggest that HACE1 is involved in optimal expression and nuclear localization and/or retention of NRF2 under oxidative stress, and that reduced NRF2 activity in HACE1-deficient cells may be attributed to defective NRF2 protein accumulation and nuclear localization. We then asked whether NRF2 perturbation is responsible for the hypersensitivity of Hace1 KO cells to oxidative stress. In keeping with this, ectopic expression of NRF2 in Hace1 KO MEFs significantly reduced their sensitivity to oxidative stress (Fig. 2E and Fig. S2E), suggesting that HACE1 protects against oxidative stress by promoting NRF2.
HACE1 Promotes NRF2 Protein Stabilization and Synthesis.
We next asked how HACE1 promotes NRF2 accumulation. We first assessed NRF2 degradation rates under oxidative stress in control siRNA versus HACE1 kd HEK293T cells subjected to cycloheximide challenge to block translation. We found that the degradation rate of NRF2, but not of c-Jun and c-Myc, which also have short half-lives, was markedly increased in HACE1 kd compared with control cells (Fig. S2 F and G). Proteasome inhibition with MG132 only partially rescued NRF2 accumulation in HACE1 kd cells under AR treatment (Fig. S2H). This suggests that HACE1 promotes NRF2 induction through NRF2 stabilization and a degradation-independent mechanism. Nrf2 mRNA levels were not reduced in Hace1 KO compared with WT MEFs (Fig. S2I), excluding a transcriptional mechanism for the latter. We therefore tested whether HACE1 promotes NRF2 protein synthesis, as NRF2 nuclear localization under oxidative stress is dependent on de novo NRF2 synthesis (22). To compare NRF2 synthesis rates between HACE1 kd and control cells, we used the methionine analog azidohomoalanine (AHA) to monitor newly synthesized proteins as described (23). Nontreated and AR-treated cells were pulsed with AHA for 1 h, and then lysates were isolated and assessed for levels of AHA-labeled NRF2 and the control proteins, eukaryotic Elongation Factor 2 (eEF2) and GAPDH (Fig. 2F). The protein synthesis rate of NRF2 but not of eEF2 or GAPDH was greatly reduced in HACE1 kd cells compared with control cells, under both resting and oxidative stress conditions, providing compelling evidence that HACE1 also supports NRF2 accumulation by promoting NRF2 protein synthesis.
To demonstrate which domains of HACE1 influence NRF2 accumulation, we overexpressed HACE1 or various HACE1 mutants in HEK293 cells treated with the NRF2 inducer tert-butylhydroquinone (tBHQ) (22) and monitored NRF2 levels by Western blotting (Fig. S2J). HACE1 promoted NRF2 accumulation under both resting and tBHQ conditions in an E3 ligase-independent manner, as the E3 ligase dead mutant, HACE1–C876S (CS) (15), equivalently induced NRF2 levels (Fig. S2J). However, this was not observed when either the HACE1 N-terminal ankyrin repeats or the C-terminal HECT domain were deleted (Fig. S2J). These data indicate that HACE1 induces NRF2 accumulation independently of its E3 ligase activity, and that the ankyrin repeats and HECT domain are necessary for its NRF2 promoting activity.
HACE1 Is Involved in Regulation of Redox Balance in Brain Tissues.
To determine the physiological relevance of these findings, we asked if HACE1 plays a role in regulating redox and the antioxidative stress response in vivo. Given the known role of oxidative stress in neurodegeneration and expression of HACE1 in brain (15), we measured ROS, antioxidant levels, and oxidative stress markers in brains of Hace1 KO mice. In line with our findings in other tissues (19), Hace1 KO brain tissue exhibited increased ROS levels (Fig. 3A). We also observed reduced GSH levels in Hace1 KO compared with WT control brains (Fig. 3B). By immunohistochemistry (Fig. 3A), there was a significant reduction in levels of NQO1 in Hace1 KO brain tissue, which was accompanied by decreased Nqo1 mRNA levels (Fig. 3C). NQO1 is a well-established marker of the antioxidative stress response and NRF2 activity (5, 13), supporting a role for HACE1 in this pathway in vivo. Moreover, there was increased oxidative damage in Hace1 KO mouse brains as they exhibit higher levels of protein carbonylation (Fig. 3D) and DNA oxidation (Fig. 3E) compared with Hace1 WT brains. Together, these data provide strong evidence for increased oxidative stress accompanied by reduced antioxidant GSH and NQO1 levels in brain tissues of Hace1 KO mice, further supporting a role for HACE1 in promoting ROS detoxification in vivo.
