

Significance
Physiological concentrations of hydrogen sulfide (H2S) exert potent prosurvival actions. We demonstrate that the cytoprotective actions of H2S are mediated in part via a second gaseous signaling molecule, nitric oxide (NO). We found that cystathionine γ-lyase (CSE) KO mice with reduced H2S levels exhibit increased oxidative stress and an exacerbated response to myocardial ischemia/reperfusion injury. CSE KO mice also exhibit reduced levels of NO and reduced NO synthesis via endothelial NO synthase (eNOS). Both oxidative stress and myocardial injury in CSE KO mice were attenuated by exogenous H2S therapy, with increased eNOS function and restoration of NO levels. These findings provide insight into H2S-mediated cytoprotetion and important information regarding the translation of H2S therapy to the clinic.
Keywords: eNOS uncoupling, myocardial infarction, cystathionase, Cth, nitrite
Abstract
Previous studies have demonstrated that hydrogen sulfide (H2S) protects against multiple cardiovascular disease states in a similar manner as nitric oxide (NO). H2S therapy also has been shown to augment NO bioavailability and signaling. The purpose of this study was to investigate the impact of H2S deficiency on endothelial NO synthase (eNOS) function, NO production, and ischemia/reperfusion (I/R) injury. We found that mice lacking the H2S-producing enzyme cystathionine γ-lyase (CSE) exhibit elevated oxidative stress, dysfunctional eNOS, diminished NO levels, and exacerbated myocardial and hepatic I/R injury. In CSE KO mice, acute H2S therapy restored eNOS function and NO bioavailability and attenuated I/R injury. In addition, we found that H2S therapy fails to protect against I/R in eNOS phosphomutant mice (S1179A). Our results suggest that H2S-mediated cytoprotective signaling in the setting of I/R injury is dependent in large part on eNOS activation and NO generation.
Hydrogen sulfide (H2S), historically known for its odorous smell and toxicity at high concentrations, has recently been classified as a physiological signaling molecule with robust cytoprotective actions in multiple organ systems (1–3). H2S is produced enzymatically in mammalian tissues by three different enzymes: cystathionine γ-lyase (CSE), cystathionine beta-synthase (CBS), and 3-mercatopyruvate sulfurtransferase (3-MST). CSE, involved in the cysteine biosynthesis pathway, coordinates with l-cystine to produce H2S within the vasculature and is known to regulate blood pressure, modulate cellular metabolism, promote angiogenesis, regulate ion channels, and mitigate fibrosis and inflammation (4). Endothelial nitric oxide synthase (eNOS) catalyzes the production of nitric oxide (NO) from l-arginine within the endothelium to regulate vascular tone via cGMP signaling in vascular smooth muscle, mitochondrial respiration, platelet function, inflammation, and angiogenesis. The biological profiles of H2S and NO are similar, and both molecules are known to protect cells against various injurious states that result in organ injury. Although H2S and NO are thought to modulate independent signaling pathways, there is limited evidence of cross-talk between these two molecules (5, 6).
H2S therapeutics and endogenous overexpression of CSE have been shown to attenuate ischemia/reperfusion (I/R) injury (7, 8). Similarly, NO therapy and eNOS gene overexpression are also protective in ischemic disease states (9). Given the potent antioxidant actions of H2S (10, 11) and the effects of exogenous H2S therapy on NO bioavailability (5, 8), we investigated the effects of genetic deletion of the cystathionase gene (Cth, i.e., CSE KO) on the regulation of eNOS function and NO bioavailability.
Results
Sulfide Levels are Reduced in CSE KO Mice.
Whole blood and heart specimens were collected from WT and CSE KO mice to measure H2S levels using a high-sensitivity gas chromatography chemiluminescence technique. Our measurements confirmed significantly lower H2S and sulfane sulfur (the reaction of H2S with oxygen gives rise to sulfane sulfur) levels in the blood and heart in CSE KO mice compared with WT mice (Fig. 1 A–D). To confirm the genotype of the CSE KO mice, we measured gene expression of the three H2S-producing enzymes in the heart. CSE mRNA was absent in the CSE KO mice (Fig. 1E); in contrast, there was no difference in the relative mRNA for CBS and 3-MST between WT and CSE KO mice (Fig. 1 F and G).
Fig. 1.

