

Significance
Soluble guanylate cyclase (sGC) is the primary nitric oxide (NO) receptor in mammals and a central component of the NO-signaling pathway. Disruptions in NO signaling have been linked to hypertension, neurodegeneration, and heart disease. The mechanistic details underlying the modulation of sGC activity remain largely unknown. Determining the structure of full-length sGC is a prerequisite to understanding its function and for the design and improvement of therapeutics for treatment of related diseases. We use electron microscopy to determine the quaternary structure of the protein. Furthermore, we found that both ligand-free and ligand-bound sGC are highly flexible. This structural information provides a significant step forward in understanding the mechanism of sGC activation and will ultimately empower the development of next-generation therapeutics.
Abstract
Soluble guanylate cyclase (sGC) is the primary nitric oxide (NO) receptor in mammals and a central component of the NO-signaling pathway. The NO-signaling pathways mediate diverse physiological processes, including vasodilation, neurotransmission, and myocardial functions. sGC is a heterodimer assembled from two homologous subunits, each comprised of four domains. Although crystal structures of isolated domains have been reported, no structure is available for full-length sGC. We used single-particle electron microscopy to obtain the structure of the complete sGC heterodimer and determine its higher-order domain architecture. Overall, the protein is formed of two rigid modules: the catalytic dimer and the clustered Per/Art/Sim and heme-NO/O2-binding domains, connected by a parallel coiled coil at two hinge points. The quaternary assembly demonstrates a very high degree of flexibility. We captured hundreds of individual conformational snapshots of free sGC, NO-bound sGC, and guanosine-5′-[(α,β)-methylene]triphosphate-bound sGC. The molecular architecture and pronounced flexibility observed provides a significant step forward in understanding the mechanism of NO signaling.
Nitric oxide (NO) has emerged as an integral signaling molecule in biology. Soluble guanylate cyclase (sGC), the primary receptor of NO in mammals, binds NO via an FeII heme cofactor leading to a several hundred-fold increase in 3,5-cyclic guanosine monophosphate (cGMP) synthesis. cGMP then acts as a second messenger, targeting phosphodiesterases, ion-gated channels, and cGMP-dependent protein kinases. These target proteins go on to regulate many critical physiological functions including vasodilation, platelet aggregation, neurotransmission, and myocardial functions (1, 2). Disruptions in NO signaling have been linked to hypertension, erectile dysfunction, neurodegeneration, stroke, and heart disease (3, 4). sGC has been the focus of small-molecule modulators of activity for therapeutic advantage. Riociguat, which is a stimulator of sGC, has recently been approved for treatment of pulmonary hypertension (5). However, the mechanistic details underlying the modulation of sGC catalytic activity by NO and other small molecules remain largely unknown. Determining the structure of the full-length sGC, free and in complex with NO, is therefore a prerequisite to understanding its function and for the design and improvement of therapeutics for treatment of diseases involving the NO/cGMP pathway.
The most extensively studied and physiologically relevant isoform of sGC is the 150-kDa heterodimer containing one α1 and one β1 subunit. Each subunit is comprised of four modular domains: the N-terminal heme-NO/O2–binding (H-NOX), the Per/Arnt/Sim (PAS), the helical, and the C-terminal catalytic domain (Fig. 1). No structure of the complete holoenzyme is available to date, and its absence precludes answering key questions such as how NO occupancy of the N-terminal β H-NOX sensor domain is communicated to the C-terminal cyclase domain. Atomic models of isolated sGC domains have been obtained by X-ray crystallography or homology modeling (6–9) (Fig. 1). The arrangement of and interactions between these domains have been further studied using a variety of techniques, including mutational and truncation studies, Förster resonance energy transfer (FRET), resonance Raman spectroscopy, chemical cross-linking, small-angle X-ray scattering (SAXS), and hydrogen deuterium exchange (HDX) (10–14). These studies have been used to propose a variety of models for the mechanisms of action of sGC, but the lack of a comprehensive 3D structure of the sGC holoenzyme has so far impeded a confident assignment of domain hierarchy.
Fig. 1.

sGC domain organization and X-ray crystallographic models. Each subunit contains four modular domains; α1 domains are shown in shades of gray, and β1 domains are shown in color. The H-NOX domain of the β1 subunit contains the heme cofactor, shown in red. Structures for Rattus norvegicus are modeled based on previously solved crystal structures of homologous domains (Materials and Methods). The H-NOX structures are modeled from a standalone Nostoc sp. PCC 7120 H-NOX domain (PDB: 2O09) (6). The PAS and helical domains are modeled on individual domain truncations. The PAS domain is based on the PAS domain from Manduca sexta (PDB: 4GJ4) (7), and the helical domain is based on the β1 R. norvegicus structure (3HLS) (8). The catalytic domain is the Homo sapiens α1β1 crystal structure (PDB: 3UVJ) (9).