Fig. 3.

HACE1 loss is associated with reduced antioxidants and increased oxidative stress in brain. (A) The striatal regions of Hace1 WT and KO brains were stained using dihydroethidium (DHE), anti-NQO1 antibodies, or H&E as indicated. Sections were imaged using a fluorescent (DHE) or light microscope (NQO1 and H&E). Staining was quantified using ImageJ. n = 3; *P < 0.05. (B) GSH levels were measured in brain lysates from Hace1 WT and KO mice. Values were normalized to WT. *P < 0.05. (C) Nqo1 mRNA levels in mouse brains determined by qRT-PCR. *P < 0.01. (D) Protein carbonylation was measured in Hace1 WT and KO mouse brains. *P < 0.05. (E) Hace1 WT and KO mouse brains were subjected to anti–8-oxo-dGuo immunohistochemistry. Staining in the cortex was quantified using ImageJ. *P < 0.001.
HACE1 Is an Oxidative Stress Response Gene.
Next we asked whether the HACE1 gene itself responds to oxidative stress, as predicted if HACE1 is linked to antioxidative stress responses. Because HACE1 expression is high in both neural tissue and kidney, we used SH-SY5Y neuroblastoma and HEK293T cells, respectively, and challenged them with H2O2 or AR as exogenous sources of oxidative stress. Transcripts of HACE1 and NQO1 (as an oxidative stress marker) were both induced by H2O2 and AR in SH-SY5Y cells (Fig. S3A), and by AR but not by H2O2 in HEK293T cells (Fig. S3B). In agreement, both HACE1 promoter activity (Fig. S3C) and protein expression (Fig. S3D) were induced by AR in HEK293T cells. Together, these results indicate that HACE1 is an oxidative stress response gene, providing further support to the premise that it has evolved a functional role in redox control.
HACE1 Protects Against mHTT Toxicity by Promoting NRF2.
Oxidative stress is a common feature in neurodegenerative diseases and is implicated in the pathogenesis of HD (2). We therefore hypothesized that in addition to its tumor suppressor function, HACE1 may also play a protective role in neurodegeneration. We chose HD as a model disease to test this, and first characterized Hace1 mRNA levels in immortalized neuronal progenitor cells obtained from the striatum of previously established HD knock-in mice (24). These cells express either full-length WT (Q7) or mHTT (Q111) driven by the endogenous HTT promoter, designated STHdhQ7 (WT) and STHdhQ111 (mHTT) cell lines, respectively. Using a publically available dataset and quantitative (q) RT-PCR, we found that Hace1 mRNA expression is reduced in STHdhQ111 compared with STHdhQ7 cells (Fig. S4 A and B). In addition, HACE1 protein expression is lower in STHdhQ111 versus STHdhQ7 cells under both control and serum starvation (SS) conditions (Fig. S4C). The latter is known to trigger mHTT toxicity in vitro (25), which we confirmed experimentally (Fig. S4D). Accordingly, STHdhQ111 cells exhibited higher basal and SS-induced ROS levels compared with STHdhQ7 cells (Fig. S4E), pointing to the possibility that ROS deregulation is involved in mHTT toxicity in this system. Indeed, mHTT-expressing STHdhQ111 cells exhibited reduced GSH levels (Fig. S4F) and enhanced sensitivity to H2O2 challenge compared with STHdhQ7 cells (Fig. S4G). GSH supplementation rescued the STHdhQ111 cells from enhanced sensitivity to H2O2 (Fig. S4H), indicating that GSH deficiency contributes to their hypersensitivity. STHdhQ111 cells thus mimic the phenotypes observed in HACE1-deficient MEFs, suggesting that reduced Hace1 levels may be involved in hypersensitivity of STHdhQ111 cells to oxidative stress. To directly assess this, we examined the impact of HACE1 levels on mHTT-associated oxidative stress and toxicity. We generated stable STHdhQ111 cell lines ectopically expressing vector control (murine stem cell virus, MSCV) or HA–HACE1 (Fig. S5A) and found that HACE1 expression increased GSH levels and decreased ROS under both resting and SS conditions (Fig. 4 A and B). Moreover, stable expression of HACE1 decreased mHTT-induced cellular sensitivity to SS and H2O2 challenge (Fig. 4 C and D). This was confirmed by transient expression of GFP–HACE1 in STHdhQ111 cells, which led to increased protection against H2O2 challenge (Fig. S5B). Furthermore, stable expression of the E3 ligase dead HACE1–C876S mutant in STHdhQ111 cells (Fig. S5A) led to enhanced resistance to SS and H2O2 challenge compared with control MSCV cells (Fig. 4 C and D) as well, further supporting the notion that the protective effects of HACE1 are independent of HACE1 E3 ligase activity. Taken together, these results suggest that reduced HACE1 levels contribute to the increased sensitivity toward ROS challenge observed in mHTT-expressing cells, and that HACE1 re-expression can protect cells against mHTT-induced toxicity by increasing their cellular antioxidant capacity in an E3 ligase-independent manner.