Mice lacking the H2S-producing enzyme CSE display diminished free H2S and sulfur stores. (A–D) Circulating free H2S and sulfane sulfur levels (A and B) and cardiac free H2S and sulfane sulfur levels (C and D) measured in WT and CSE KO mice. (E–G) Cardiac mRNA expression of CSE (E), CBS (F), and 3-MST (G) standardized to 18s rRNA. Circles inside bars denote the number of animals per group.
Oxidative Stress is Increased in H2S Deficient Mice.
Exogenous H2S therapy has been shown to exert potent antioxidant actions during ischemic conditions (10). To examine whether endogenous insufficiency of H2S promotes oxidative stress, we evaluated the extent of oxidative stress in two organ systems. Measurement of malondialdehyde (MDA), which is commonly used as an index for lipid peroxidation, revealed significantly higher baseline levels in both the heart (Fig. 2A) and liver (Fig. 2B) in CSE KO mice compared with WT controls. Levels of protein carbonyl groups, an indicator of the oxidative modification of proteins, were also significantly higher in the heart (Fig. 2C) and liver (Fig. 2D) in the CSE KO mice.
Fig. 2.

Oxidative stress is exacerbated in CSE KO mice. (A and B) Heart and liver MDA levels. (C and D) Heart and liver carbonyl protein content. (E and G) Representative photomicrographs of DMPO-stained heart (E) and liver (G) depicting immunodetectable DMPO-adducted biomolecules indicative of oxidative stress and resultant biomolecular free radical formation. (F and H) Quantification of heart (F) and liver (H) tissue images. Circles inside bars denote number of animals per group. MFI, mean fluorescence intensity.
In vivo immune-spin trapping of biomolecular free radicals using the EPR nitrone spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) revealed enhanced immunodectable, covalently bound, DMPO-protein/lipid adducts in the heart (Fig. 2 E and F) and liver (Fig. 2 G and H) of CSE KO mice, further confirming elevated oxidative stress in these tissues.
Because oxidative stress is associated with mitochondrial dysfunction, we examined mitochondrial respiration in WT, CSE KO, and CSE KO + H2S donor-treated mice. Heart mitochondria were isolated and used for respiration measurements (Fig. S1A). The CSE KO mice displayed a marked reduction in state 3 respiration compared with the WT mice, which was restored with H2S therapy (Fig. S1A). State 3 respiration indicates electron transport efficiency and subsequently, the rate of ATP synthesis. In a similar fashion, the respiratory control ratio (RCR; i.e., state 3/state 4) was reduced in CSE KO mice, but restored with an H2S donor (Fig. S1B). The RCR is an index of the efficiency of the electron transport in regards to ATP generation (higher RCR = increased coupling of oxidative phosphorylation). We also evaluated calcium-induced mitochondrial swelling, and found no difference in either rate of swelling or time to swelling between the CSE KO and WT mice (Fig. S1 C and D).
CSE KO Mice Display Impaired eNOS Function.
We investigated whether H2S deficiency alters eNOS function under basal conditions. Myocardial eNOS expression and phosphorylation was determined by Western blot analysis in CSE KO and WT mice. CSE KO mice demonstrated significantly lower phosphorylation of the eNOS activation site, P-eNOSS1177, compared with WT mice (Fig. 3 A and B). Moreover, the eNOS inhibitory site, P-eNOST495, was phosphorylated to a significantly greater degree in CSE KO mice (Fig. 3 A and C). There was no difference in total eNOS expression (Fig. 3 A and D), and also no significant difference in iNOS or nNOS myocardial expression (Fig. S2).
Fig. 3.

CSE KO mice exhibit altered eNOS phosphorylation status. (A) Representative immnoblots of eNOS from either WT or CSE KO hearts. (B–D) Relative intensity of P-eNOSS1177 (B), P-eNOST495 (C), and total eNOS (D) protein expression. (E–G) Levels of eNOS cofactors BH4 (E) and BH2 (F), and their ratio (G), in WT and CSE KO cardiac tissue. Circles inside bars denote the number of animals per group.
We next measured cardiac levels of the eNOS cofactor tetrahydropbiopterin (BH4) by HPLC, and found significantly lower cardiac BH4 levels in the CSE KO mice compared with the WT mice (Fig. 3 E–G). Our findings of no significant differences in inducible nitric oxide synthase (iNOS), neuronal nitric oxide synthase (nNOS), protein kinase B (AKT), and phosphorylated protein kinase B (P-AKTT308) expression between CSE KO mice and WT mice (Fig. S2) suggest an alteration in eNOS activity is most likely the key mechanism accounting for alterations in NO bioavailability.
H2S Deficiency Reduces NO Bioavailability and cGMP Levels.