Here, we used EM to determine the first structure of the heterodimeric sGC holoenzyme. Fitting of the domain crystal structures into the EM reconstruction provides a detailed model for the higher-order architecture and quaternary organization of sGC and is consistent with all reported biochemical data. We obtained hundreds of individual 3D reconstructions of full-length Rattus norvegicus sGC using automated high-throughput single-particle electron microscopy (15, 16). The structures correspond to various snapshots of the enzyme and describe the conformational trajectory of this highly flexible protein. sGC is assembled from two ridged units: the smaller unit comprises the dimeric catalytic domain, and the larger unit is built from the clustering of the PAS and H-NOX domains. The helical domains form a dimeric parallel coiled coil that flexibly connects the two modules. These modules swing freely in relation to each other thereby allowing the structure to access a wide range of conformations. Strikingly, some of these conformations allow the N-terminal H-NOX domain to contact the C-terminal catalytic domain indicating the possibility of a direct allosteric control mechanism. We also obtained reconstructions of sGC in complex with NO as well as with guanosine-5′-[(α,β)-methylene]triphosphate (GPCPP), a noncyclizable analog of the natural substrate GTP, both in the presence and absence of NO. The overall domain architecture and range of the accessible conformations of these complexes are similar to the unbound sGC state, suggesting that ligand binding induces small-scale intradomain conformational changes mediated by flexibility transitions at two key linker points.
Results
sGC Is Separated into Two Main Globular Densities and Exhibits Continuous Flexibility.
Visualized through negative-stain EM, purified R. norvegicus sGC is distinguishable as small highly flexible particles (Fig. S1). We used the random conical tilt (RCT) (17, 18) strategy to sort out the multitude of conformations adopted by the holoenzyme. This ab initio method allows for 2D classes to be reconstructed independently into 3D electron density maps. This reference-free strategy was key because no other validated model of the full-length sGC is currently available.
Particles were chosen and sorted using iterative reference-free and reference-based techniques (Materials and Methods). Approximately 43,000 particles were clustered into 128 2D class averages, revealing that sGC occupies a multitude of conformations and is comprised of two globular densities connected by a thin stalk. The detail in the obtained class averages revealed a distinctly heart-shaped smaller density and four lobes in the larger density. From these 128 classes, 94 3D maps were reconstructed revealing a set of structures with a consistent interpretation (Fig. 2). When in an “extended” conformation the maximum vertical length of the protein is 135 Å from the cleft of the smaller heart-shaped density to the top peak of the four-lobed density. The dimensions of the heart-shaped density are 60 × 40 × 40 Å, and the four-lobed cluster is 90 × 40 × 45 Å (length × width × height) (Fig. 3). The relative location and orientation of these two regions of density vary widely among the conformational snapshots obtained: they are 40 Å apart in the extended conformation, whereas they directly interact in the bent conformation. The reconstructions obtained describe the conformational trajectory between these two extreme conformations and suggest that sGC has a continuous range of motion enabled by the flexible linkers connecting the thin stalk to the two globular densities.
Fig. 2.

A continuous range of conformations is observed for the sGC holoenzyme, as illustrated here by 18 different conformational snapshots. Three-dimensional reconstructions are shown in green directly above the corresponding 2D class averages. The 3D maps were aligned relative to each other based on the heart-shaped catalytic domain. For consistency, models are displayed such that the four-lobed density is flexing toward the right, although we suggest that the range of motions allows the four-lobed cluster to make contact with both sides of the heart-shaped catalytic domain. (A) Nine snapshots of sGC exhibiting in-plane flexing of the four-lobed cluster. (B) Nine conformational snapshots exhibiting out-of-plane flexing of the four-lobed cluster. The thin stalk connecting the main densities is readily observed in the 2D class averages but sometimes only weakly observed in the 3D density maps, which are reconstructed from images tilted to a high angle (Materials and Methods).
Fig. 3.

Two selected sGC maps illustrating two extreme sGC conformations. (A) An extended structure showing the heart-shaped domain and the four-lobed cluster separated by 40 Å. (B) A “bent” structure where these two densities directly interact.
Dimeric Catalytic Domain Corresponds to the Smaller Heart-Shaped Density.