Fig. 4.

HACE1 mitigates mHTT-associated oxidative stress and toxicity. (A) GSH levels in the indicated STHdh cells lysates were measured and presented relative to MSCV. n = 3; *P < 0.05. (B) STHdh cells were grown in complete media or serum starved for 6 h. ROS were measured using DCFDA. (Left) Average geometric means relative to MSCV. n = 3; *P < 0.05. (Right) Typical FACS histograms. (C) The indicated STHdh cells were serum starved for 24 h. Cell death was measured using PI staining. n = 3; *P < 0.05. (D) The indicated STHdh cells were treated with H2O2 (400 μM; 24 h) and cell death was measured using PI staining. n = 3; *P < 0.05. (E) NRF2 activity in STHdh cells transfected with the indicated constructs and treated with H2O2 (200 μM) for 8 h was measured using ARE–LUC assay. n = 3; *P < 0.05.
We next asked whether NRF2 is involved in the ability of HACE1 to protect against mHTT toxicity. In agreement with previous reports (26), we found that NRF2 activity was substantially reduced in STHdhQ111 cells under basal conditions and following H2O2 treatment compared with STHdhQ7 cells (Fig. S5C). This was not rescued by Apocynin or ML171, inhibitors of NADPH oxidase activity (Fig. S5D), suggesting that the reduced NRF2 activity occurs independently of NADPH oxidase activity (19). Similar to Hace1 KO MEFs, NRF2 accumulation was reduced under oxidative stress in mHTT-expressing cells (Fig. S5E). Ectopic HACE1 expression was able to increase NRF2 activity in STHdhQ111 cells under both basal and H2O2-treated conditions (Fig. 4E). Moreover, NRF2 kd using two independent siRNAs (Fig. S5F) significantly increased sensitivity of STHdhQ111–HACE1 cells to H2O2 treatment. These results provide compelling evidence that HACE1 protects neuronal progenitor cells from mHTT-induced oxidative stress by promoting NRF2 activity and the antioxidant response.
HACE1 Is Reduced in HD Striatum.
We next asked if HACE1 expression is altered in the brain of the HD YAC128 mouse model. We found that HACE1 levels are not altered in this HD mouse model (Fig. S6 A–C). Although the YAC128 mouse is one of the most comprehensive and well-established models of HD, no mouse model recapitulates all molecular manifestations of human HD. In fact, it is reported that YAC128 mice fail to show concordant gene expression changes with human HD until the mice are 24 mo of age (27), whereas our studies of altered HACE1 expression in YAC128 mice were restricted to mice 12 mo of age or younger. Additionally, in a recent study of 12 genes that were differentially expressed in the striatum of YAC128 versus control mouse striatum, only four of these 12 genes were concordantly changed when assessed in human HD versus control striatum (28). This highlights potential differences in transcriptional responses to mHTT expression in brains across different species. We therefore sought to directly evaluate HACE1 levels in human HD brain tissues.
Interrogating publicly available human gene expression data (29), we found that HACE1 mRNA is specifically reduced in the striatum, the most affected region of the brain in HD (30), but not in the cerebellum or cortex of HD patients (Fig. S6D). We next measured HACE1 protein levels in striatal and cortical tissues obtained from control and HD brains (Table S1). Notably, HACE1 protein levels were demonstrably lower in HD compared with control striatal samples, but this was not observed in cortical samples (Fig. 5 A and B and Fig. S6E) and was not a result of differential postmortem intervals (Fig. S6F). Furthermore, low HACE1 levels correlated with dramatically reduced expression of the known NRF2 target, GSS (Fig. S7). We also found that levels of NQO1, another NRF2 target, are increased in HD cortex compared with controls, but not in the striatum (Fig. S7), suggesting that reduced HACE1 levels in HD striatum result in blunted NRF2 activation and, therefore, diminished NQO1 induction. Together, our findings suggest that low HACE1 expression may contribute to reduced expression and/or induction of NRF2 targets in HD striatum, and therefore to deficits in the oxidative stress response and selective vulnerability of striatal cells to mHTT-induced toxicity.