We measured NO metabolites (nitrite and nitrosylated protein, RXNO) in plasma and myocardial tissue to evaluate NO bioavailability. We observed a significant reduction in circulating and myocardial nitrite levels in CSE KO mice compared with WT mice (Fig. 4 A and B). Similarly, RXNO measurements in plasma and heart tissue revealed a threefold reduction in CSE KO mice (Fig. 4 C and D). After generation by eNOS, NO activates soluble guanylyl cyclase (sGC) to form cyclic guanosine 5′-monophosphate (cGMP). Circulating cGMP levels were significantly diminished in CSE KO mice (Fig. 4E).
Fig. 4.

NO bioavailability and signaling is mitigated in CSE KO mice. (A) Plasma nitrite. (B) Cardiac nitrite. (C) Plasma RXNO. (D) Cardiac RXNO. (E) Plasma cGMP. (F) Cardiac cGMP. Circles inside bars denote the number of animals per group.
H2S Therapy Restores eNOS Function and NO Bioavailability in CSE KO Mice.
To determine whether exogenous H2S could restore eNOS function and NO bioavailability in CSE KO mice, we utilized the H2S donor, diallyl trisulfide (DATS) in CSE KO mice. Treatment with DATS restored eNOS P-eNOSS1177 to near-WT levels (Fig. 5C). After administration of DATS, cardiac nitrite levels increased significantly in CSE KO mice (Fig. 5F). Further evidence for increased NO bioavailability resulting from H2S therapy was provided by a 6.3-fold increase in circulating RXNO levels (Fig. 5G) and a 6-fold increase in myocardial RXNO levels (Fig. 5H).
Fig. 5.

H2S therapy activates eNOS and augments NO bioavailability in CSE KO mice. (A) Representative immnoblots of eNOS. (B–D) Relative intensity of P-eNOSS1177 (B), P-eNOST495 (C), and total eNOS (D) protein expression in CSE KO hearts ± H2S donor. (E–H) Levels of plasma nitrite (E), cardiac nitrite (F), plasma RXNO (G), and cardiac RXNO (H). Circles inside bars denote the number of animals studied per group.
H2S Therapy Protects Against Exacerbated I/R Injury in CSE KO Mice.
We next subjected CSE KO mice to cardiac and hepatic I/R injury. Mice were subjected to myocardial ischemia for 45 min, followed by 24 h of reperfusion. The CSE KO mice displayed a significant (P < 0.01) increase in myocardial infarct size (INF) per area-at-risk (AAR) compared with the WT mice (62 ± 2% vs. 42 ± 3%) (Fig. 6A). This exacerbation of myocardial I/R injury in CSE KO mice was completely reversed to WT levels with acute H2S therapy (Na2S, 100 μg/kg at 5 min before reperfusion), evident by a significant reduction in myocardial infarct size and plasma troponin-I levels (Fig. 6 A and B).
Fig. 6.

Ischemia/reperfusion injury is exacerbated in H2S-deficient mice, but rescued with H2S therapy. (A) Bar graphs of myocardial AAR/LV, INF/AAR, and INF/LV. (B) Cardiac troponin-I levels after 24 h of reperfusion. (C and D) Circulating ALT and AST and hepatic 8-isoprostane levels after 45 min of hepatic ischemia and 5 h of reperfusion. Circles inside bars denote the number of animals per group. LV, left ventricle.
We next subjected another cohort of mice to 45 min of hepatic ischemia and 5 h of reperfusion, then measured liver transaminases, asparate aminotransferase (AST) and alanine aminotransferase (ALT), as markers of hepatic injury. AST and ALT levels were significantly increased after hepatic I/R injury in CSE KO mice compared with WT mice (Fig. 6C). This exacerbated hepatic injury was largely reversed after H2S treatment with Na2S (500 μg/kg) administered i.v. 5 min before reperfusion (Fig. 6C). Hepatic 8-isoprostane measurements indicated that oxidative stress was increased in CSE KO mice after I/R and corrected to WT levels with H2S therapy (Fig. 6D).
H2S Therapy Fails to Reduce Myocardial I/R Injury in eNOS Phospho-Dead Mutant Mice.
Exogenous H2S therapy has been demonstrated to attenuate myocardial I/R injury in mice (7, 8, 12). To determine whether the protective mechanism is dependent on eNOS, we first used a mouse model with global genetic ablation of eNOS. Again, mice were subjected to 45 min of ischemia, followed by 24 h of reperfusion. The H2S donor, Na2S (100 μg/kg), or vehicle (0.9% NaCl) was administered 5 min before reperfusion. There was no significant change in infarct size in the H2S-treated group compared with the vehicle-treated group (Fig. 7A).