The crystal structure of the isolated catalytic domain from Homo sapiens sGC (which shares 99% sequence identity with R. norvegicus sGC) bears a striking resemblance to the smaller heart-shaped domain in our density maps (9). The “fit-in-map” function of UCSF Chimera (University of California, San Francisco Chimera, 19) was used to automatically dock the crystal structure into the EM density maps which yielded fit correlations between 0.97 and 0.99. In the docked position, the N-terminal end of the domain points toward the connection with the coiled-coil stalk (Fig. 4).
Fig. 4.

The higher-order domain architecture of sGC. Homology models of the individual domains were fit into representative EM maps exhibiting two conformational extremes: the extended conformation (A and B) and a bent conformation (C and D). The catalytic dimer fits in the heart-shaped density, and a parallel coiled coil connects it to the PAS/H-NOX cluster. The heme-associated helix (red) of the β1 H-NOX domain is positioned to interact with PAS dimer. The β1 H-NOX domain is also poised to interact directly with the α1 catalytic domain in the bent conformation. (A and C) Space-filling representation of the complete sGC structure emphasizing the striking consistency with the EM density. (B and D) Fit of the homology models (displayed as ribbons) within the EM density map.
Helical Coiled-Coil Domain Forms the Elongated Stalk and Provides Two Points of Flexible Movement and Rotation.
The structure of the rat sGC β1 monomer helical domain was previously determined and is formed by a long 13-turn helix followed by a shorter 2.5-turn helix (8). Although crystallized as an antiparallel homodimer, its arrangement in native sGC is considered to be a parallel heterodimer (8, 12, 20). The thin stalk of density observed in our 3D reconstructions accounts for this coiled coil, and a parallel arrangement of the helices is confirmed as it is the only way all of the domains can fit into the overall density with the termini aligned correctly. This organization is further supported by the fact that the length of the linkers anchoring the helical domain to the rest of the structure is insufficient to allow an antiparallel arrangement, as previously suggested (8). The helical domains were modeled as a coiled coil using homology modeling and structural prediction software (Materials and Methods). The short, C-terminal helices of the helical domains are positioned directly at the N-terminal ends of catalytic domains. These results are consistent with HDX data, which demonstrated that two discrete regions of the catalytic domain are protected by the helical domain (13). These attachment sites between the helical and catalytic domains provide a flexible linker, allowing movement between the two domains.
PAS and H-NOX Domains Cluster Together in the Larger Four-Lobed Density.
The remaining H-NOX and PAS domains fill the four lobes of the larger, 90 × 35 × 45 Å density. The overall 3D shape of this region of density closely resembles the previously reported SAXS envelope obtained for a truncated construct of Manduca sexta sGC that did not include the catalytic domain (12). The volume of the corresponding EM density is slightly smaller than the SAXS envelope (115 × 70 × 90 Å), likely due to the presence of the catalytic domain that prevents the helical domain from folding over on the PAS/H-NOX cluster.
Homology models of the H-NOX and PAS domains were built for each subunit with Modeler (21), using the crystal structure of a related bacterial H-NOX protein (PDB: 2O09) (6) and the recently crystallized M. sexta PAS domain (PDB: 4GJ4) (7), respectively. To model the dimeric organization of the PAS domains, the resulting homology models were superimposed on a histidine kinase PAS dimer (PDB: 2P04) (22), exhibiting 35% sequence identity to the rat sGC PAS domain. Finally, the PAS dimer and H-NOX domains were fit into the density while integrating the available structural and biochemical data to guide the placement (Materials and Methods).
As directly observed in the 2D class averages, the larger density consists of four distinct modules. The two slightly smaller round domains are closely associated, symmetrically organized, and paired together. We attributed these densities to the PAS dimer, which is therefore located near the projected connection point to the helical domains. The two more elongated densities on the periphery are assigned to the H-NOX domains. Our assignment is similar to a proposed model based primarily on truncation, cross-linking, and SAXS data (12). Due to the crossing of the helical domains in the coiled-coil arrangement, the β1 H-NOX domain is poised to interact with the α1 subunit of the catalytic dimer, consistent with mounting structural and biochemical data (11–13). The EM maps indicate asymmetry in the elongated lobes: one is slightly smaller than the other. We have tentatively assigned the smaller lobe to the β1 H-NOX domain, which contains 66 fewer residues than its α1 counterpart. The α1 N-terminal extension, like many other targeting sequences, is thought to be largely unstructured (14). Thus, it would not provide a well-defined density in a single-particle EM map, making it difficult to unequivocally assign which H-NOX domain occupies which lobe. The two H-NOX domains were placed symmetrically on either side of the PAS dimer, relying on the heme-associated helix of the β1 H-NOX domain to guide the placement, as its direct interactions with the PAS domain have been recently characterized (13) (Fig. 4).