Fig. 5.
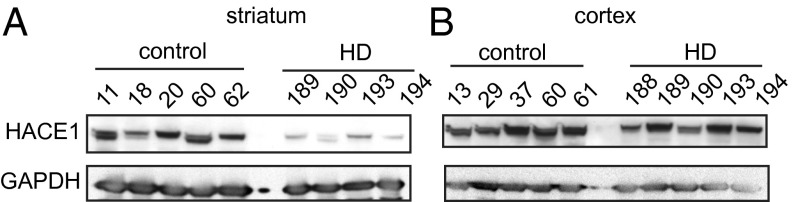
HACE1 is reduced in HD striatum. HACE1 protein levels were determined by Western blot in HD patient or control brain lysates. GAPDH was used as a loading control. A, striatum; B, cortex.
Discussion
In this study we report that HACE1 is essential for optimal activation of the NRF2 response under oxidative stress conditions by promoting NRF2 protein synthesis, stabilization, and nuclear localization. Recently, we found increased ROS levels in different peripheral tissues of Hace1 KO mice due to increased activity of ROS generating Rac1-dependent NADPH oxidase complexes (19). We show here that brains of Hace1 KO mice exhibit redox imbalance and increased oxidative stress but reduced NQO1 expression, a known NRF2 target gene and a marker of the antioxidative stress response (5, 13). Therefore, the consequences of Hace1 deficiency in mice are very similar to that of genetic loss of Nrf2 (8). These data support a model where HACE1 positively regulates the NRF2-mediated antioxidative stress response and functions to maintain redox homeostasis in brain tissues. This requires HACE1 ankyrin repeats as well as its HECT domain, but occurs independently of HACE1 E3 ligase activity and its ability to negatively regulate NADPH oxidase complexes.
Increased oxidative stress and vulnerability of the striatum to mHTT toxicity are prominent features in HD brains (31, 32). We observed a reduction in HACE1 levels in mHTT-expressing STHdhQ111 striatal neuronal progenitor cells that correlated with previously reported diminished NRF2 activity (26) and increased oxidative stress, which could be rescued by reintroduction of HACE1. Reduced HACE1 levels and induction of NRF2 target genes were observed in the HD striatum but not in the HD cortex (Fig. 5 and Fig. S7). Although other brain tissues are affected in HD, including cortex (33), the earliest and most dramatic changes are typically observed in the striatum (32). This may be due to physiological differences between striatal and cortical neurons, such as increased susceptibility of striatal neurons to excitotoxicity (34), a process that is thought to lead to ROS production. There is also evidence that mHtt expression in the striatum is sufficient to cause altered expression of oxidative stress genes (35), further indicating that the effects of mHtt expression could be cell type specific. In keeping with this, previous gene expression analyses reported that 90% of genes (including HACE1) that were found to be differentially expressed in HD versus control striatum were not differentially expressed in HD versus control cortex (29). Alternatively, cortical neurons may have evolved alternative mechanisms to retain HACE1 expression, even under mHtt-mediated stress, as these cells may be particularly sensitive to HACE1 loss.
HACE1 was recently reported as a putative NRF2 target gene (36), and HACE1 is induced by oxidative stress, raising the possibility that reduced NRF2 activity may explain why HACE1 expression is diminished in mHTT-expressing STHdhQ111 striatal progenitor cells and in HD striatum. Conversely, we found that NQO1 levels are not altered in HD striatum and are actually increased in HD cortex. This suggests redundant mechanisms of NQO1 regulation in vivo, potentially involving other transcription factors such as FBJ Murine Osteosarcoma Viral Oncogene Homolog (c-FOS) (37). Restoring HACE1 expression to STHdhQ111 cells increased their NRF2 activity, rescued redox balance, and enhanced survival under oxidative stress conditions, suggesting that HACE1 loss may contribute to the selective vulnerability of striatal cells to mHTT toxicity.
In summary, we have demonstrated that the HACE1 tumor suppressor is an important component of the antioxidative stress response, and functions by facilitating NRF2 activity. Our results support the notion that enhancing HACE1 expression may be therapeutically beneficial in HD and potentially other neurodegenerative diseases.