Fig. 7.
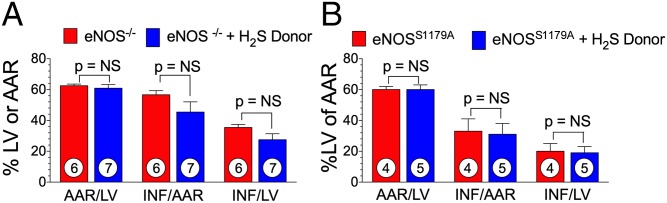
H2S therapy fails to protect against myocardial I/R injury in eNOS mutant mice. Bar graphs of myocardial AAR/LV, INF/AAR, and INF/LV for eNOS KO mice with or without an H2S donor (A) and eNOSS1179A phosphomutant mice with or without an H2S donor (B). Circles inside bars denote the number of animals per group.
We then examined whether H2S therapy could protect against myocardial I/R injury in a mutant mouse model expressing a transgene encoding non-phosphorylatable eNOS (S1179A) on the eNOS−/− background. S1179A mutant mice and WT control mice were subjected to the same myocardial I/R protocol, and the H2S donor, DATS (200 μg/kg), was administered 5 min before reperfusion. Similarly, H2S was unable to reduce injury in eNOS phospho-dead mice (Fig. 7B). These results indicate that the cardioprotective actions of H2S are dependent on eNOS phosphorylation.
Discussion
It is well established that both H2S and NO exert potent cytoprotective effects in the setting of cardiovascular disease in various animal model systems (5, 7, 13–15). In addition, H2S has been reported to be cytoprotective in some organ systems, such as the central nervous system and gastrointestinal tract, independent of NO (3, 11, 16–18). Although originally considered separate signaling pathways, recent evidence suggests cross-talk between H2S and NO signaling in the cardiovascular system. Previous studies have reported that vascular function, inflammation, angiogenesis, and ischemic injury are regulated by cross-talk between the H2S and NO signaling pathways (8, 19–21). We postulate that the cross-talk between H2S and NO is mediated via the phosphorylation of eNOS (enzymatic production of NO).
In mammalian tissues, CSE is the dominant enzyme for H2S formation (22). Yang et al. (23) reported that CSE genetic ablation in mice results in significant depletion of H2S levels in peripheral tissue and blood. The loss of CSE-derived H2S in these mice also resulted in an age-dependent hypertension (23). In the present study, we used the same CSE KO mouse to evaluate cross-talk between CSE and eNOS. Consistent with findings of Yang et al. (23), loss of the CSE enzyme resulted in marked depletion of H2S and sulfane sulfur, a storage intermediate of H2S, in the circulation and the heart. We previously showed that treatment with exogenous H2S or genetic overexpression of CSE in cardiomyocytes results in increased endogenous H2S production, and profound protection against ischemia-induced heart failure and myocardial I/R injury (14). A recent clinical study reported lower circulating sulfide levels in patients suffering from congestive heart failure (24). Using a mouse model lacking CSE, we have demonstrated that a deficiency in endogenous H2S results in increased myocardial injury. The administration of an H2S donor immediately before reperfusion in CSE KO mice reversed injury to WT levels. Similarly, CSE ablation in a model of hepatic I/R resulted in markedly elevated levels of the circulating liver enzymes AST and ALT, indicating significant liver injury, which was attenuated after acute administration of H2S.