The PAS dimer was oriented so that the C termini face toward the helical domain such that the ∼20 residue linker region would be sufficient to connect to the N termini of the helical domain. As the PAS dimer is nearly symmetrical, the two reported cross-links (12) between the α1 PAS (homologous to G337 and T340 in rat) and β1 H-NOX domain (K170) were used to guide the placement of the α and β subunits of the PAS domains relative to the H-NOX domains (Fig. 4).
Ligand Binding Retains the Conformational Flexibility of sGC.
We also investigated the structure of sGC in complex with various ligands to assess their influence on the structure and flexibility of the enzyme. Three additional RCT datasets were collected for (i) sGC in complex with NO, (ii) sGC in complex with the noncyclizable GTP analog, GPCPP, and (iii) sGC in complex with NO and GPCPP. The experimental conditions were identical to the ones used for the isolated sGC dataset (Materials and Methods). We obtained 81, 49, and 58 reconstructions for the three experiments, respectively. All structures are consistent with the overall shape and architecture of the sGC holoenzyme: a smaller heart domain connected by a thin density to a four-lobed cluster (Fig. S2). To understand and compare the range of motion of the domains in each of these experiments, the maps for all 3D reconstructions were aligned based on the catalytic domain as described before. The aligned maps for each separate experiment were then merged together, yielding a density representing the conformational space sampled by the various sGC conformational snapshots (Fig. 5). Overall, the merged maps indicate a similar range of accessible conformations under all experimental conditions examined.
Fig. 5.
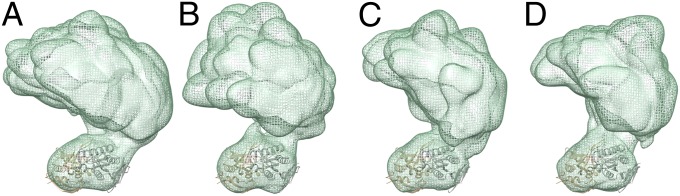
Merged maps illustrating the range of motion available to sGC when free or ligand-bound. Maps were aligned to the catalytic domain and show a similar range of motion under several experimental conditions. (A) The sGC holoenzyme. (B) NO-bound sGC. (C) GPCPP-bound sGC. (D) GPCPP and NO-bound sGC.
Discussion
The structural analysis presented here not only provides a comprehensive assignment of the higher-order domain architecture within the uniquely shaped sGC holoenzyme, but it represents a turning point in the understanding of the overall structure. The models proposed thus far assumed that the holoenzyme exists in a single conformation, making it difficult to diagram a model that reconciled the numerous interdomain contacts known to occur based on the literature. Using EM we show that sGC is instead a highly dynamic protein, sampling a wide range of conformations. NO addition does not lock the protein into a defined conformation, but instead sGC appears to retain its large-scale flexibility. Because the low pH of the uranyl formate negative stain used could potentially have a perturbing effect (23) on possible interactions between the N- and C-terminal halves of sGC, we examined both free sGC and NO-bound sGC using the neutral negative-stain nanoW (pH = 6.8). The 2D class averages resulting from images obtained in nanoW revealed a similar range of conformations both for the free and NO-bound sGC, suggesting the range of conformations is not perturbed by the low pH of the stain. Consequently, we propose that the activation mechanism involves a combination of small-scale intradomain reorientations mediated by local changes in flexibility at interdomain articulation points. Specifically, we suggest that flexibility accessible to the PAS–helical and helical–catalytic interdomain linkers is involved in activating the cyclase domain. Furthermore, we hypothesize that the dynamic architecture of sGC stems from evolutionary adaptation to balance a large surface area to intercept small-molecule signals with the ability to rapidly and precisely transmit signals between domains.
The structural interpretation provided by the EM analysis coalesces numerous pieces of biochemical and structural data and consolidates proposed models to visualize the domain arrangement and interactions. We clearly establish here that the PAS and H-NOX domains directly interact, as previously suggested (12, 13). Moreover, we show that these two domains form a tight cluster, sharing large surfaces of interactions, and allow each H-NOX domain to interact with both the α1 and β1 PAS domain. This close configuration would allow small-scale changes in the H-NOX domain to be quickly recognized by the adjacent PAS. A growing body of evidence also supports a direct interaction between the two opposite ends of the protein, the N-terminal H-NOX and C-terminal catalytic domain (10, 13, 24). The EM data demonstrates how this contact is made possible due to the marked flexibility of the enzyme.