Materials and Methods
For determining GHS levels the Glutathione assay kit was used (Cayman Chemicals). For determining protein carbonylation the Protein Carbonyl Colorimetric Assay Kit (Cayman Chemicals) was used. Protocols for ROS measurements, mouse strains, cell culture and transfection, Western blotting, cell fractionation, cell death assays, qRT-PCR, and NQO1 and DNA oxidation staining can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
B.R. was supported by a fellowship from the International Human Frontier Science Program Organization. A.L.S. and D.E.E. were supported by Canadian Institutes of Health Research (CIHR) postdoctoral fellowships. This work was supported in part by funds from CIHR (MOP-123416) (to P.H.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314421111/-/DCSupplemental.
References
- 1.Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22(17):R741–R752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Johri A, Beal MF. Antioxidants in Huntington’s disease. Biochim Biophys Acta. 2012;1822(5):664–674. doi: 10.1016/j.bbadis.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 4.Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274(47):33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 5.Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 6.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10(11):549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Dinkova-Kostova AT, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99(18):11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73(13):4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 9.Sykiotis GP, Bohmann D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci Signal. 2010;3(112):re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34(6):663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 12.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277(45):42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 13.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purdom-Dickinson SE, Sheveleva EV, Sun H, Chen QM. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol Pharmacol. 2007;72(4):1074–1081. doi: 10.1124/mol.107.035360. [DOI] [PubMed] [Google Scholar]
- 15.Anglesio MS, et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum Mol Genet. 2004;13(18):2061–2074. doi: 10.1093/hmg/ddh215. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med. 2007;13(9):1060–1069. doi: 10.1038/nm1621. [DOI] [PubMed] [Google Scholar]
- 17.Torrino S, et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell. 2011;21(5):959–965. doi: 10.1016/j.devcel.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Castillo-Lluva S, Tan CT, Daugaard M, Sorensen PH, Malliri A. The tumour suppressor HACE1 controls cell migration by regulating Rac1 degradation. Oncogene. 2013;32(13):1735–1742. doi: 10.1038/onc.2012.189. [DOI] [PubMed] [Google Scholar]
- 19.Daugaard M, et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat Commun. 2013;4:2180. doi: 10.1038/ncomms3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: Reassessment of the ARE consensus sequence. Biochem J. 2003;374(Pt 2):337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh K, et al. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8(4):379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi A, et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26(1):221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Somasekharan SP, et al. Identification and quantification of newly synthesized proteins translationally regulated by YB-1 using a novel Click-SILAC approach. J Proteomics. 2012;77:e1–e10. doi: 10.1016/j.jprot.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Trettel F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9(19):2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 25.Kong PJ, et al. Increased expression of Bim contributes to the potentiation of serum deprivation-induced apoptotic cell death in Huntington’s disease knock-in striatal cell line. Neurol Res. 2009;31(1):77–83. doi: 10.1179/174313208X331572. [DOI] [PubMed] [Google Scholar]
- 26.Jin YN, et al. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS ONE. 2013;8(3):e57932. doi: 10.1371/journal.pone.0057932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhn A, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16(15):1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- 28.Becanovic K, et al. Transcriptional changes in Huntington disease identified using genome-wide expression profiling and cross-platform analysis. Hum Mol Genet. 2010;19(8):1438–1452. doi: 10.1093/hmg/ddq018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hodges A, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15(6):965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 30.Ross CA, Tabrizi SJ. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 31.Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90(3):905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- 32.Tabrizi SJ, et al. TRACK-HD Investigators Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: The 12-month longitudinal analysis. Lancet Neurol. 2011;10(1):31–42. doi: 10.1016/S1474-4422(10)70276-3. [DOI] [PubMed] [Google Scholar]
- 33.Estrada-Sanchez AM, Rebec GV. Role of cerebral cortex in the neuropathology of Huntington's disease. Front Neural Circuits. 2013;7:19. doi: 10.3389/fncir.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brustovetsky T, Purl K, Young A, Shimizu K, Dubinsky JM. Dearth of glutamate transporters contributes to striatal excitotoxicity. Exp Neurol. 2004;189(2):222–230. doi: 10.1016/j.expneurol.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 35.Thomas EA, et al. In vivo cell-autonomous transcriptional abnormalities revealed in mice expressing mutant huntingtin in striatal but not cortical neurons. Hum Mol Genet. 2011;20(6):1049–1060. doi: 10.1093/hmg/ddq548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malhotra D, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38(17):5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA. 1996;93(25):14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.