I/R injury involves multiple pathological mechanisms, including free radical accumulation and reduced bioavailability of NO. The controlled regulation of NO synthesis by eNOS is essential for cardiovascular health. It is well established that eNOS can undergo posttranslational modifications, including multisite phosphorylation, which tightly regulates NO production (25–27). Specifically, phosphorylation of the amino acids S1177 and T495 regulates eNOS activity, thereby enhancing or inhibiting NO production (25–27). In the present study, evaluation of eNOS S1177 and T495 revealed that compared with WT mice, CSE KO mice exhibited markedly lower phosphorylation at the active site, eNOSS1177, and greater phosphorylation at the inhibitory site, eNOST495. This altered phosphorylation of eNOS was coupled with concomitant reductions in both circulating and myocardial levels of nitrite, nitrosylated proteins, and plasma cGMP, corroborating decreased NO bioavailability and signaling. These results are consistent with the previous finding of marked reductions in cGMP in the plasma, aorta, and mesenteric artery in CSE KO mice (28). Diminished cGMP and reductions in protein kinase G activity are also likely to contribute to the exacerbated tissue injury in the heart and liver observed in CSE KO mice. When CSE KO mice were treated with the H2S donor DATS for 7 d, phosphorylation at the active site of eNOSS1177 was enhanced compared with controls. Treatment with DATS significantly enhanced cardiac nitrite and both plasma and cardiac levels of nitrosylated proteins in the CSE KO mice. Previous studies from our laboratory demonstrated that in a murine model of myocardial I/R, treatment with DATS increases eNOS phosphorylation at S1177 compared with vehicle. This resulted in elevated circulating nitrate and nitrite and promoted increased protection on the heart (8). Similarly, 12 wk of H2S therapy during pressure overload heart failure in mice similarly enhanced eNOS phosphorylation at the active site S1177, increasing cardiac nitrite and ultimately preserving cardiac function (5).
In the present study, we examined the effect of H2S in eNOS-ablated and an eNOS phospho-dead mutant mouse model. There was no change in infarct size in either the total eNOS KO mice or the eNOSS1179A mice that received H2S, compared with vehicle controls. Previous studies reveal not only that H2S is cardioprotective in the setting of myocardial I/R (7, 8), but also that it activates eNOS and augments NO bioavailability (8, 29). These results further corroborate the regulation of H2S cardioprotection through eNOS phosphorylation at the S1177 active site.
Previous studies have demonstrated that phosphorylation of S1177 is mediated through an Akt-dependent mechanism (30, 31). Moreover, numerous factors, including insulin (31), corticosteroids (32), bradykinin (33), and H2S (5), stimulate NO production through Akt-induced phosphorylation of eNOSS1177. In the present study, examination of Akt phosphorylation revealed no difference between CSE KO and WT mice perhaps suggesting a mechanism independent of AKT. Tetrahydrobiopterin (BH4), an essential cofactor, has been shown to regulate eNOS activity. When BH4 availability is limiting, electron transport within the active site becomes “uncoupled” from l-arginine oxidation, causing a reduction of oxygen to superoxide (34). Depletion in BH4 and eNOS uncoupling has been correlated with cardiac dysfunction in murine models of hypertension and transverse aortic constriction (35, 36). Compared with WT mice, CSE KO mice had significantly lower cardiac BH4 levels, and a trend toward higher dihydrobiopterin (BH2) levels. Moreover, CSE KO mice exhibited significantly elevated levels of MDA, protein carbonyl, and tissue free radicals (markers of oxidative stress) in the heart and marked increases of MDA and carbonyl in the liver. Oxidation of BH4 can result in uncoupled eNOS, which can lead to increased oxidative stress (37). However, oxidative stress also may be related in part to changes in cellular respiration via impaired mitochondrial function. The elevated oxidative stress in CSE KO mice may be related in part to the absence of the enzyme responsible for the conversion of homocysteine to cystathionine. Although mitochondrial function and eNOS function were restored with H2S therapy, the elevated homocysteine levels may be partially responsible for the increased oxidative stress in CSE KO mice.
Cellular respiration is controlled by NO interacting with complexes I and IV of the electron transport chain (38). Examination of basal respiration revealed significantly lower state 3 respiration levels and RCR in CSE KO mice compared with WT mice, indicating mitochondrial dysfunction. Much of the cellular damage observed in cardiovascular disease is the result of mitochondrial events, such as Ca2+ overload, which leads to overproduction of reactive oxygen species (ROS) (39, 40). In the presence of NO, produced enzymatically or directly via the reduction of nitrite, the inhibition of complex I and IV has been reported to be cytoprotective. In the present study, the loss of the CSE-derived H2S resulted in a significant increase in ROS. Moreover, these mice exhibited a reduction in nitrite levels owing to lower phosphorylation of eNOSS1177 and reduced eNOS activity compared with WT mice. This combination of insufficient NO bioavailability and increased ROS may explain in part why CSE KO mice are more susceptible to myocardial I/R injury.
In this study, we used various H2S-releasing compounds to confirm that the effects of H2S on eNOS were not limited to a particular H2S donor compound. For example, polysulfide compounds (i.e., DATS) have previously been shown to be antioxidants and modify thiol proteins (41, 42). It is possible that some of the effects of DATS that we observed are independent of H2S release and are a result of DATS-mediated protein modifications. In addition, H2S may alter the overall redox status, leading to increased eNOS coupling (phosphorylation at S1177).