As previously mentioned, the two cross-links reported to form between the β1 H-NOX and α1 PAS domain were used to guide the orientation of the PAS dimer in relation to the H-NOX domain. The other cross-links found between domains in the study by Fritz et al. (12) are largely consistent with our data. For instance, several cross-links were found between the β1 H-NOX domain and the α1 helical domain. In agreement with these results, the orientation of the β1 H-NOX in our reconstructions positions the β1 residues contributing to these cross-links on the bottom surface of the four-lobed density poised to transiently connect with the helical domain (Fig. 6). Our results also suggest that these cross-links do not occur simultaneously, but instead illustrate several possible interactions as a result of the numerous different conformations that sGC can adopt. Furthermore the similarly positioned cross-links between the bottom of the α1 H-NOX and β1 helical domain support the idea that the PAS/H-NOX cluster bends toward both sides of the catalytic domain, allowing each H-NOX domain to contact the catalytic domain of the opposite subunit. A single cross-link was found between the PAS and helical domain, which is consistent with the out-of-plane motions observed in our studies. Finally, the detection of two cross-links between the two H-NOX domains is more surprising. However, we note that the residues contributing to these cross-links are in the extension region of the α1 subunit, and it is probable that this region is an unstructured extension (14) that may extend so as to come into proximity of the β1 H-NOX domain during cross-linking.
Fig. 6.

Previously found cross-links mapped onto model. Domains colored as previously described. Cross-links are shown as blue dashed lines. Residues contributing to cross-links within the PAS/H-NOX cluster are shown in magenta. Residues contributing to cross-links between the PAS/H-NOX cluster and the coiled coil are shown in green. The conformational space sampled by sGC in our reconstructions allows rationalizing a large amount of available cross-linking data when taking into account the fact that not all cross-links with the helical domains occur simultaneously.
The mechanism behind the NO activation of sGC remains a central question in the NO/cGMP-signaling pathway. The allosteric pathway by which NO binding is transmitted to the catalytic domain remains unclear. Comparisons of unliganded and NO-bound sGC reveal that both signaling states share a similar dynamic range of accessible conformations with two discreet points of articulation. The NO-bound state is clearly not locked into one static conformation. Instead, the signaling mechanism may leverage transitions in the degree of flexibility in the two key points of articulation: the PAS–helical linker and the helical–catalytic domain linker. The conformational flexibility accessible to these linkers supports a role for allosteric communication through the PAS and helical domains. The functional importance of the PAS and helical domains in signal communication was previously revealed by mutational screens (25). In addition, the flexibility of the PAS–helical junction interestingly parallels the results of a recent HDX study that revealed NO-induced increases in conformational flexibility at the linker between the PAS and helical domains (14). Indeed, in several archetypal PAS domains, unfolding at the helical termini is thought to be important for signal communication (26, 27). Here, the putative order/disorder transitions within the PAS–helical linker may be obscured by the intrinsic dynamics of sGC revealed in our series of conformational snapshots. Interestingly, in our maps of both unliganded and NO-bound sGC we observe that the H-NOX domain can make direct contact with the catalytic domain, which could contribute to allosteric control of the catalytic domain by the H-NOX. Busker et al. (28) recently proposed a mechanism whereby sGC activation is mediated by both direct interaction between the H-NOX and catalytic domains as well as allosteric interactions through the intervening PAS and helical domains (28). The apparent crucial role of flexible linkers as well as their dynamic fluctuations observed for sGC are common themes among signaling proteins (29).
We propose that the biological drive for this uniquely shaped, dynamic protein stems from a necessity to react rapidly and accurately to a myriad of external signals to carefully control cGMP production. From an evolutionary standpoint, we speculate that individual ancestral proteins are now organized as successive domains belonging to a single polypeptide chain while preserving the flexibility to enable rapid signal transduction. Sequence profile analysis of bacterial H-NOX domains showed that they are found in predicted operons encoding genes for two component systems and diguanylate cyclases or phosphodiesterases. Furthermore, fused PAS/coiled-coil domain genes are frequently in close proximity to H-NOX genes in bacterial genomes (30). In sGC, multiple protein domains must transfer signals to each other via direct and allosteric signaling mechanism involving many regulatory interfaces. In addition to the heme-based NO-binding site on the H-NOX domain and the active site in the catalytic domain, it has been suggested that sGC has an additional NO-binding site, an allosteric nucleotide-binding site in the catalytic domain (31), a site for heme-independent small-molecule activator binding [such as YC-1 (5-[1-(phenylmethyl)-1H-indazol-3-yl]-2-furanmethanol; 32, 33)], as well as an internal cavity in the PAS domain that may bind ligands (7). All of these sites must be accessible to allow modulation of the sGC catalytic activity through integration of the external signals. For this reason, a more globular structure that ensures direct rapid domain to domain signaling might not be sufficient to allow the necessary surfaces to remain exposed. We speculate that the complexity and molecular crowding within eukaryotic cells favored the evolution of a modular protein possessing domains that can subtly rearrange in relation to one another to rapidly transfer signals. This allows sGC to react within milliseconds in vivo to picomolar levels of NO gas, triggering a 100-fold increase in cGMP synthesis, as well as to a myriad of other external signals further modulating its enzymatic activity.
In summary, combining EM with previous biochemical and structural data has enabled us to describe the architecture and domain organization of the full-length sGC holoenzyme. The large exposed surface area of the protein is such that it can optimize its response to various external small-molecular signals. At the same time, the overall dynamics of the sGC holoenzyme allow for the β1 H-NOX sensor domain to directly contact the PAS dimer, the α1 helical, and the α1 catalytic domain. These numerous direct contacts support a model by which NO-binding is communicated to the catalytic domain through the PAS–helical domain as well as directly by the H-NOX domain. The molecular architecture and pronounced flexibility observed provides a significant step forward in understanding the mechanism of NO signaling.
Materials and Methods
Cloning, Expression, and Purification of sGC.
Full-length R. norvegicus α1 β1 sGC was expressed and purified from Sf9 cells using the Bac-to-Bac baculovirus expression system (Invitrogen), as previously described (13).
Activators Binding.
Full-length α1 β1 sGC in 50 mM Hepes, 50 mM NaCl, 5 mM DTT, pH 7.5 was preequilibrated with 300 μM GPCPP. NO-bound sGC was formed by incubating with excess Diethylamine NONOate (DEA/NO) (300 μM).
Negative-Stain EM Specimen Preparation.
For all samples, 3 µL of the sample was applied to a glow-discharged C-flat grid with 2-µm–diameter holes overlaid by a thin (∼1.5 nm) layer of continuous carbon. The specimen was stained with a solution containing 2% (wt/vol) uranyl formate as previously described (34).
Electron Microscopy.
Data were acquired using a Tecnai F20 Twin transmission electron microscope operating at 200 kV with a dose of ∼35 e−/Å2 with a nominal focus range from 1.2 to 2.2 below true focus. Tilt pair micrographs at 0° and −55° at a nominal magnification of 80,000× (pixel size = 0.106 nm) were collected using the automated software Leginon (15). All images were recorded on a 4 × 4-k Tietz F416 CMOS detector using Leginon software in a single session for each experiment ranging from 1 to 3 d. For ligand-free sGC, a total of 1,102 micrograph pairs (2,204 micrographs) were collected; for NO-bound sGC, 1,118 pairs were collected; for GPCPP-bound sGC, 1,096 pairs were collected; and for GPCPP and NO-bound sGC, 826 pairs were collected.
Image Processing and Reconstruction.
Experimental data were processed using the Appion software package (16), which interfaces with the Leginon database infrastructure. The contrast transfer function (CTF) for each micrograph was estimated using CTFFIND3 (35). An iterative series of reference-free and reference-based sorting and alignment methods were used to effectively deconvolute heterogeneity within the sample. For ligand-free sGC, initial particle picking was done on untilted micrographs using a reference free difference of Gaussians approach (36). These particles were sorted, aligned, and clustered using Xmipp Clustering 2D Alignment (CL2D) (37) to generate templates. These templates were used as references to repick particles on untilted and tilted micrographs which yielded a total of 46,688 particle pairs that were extracted at a box size of 224 pixels. The new untilted particles were aligned and classified using Xmipp CL2D, and the 2D averages were visually inspected to remove classes containing predominantly false positive picks or badly aligned particles. The remaining 2D class averages were used to create a new set of templates. These 32 templates were used for iterative rounds of multireference alignment using SPIDER (38) and multivariate statistical analysis using IMAGIC (39). RCT reconstructions (17, 18) were carried out using the initial model pipeline within Appion, which gave a total of 114 independent 3D maps, which ranged in resolution from 25 to 40 Å based on the 0.5 Fourier shell correlation criteria. The maps were visually inspected for a recognizable catalytic domain and H-NOX/PAS cluster, which resulted in 94 acceptable reconstructions. Alignment of the volumes to the catalytic domain was performed automatically in Chimera (19) using the fit-in-map function and the crystal structure of the catalytic domain (9), low-pass-filtered to 27 Å. Volumes were merged together according to the formalism: output-pixel = max {input-pixelvolume1, input-pixelvolume2} to create the merged map. All electron density maps were displayed and rendered using Chimera.