The findings in this study reveal that cytoprotection elicited by CSE-derived H2S is eNOS/NO-dependent. Future studies will aim to more fully understand the mechanisms related to cross-talk between CSE and eNOS-derived NO in the context of clinical syndromes.
Methods
Animals.
CSE-ablated (KO) mice (Sv129/C57 background) and eNOS KO (eNOS−/−) mice were developed as described previously (5). The eNOS phospho-dead mutant mouse model expresses a bovine eNOS cDNA with a single-point mutation (S1179A) under the control of the human eNOS promoter. This transgenic mouse was then crossed into the eNOS−/− background. This model has been previously described in detail (43). Male mice age 14–16 wk were used during the course of this present study. All animals received humane care in compliance with the National Society of Medical Research’s Principles of Laboratory Animal Care and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (44), and all protocols were approved by Emory University’s Institutional Animal Care and Use Committee.
Mouse DMPO Protocol.
Between 14–16 wk of age, WT and CSE KO mice received DMPO (Dojindo) dissolved in pyrogen-free saline [1.5 g/kg i.p. total in three doses (0.5 g/kg) over 24 h]. At sacrifice, hearts and livers were perfused with PBS (pH 7.4), removed, and fixed for immunohistochemical analysis (see below).
H2S Donors.
Na2S was produced and formulated to pH neutrality by Ikaria using H2S gas (Matheson) as a starting material. DATS was prepared and dosed as described previously (29).
Myocardial I/R Protocol and Myocardial Infarct Size Determination.
Mice were subjected to 45 min of myocardial ischemia and 24 h of reperfusion. Surgical procedures were performed as described previously (45).
Liver I/R Protocol.
Mice were subjected to 45 min of regional hepatic ischemia and 5 h of reperfusion as described previously (46).
AST and ALT Assay.
Blood from mice subjected to 45 min of hepatic ischemia and 5 h of reperfusion was collected and used to determine liver transaminase values as described previously (46).
Cardiac Troponin-I Assay.
Serum was obtained from mice after 45 min of LCA ischemia and 24 h of reperfusion to measure the cardiac-specific isoform of troponin-I using a mouse-specific ELISA kit (Life Diagnostics).
8-Isoprostane Assay.
After 45 min of hepatic ischemia and 5 h of reperfusion, mouse livers were rapidly excised and homogenized. 8-Isoprostane was assayed using an ELISA kit (Cell Biolabs) according to the manufacturer’s recommendations.
Cardiac Mitochondrial Isolation.
Mitochondria were isolated and prepared as described previously (8).
Mitochondrial Respiration Measurement.
Oxygen consumption and respiration were determined as described previously (8).
Measurement of Hydrogen Sulfide and Sulfane Sulfur.
H2S and sulfane sulfur levels were measured in heart and blood by gas chromatography chemilumunescence (Agilent 7890 GC gas chromatography system and G660XA Series chemiluminescence detector). Free H2S in fresh blood and tissue was liberated by incubating in 1 M sodium citrate solution at 37 °C for 10 min. Sulfane sulfur was released by incubating 100 μL of sample in an equal volume of 15 mM DTT for 50 min, followed by the addition of 400 μL of 1 M sodium citrate at 37 °C. The resultant headspace gases were analyzed using the GC system.
Measurement of NO Metabolites.
Nitrite concentrations were quantified by ion chromatography (ENO20 Analyzer; Eicom). RXNO levels were measured by chemiluminescence detection (CLD 88Y; Eco Physics) achieved by an acidified sulfanilamide reaction with the biological samples into a tri-iodide–containing mixture purged continuously with helium.
cGMP RIA.
cGMP standards (Sigma-Aldrich) and samples were acetylated by adding 8 μL of 5 N KOH and 2 μL of acetic anhydride in a volume of 200 μL, followed by incubation at room temperature for 30 min. The tubes were then placed on ice to stop the reaction. All determinations were performed in duplicate. Each tube contained 50 μL of acetylated standards or samples, 50 μL of iodinated cGMP (14,000–16,000 cpm), and 50 μL of cGMP antibody (Sigma-Aldrich). The reaction mixture was incubated overnight at 4 °C, followed by precipitation of the cGMP–antibody complex by 12% PEG8000. Radioactivity was counted in a gamma counter.
Western Blot Analysis.
Myocardial tissue was used for Western blot analysis, performed as described previously (5). The following primary antibodies were used: GAPDH, α-tubulin, P-eNOSS1177, P-eNOST495, iNOS, nNOS, total AKT, and P-AKTT308 (Cell Signaling), and total eNOS (BD Biosciences).
HPLCy Analysis of BH4 and BH2.
Cardiac BH4 and BH2 quantifications were determined as described previously (47).
Determination of Protein Carbonyl Content.
Protein carbonyl content from heart and liver was measured as described previously (48).
Measurement of MDA Levels.
MDA levels were measured in heart and liver protein as described previously (48).
Immunohistochemistry.
Samples for immunohistochemistry analysis were prepared as described previously (49).
Statistical Analysis.
All data in this study are expressed as mean ± SEM. Differences in data between groups were compared using Prism 4 (GraphPad Software) with the Student unpaired two-tailed t test when comparing two groups and one-way ANOVA when comparing three or more groups. If a significant difference was found on ANOVA, then Tukey’s multiple-comparison test was used for post hoc analysis. A P value of <0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Valeria Hebert, Marah Condit, and Benjamin Predmore, as well as the University of Pittsburgh Center for Biological Imaging, for expert technical assistance. This work was supported by Grants from the National Heart, Lung, and Blood Institute (1R01 HL092141, 1R01 HL093579, 1U24 HL 094373, and 1P20 HL113452 to D.J.L., and 5R01 HL 098481 to J.W.C.). These studies were also supported by the Canadian Institutes of Health Research (R.W.). We are also grateful for the generous financial support from the Carlyle Fraser Heart Center of Emory University and the Louisiana State University Health Foundation in New Orleans.
Footnotes
Conflict of interest statement: D.J.L. is a participant in a pending US patent, filed through the National Institutes of Health (patent no. 60/511, 244), regarding the use of sodium nitrite in cardiovascular disease. D.J.L. and J.W.E. are founders of and scientific advisors for Sulfagenix, a biotechnology company that is currently developing hydrogen sulfide-based therapeutics for human disease conditions. The conflicts are considered significant.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321871111/-/DCSupplemental.
References
- 1.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20(21):6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6(11):917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 3.Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346) Br J Pharmacol. 2010;159(6):1236–1246. doi: 10.1111/j.1476-5381.2009.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol Rev. 2012;92(2):791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 5.Kondo K, et al. H2S protects against pressure overload-induced heart failure via up-regulation of endothelial nitric oxide synthase. Circulation. 2013;127(10):1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali MY, et al. Regulation of vascular nitric oxide in vitro and in vivo: A new role for endogenous hydrogen sulphide? Br J Pharmacol. 2006;149(6):625–634. doi: 10.1038/sj.bjp.0706906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elrod JW, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104(39):15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Predmore BL, et al. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am J Physiol Heart Circ Physiol. 2012;302(11):H2410–H2418. doi: 10.1152/ajpheart.00044.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elrod JW, Calvert JW, Gundewar S, Bryan NS, Lefer DJ. Nitric oxide promotes distant organ protection: Evidence for an endocrine role of nitric oxide. Proc Natl Acad Sci USA. 2008;105(32):11430–11435. doi: 10.1073/pnas.0800700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295(2):H801–H806. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18(10):1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 12.Peake BF, et al. Hydrogen sulfide preconditions The db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am J Physiol Heart Circ Physiol. 2013;304(9):H1215–H1224. doi: 10.1152/ajpheart.00796.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calvert JW, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105(4):365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calvert JW, et al. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122(1):11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefer AM, Peck RC. Cardioprotective effects of enalapril in acute myocardial ischemia. Pharmacology. 1984;29(2):61–69. doi: 10.1159/000137993. [DOI] [PubMed] [Google Scholar]
- 16.Fiorucci S, et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129(4):1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 17.Chan MV, Wallace JL. Hydrogen sulfide-based therapeutics and gastrointestinal diseases: Translating physiology to treatments. Am J Physiol Gastrointest Liver Physiol. 2013;305(7):G467–G473. doi: 10.1152/ajpgi.00169.2013. [DOI] [PubMed] [Google Scholar]
- 18.Campolo M, et al. A hydrogen sulfide-releasing cyclooxygenase inhibitor markedly accelerates recovery from experimental spinal cord injury. FASEB J. 2013;27(11):4489–4499. doi: 10.1096/fj.13-234716. [DOI] [PubMed] [Google Scholar]