The data involving sGC bound to ligands were processed in a similar manner to ligand-free sGC. New, separate templates and references were used for each experiment. For sGC + NO, 35,399 particle pairs led to 89 3D maps; for sGC + GPCPP, 51,471 particle pairs led to 49 3D maps; and for sGC + NO + GPCPP, 34,643 particle pairs led to 58 3D maps.
Homology Models.
Coiled coil: The existing crystal structure of the rat β1 helical monomer (PDB: 3HLS) (8) was used to model the α1 monomer using the SWISS-MODEL workspace (40). The α1 β1 coiled coil was constructed using the ClusPro 2.0 server, selecting the highest populated class with a parallel orientation (41).
PAS Dimer: First, individual rat α1 and β1 subunits we modeled using the crystal structure of the β1 M. sexta PAS domain (PDB: 4GJ4) (7) in the SWISS-MODEL workspace (40). The dimeric arrangement was modeled using a Nostoc punctiforme PAS dimer as a scaffold (22), further resolving structural elements using energy minimization in Coot (Crystallographic Object-Oriented Toolkit) (42).
H-NOX: Homology models of the sGC α1 and β1 H-NOX monomers were constructed from the structure of a bacterial H-NOX (PDB: 2O09) using Robetta (43) (14).
Supplementary Material
Acknowledgments
We thank Anchi Cheng and Sargis Dallakyan (National Resource for Automated Molecular Microscopy) for technical support. We also thank Jean-Christophe Ducom, head of the high-performance computing facility for his assistance. Finally, we thank members of the Automated Molecular Imaging group for comments on the manuscript. M.G.C. received financial support from an American Heart Association predoctoral fellowship. E.S.U. received financial support from the National Institutes of Health Postdoctoral Fellowship 5F32GM093564. The work presented here was conducted at the National Resource for Automated Molecular Microscopy, which is supported by the National Institute of General Medical Sciences (9 P41 GM103310).
Footnotes
Data deposition: The structures have been deposited in the Electron Microscopy Data Bank (accession nos. EMD-5861–EMD-5884).
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1400711111/-/DCSupplemental.
References
- 1.Lucas KA, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52(3):375–414. [PubMed] [Google Scholar]
- 2.Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27(11):2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bredt DS. Endogenous nitric oxide synthesis: Biological functions and pathophysiology. Free Radic Res. 1999;31(6):577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- 4.Friebe A, Mergia E, Dangel O, Lange A, Koesling D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc Natl Acad Sci USA. 2007;104(18):7699–7704. doi: 10.1073/pnas.0609778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conole D, Scott LJ. Riociguat: First global approval. Drugs. 2013;73(17):1967–1975. doi: 10.1007/s40265-013-0149-5. [DOI] [PubMed] [Google Scholar]
- 6.Ma X, Sayed N, Beuve A, van den Akker F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007;26(2):578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purohit R, Weichsel A, Montfort WR. Crystal structure of the Alpha subunit PAS domain from soluble guanylyl cyclase. Protein Sci. 2013;22(10):1439–1444. doi: 10.1002/pro.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma X, Beuve A, van den Akker F. Crystal structure of the signaling helix coiled-coil domain of the beta1 subunit of the soluble guanylyl cyclase. BMC Struct Biol. 2010;10:2. doi: 10.1186/1472-6807-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allerston CK, von Delft F, Gileadi O. Crystal structures of the catalytic domain of human soluble guanylate cyclase. PLoS ONE. 2013;8(3):e57644. doi: 10.1371/journal.pone.0057644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haase T, Haase N, Kraehling JR, Behrends S. Fluorescent fusion proteins of soluble guanylyl cyclase indicate proximity of the heme nitric oxide domain and catalytic domain. PLoS ONE. 2010;5(7):e11617. doi: 10.1371/journal.pone.0011617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derbyshire ER, et al. Probing domain interactions in soluble guanylate cyclase. Biochemistry. 2011;50(20):4281–4290. doi: 10.1021/bi200341b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fritz BG, et al. Molecular model of a soluble guanylyl cyclase fragment determined by small-angle X-ray scattering and chemical cross-linking. Biochemistry. 2013;52(9):1568–1582. doi: 10.1021/bi301570m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Underbakke ES, Iavarone AT, Marletta MA. Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc Natl Acad Sci USA. 2013;110(17):6777–6782. doi: 10.1073/pnas.1301934110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Underbakke ES, et al. Nitric oxide-induced conformational changes in soluble guanylate cyclase. Structure. 