- 19.Coletta C, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA. 2012;109(23):9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altaany Z, Yang G, Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med. 2013;17(7):879–888. doi: 10.1111/jcmm.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zanardo RC, et al. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20(12):2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 22.Paul BD, Snyder SH. H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13(8):499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 23.Yang G, et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovačić D, et al. Total plasma sulfide in congestive heart failure. J Card Fail. 2012;18(7):541–548. doi: 10.1016/j.cardfail.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 25.Lin MI, et al. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem. 2003;278(45):44719–44726. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]
- 26.Boo YC, et al. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: Role of protein kinase A. J Biol Chem. 2002;277(5):3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 27.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42(2):271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 28.Bucci M, et al. cGMP-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation. PLoS ONE. 2012;7(12):e53319. doi: 10.1371/journal.pone.0053319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polhemus DJ, et al. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail. 2013;6(5):1077–1086. doi: 10.1161/CIRCHEARTFAILURE.113.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimmeler S, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 31.Gao F, et al. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: The roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002;105(12):1497–1502. doi: 10.1161/01.cir.0000012529.00367.0f. [DOI] [PubMed] [Google Scholar]
- 32.Hafezi-Moghadam A, et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8(5):473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell RM, Yellon DM. Bradykinin limits infarction when administered as an adjunct to reperfusion in mouse heart: The role of PI3K, Akt and eNOS. J Mol Cell Cardiol. 2003;35(2):185–193. doi: 10.1016/s0022-2828(02)00310-3. [DOI] [PubMed] [Google Scholar]
- 34.Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol. 2004;24(3):413–420. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- 35.Silberman GA, et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121(4):519–528. doi: 10.1161/CIRCULATIONAHA.109.883777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takimoto E, et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115(5):1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landmesser U, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111(8):1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarti P, Arese M, Forte E, Giuffrè A, Mastronicola D. Mitochondria and nitric oxide: Chemistry and pathophysiology. Adv Exp Med Biol. 2012;942:75–92. doi: 10.1007/978-94-007-2869-1_4. [DOI] [PubMed] [Google Scholar]
- 39.Brookes P, Darley-Usmar VM. Hypothesis: The mitochondrial NO+ signaling pathway, and the transduction of nitrosative to oxidative cell signals: An alternative function for cytochrome C oxidase. Free Radic Biol Med. 2002;32(4):370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 40.Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98(13):7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin MC, Hwang SW, Chan KC. Nonenzymatic antioxidant activity of four organosulfur compounds derived from garlic. J Agric Food Chem. 2002;50(21):6143–6147. doi: 10.1021/jf0204203. [DOI] [PubMed] [Google Scholar]
- 42.Grudzinski IP, Frankiewicz-Jozko A, Bany J. Diallyl sulfide—a flavour component from garlic (Allium sativum) attenuates lipid peroxidation in mice infected with Trichinella spiralis. Phytomedicine. 2001;8(3):174–177. doi: 10.1078/0944-7113-00037. [DOI] [PubMed] [Google Scholar]
- 43.Atochin D, et al. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. The Journal of Clinical Investigation. 2007;117(7):1961–1967. doi: 10.1172/JCI29877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Committee on Care and Use of Laboratory Animals (1985) Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No (NIH) 85-23. [Google Scholar]
- 45.Elrod JW, et al. Cardiomyocyte-specific overexpression of NO synthase-3 protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26(7):1517–1523. doi: 10.1161/01.ATV.0000224324.52466.e6. [DOI] [PubMed] [Google Scholar]
- 46.Duranski MR, et al. Genetic overexpression of eNOS attenuates hepatic ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;291(6):H2980–H2986. doi: 10.1152/ajpheart.01173.2005. [DOI] [PubMed] [Google Scholar]
- 47.Stokes KY, et al. Dietary nitrite prevents hypercholesterolemic microvascular inflammation and reverses endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2009;296(5):H1281–H1288. doi: 10.1152/ajpheart.01291.2008. [DOI] [PubMed] [Google Scholar]
- 48.Islam KN, et al. TGF-beta1 triggers oxidative modifications and enhances apoptosis in HIT cells through accumulation of reactive oxygen species by suppression of catalase and glutathione peroxidase. Free Radic Biol Med. 1997;22(6):1007–1017. doi: 10.1016/s0891-5849(96)00493-5. [DOI] [PubMed] [Google Scholar]
- 49.Khoo NK, et al. Obesity-induced tissue free radical generation: An in vivo immuno-spin trapping study. Free Radic Biol Med. 2012;52(11-12):2312–2319. doi: 10.1016/j.freeradbiomed.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.