2014 doi: 10.1016/j.str.2014.01.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suloway C, et al. Automated molecular microscopy: The new Leginon system. J Struct Biol. 2005;151(1):41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Lander GC, et al. Appion: An integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166(1):95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J Microsc. 1987;146(Pt 2):113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- 18.Voss NR, et al. A toolbox for ab initio 3-D reconstructions in single-particle electron microscopy. J Struct Biol. 2010;169(3):389–398. doi: 10.1016/j.jsb.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 20.Anantharaman V, Balaji S, Aravind L. The signaling helix: A common functional theme in diverse signaling proteins. Biol Direct. 2006;1:25. doi: 10.1186/1745-6150-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eswar N, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;2(2.9) doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 22.Ma X, Sayed N, Baskaran P, Beuve A, van den Akker F. PAS-mediated dimerization of soluble guanylyl cyclase revealed by signal transduction histidine kinase domain crystal structure. J Biol Chem. 2008;283(2):1167–1178. doi: 10.1074/jbc.M706218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Carlo S, Harris JR. Negative staining and cryo-negative staining of macromolecules and viruses for TEM. Micron. 2011;42(2):117–131. doi: 10.1016/j.micron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winger JA, Marletta MA. Expression and characterization of the catalytic domains of soluble guanylate cyclase: Interaction with the heme domain. Biochemistry. 2005;44(10):4083–4090. doi: 10.1021/bi047601d. [DOI] [PubMed] [Google Scholar]
- 25.Rothkegel C, et al. Dimerization region of soluble guanylate cyclase characterized by bimolecular fluorescence complementation in vivo. Mol Pharmacol. 2007;72(5):1181–1190. doi: 10.1124/mol.107.036368. [DOI] [PubMed] [Google Scholar]
- 26.Harper SM, Neil LC, Gardner KH. Structural basis of a phototropin light switch. Science. 2003;301(5639):1541–1544. doi: 10.1126/science.1086810. [DOI] [PubMed] [Google Scholar]
- 27.Lee BC, Croonquist PA, Sosnick TR, Hoff WD. PAS domain receptor photoactive yellow protein is converted to a molten globule state upon activation. J Biol Chem. 2001;276(24):20821–20823. doi: 10.1074/jbc.C100106200. [DOI] [PubMed] [Google Scholar]
- 28.Busker M, Neidhardt I, Behrends S. Nitric oxide activation of guanylate cyclase pushes the α1 signaling helix and the β1 heme binding domain closer to the substrate binding site. J Biol Chem. 2014;289(1):476–484. doi: 10.1074/jbc.M113.504472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13(9):831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angermayr M, Schwerdffeger K, Bandlow W. A nucleosome-free dG-dC-rich sequence element promotes constitutive transcription of the essential yeast RIO1 gene. Biol Chem. 2003;384(9):1287–1292. doi: 10.1515/BC.2003.143. [DOI] [PubMed] [Google Scholar]
- 31.Cary SPL, Winger JA, Marletta MA. Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proc Natl Acad Sci USA. 2005;102(37):13064–13069. doi: 10.1073/pnas.0506289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ko FN, Wu CC, Kuo SC, Lee FY, Teng CM. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84(12):4226–4233. [PubMed] [Google Scholar]
- 33.Purohit R, et al. YC-1 binding to the β subunit of soluble guanylyl cyclase overcomes allosteric inhibition by the α subunit. Biochemistry. 2014;53(1):101–114. doi: 10.1021/bi4015133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification: Powerful tools in modern electron microscopy. Biol Proced Online. 2004;6(1):23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142(3):334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 36.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: Software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166(2):205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorzano COS, et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. J Struct Biol. 2010;171(2):197–206. doi: 10.1016/j.jsb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116(1):190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 39.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116(1):17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 40.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 41.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: A fully automated algorithm for protein–protein docking. Nucleic Acids Res. 2004;32:W96–W99. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526-31. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.