

ABSTRACT
The urinary tract is one of the most common sites of infection in humans, and uropathogenic Escherichia coli (UPEC) is the main causative agent of urinary tract infections. Bacteria colonizing the urinary tract face extremely low iron availability. To counteract this, UPEC expresses a wide variety of iron acquisition systems. To exploit iron acquisition in UPEC as a global target for small-molecule inhibition, we developed and carried out a whole-cell growth-based high throughput screen of 149,243 compounds. Our primary assay was carried out under iron-limiting conditions. Hits in the primary screen were assayed using two counterscreens that ruled out iron chelators and compounds that inhibit growth by means other than inhibition of iron acquisition. We determined dose-response curves under two different iron conditions and purchased fresh compounds for selected hits. After retesting dose-response relationships, we identified 16 compounds that arrest growth of UPEC only under iron-limiting conditions. All compounds are bacteriostatic and do not inhibit proton motive force. A loss-of-target strategy was employed to identify the cellular target of these inhibitors. Two compounds lost inhibitory activity against a strain lacking TonB and were shown to inhibit irreversible adsorption of a TonB-dependent bacteriophage. Our results validate iron acquisition as a target for antibacterial strategies against UPEC and identify TonB as one of the cellular targets.
IMPORTANCE
Half of women will suffer at least one episode of urinary tract infection (UTI) during their lifetime. The current treatment for UTI involves antibiotic therapy. Resistance to currently used antibiotics has steadily increased over the last decade, generating a pressing need for the development of new therapeutic agents. Since iron is essential for colonization and scarce in the urinary tract, targeting iron acquisition would seem to be an attractive strategy. However, the multiplicity and redundancy of iron acquisition systems in uropathogenic Escherichia coli (UPEC) make it difficult to pinpoint a specific cellular target. Here, we identified 16 iron acquisition inhibitors through a whole-cell high-throughput screen, validating iron acquisition as a target for antibacterial strategies against UPEC. We also identified the cellular target of two of the inhibitors as the TonB system.
INTRODUCTION
Urinary tract infections (UTIs) are the second-most-common bacterial infection in humans after those affecting the respiratory tract, and approximately 50% of women will experience at least one episode of UTI during their lifetime. The most common etiological agent of UTIs is uropathogenic Escherichia coli (UPEC). The current routine treatment for UTIs is antibiotic therapy, most commonly trimethoprim-sulfamethoxazole (TMP-SMX) or ciprofloxacin. Resistance to these first-line antibiotics has steadily and rapidly risen over the last decade. A recent survey of more than 12 million clinical isolates across the United States from 2000 to 2011 detected resistance to ciprofloxacin and TMP-SMX in 17.1% and 24.2% of UPEC strains, respectively (1). This study tested 7 other antibiotics, including nitrofurantoin and ceftriaxone, and demonstrated that resistance to every antibiotic tested had also increased (1). Even more troubling is the rate of multidrug resistance among UPEC isolates. A recent international study documented that more than 10% of E. coli cystitis isolates are resistant to at least three different classes of antimicrobial agents (2). In the face of rising antibiotic resistance, there is a pressing need for development of new therapeutic agents against UPEC beyond existing antibiotics.
Iron is essential for bacterial growth, and the urinary tract is iron depleted. Even though the average human body contains as much as 5 g of the metal, iron metabolism is tightly controlled. Upon intestinal absorption, ferric iron is reduced to ferrous iron and transported into the enterocyte by the divalent metal ion transporter Nramp2 (3). After this, it can exit the cell bound to transferrin and is delivered to other cells by receptor-mediated endocytosis. If transferrin capacity is exceeded, iron can be chelated with lower affinity by other plasma molecules, including albumin, citrate, and certain amino acids (3). The majority of iron is found in erythrocytes complexed to heme moieties in hemoglobin. Alternatively, iron is found incorporated into iron-sulfur clusters or stored intracellularly as ferritin. Extracellular iron can also be bound to the antimicrobial peptide lactoferrin (4), which binds iron with high affinity at mucosal surfaces. Upon infection, the host exerts nutritional immunity by increasing the concentration of iron-binding proteins, further lowering the free iron concentration. For these reasons, the human body resembles an iron-depleted environment for bacteria. To colonize this hostile environment, UPEC produces an extensive array of iron acquisition systems (Fig. 1). Iron acquisition requires synthesis pathways for multiple siderophores, their corresponding uptake and processing operons, and receptors for exogenous siderophores as well. Siderophores chelate iron with extremely high affinity and are able to extract iron bound to host proteins. There are also two heme receptors that we have previously shown to be important at different stages of infection (5), as well as pathways to metabolize heme-bound iron. Finally, there are genes related to uptake of ferric citrate and ferrous iron and several outer membrane proteins of unknown function that are expressed under low-iron conditions.
FIG 1 .

Iron acquisition systems of uropathogenic E. coli. The diagram of an E. coli CFT073 bacterium depicts annotated iron acquisition systems. The inner box includes genes involved in siderophore biosynthesis and regulation. The TonB-ExbBD complex energizes transport through the outer membrane.
Many of these iron uptake systems rely on the energy generated by the proton motive force (PMF) across the bacterial inner membrane. The TonB system, composed of the three transmembrane proteins TonB, ExbB, and ExbD (represented in red in Fig. 1), harnesses this energy across the periplasm to promote transport across TonB-dependent receptors located in the outer membrane (6). The TonB system has been linked to uptake of iron sources, vitamin B12, nickel, certain carbohydrates, phages, and colicins (7, 8). Despite extensive work on the subject, the exact mechanism by which the TonB system transduces energy and the stoichiometry of the different components within the complex remain unclear. Several mechanisms of action for the TonB system have been proposed and can be divided into mechanical models, in which TonB remains constantly associated with the cell membrane, and the shuttle model, where TonB dissociates its N-terminal domain from the cell membrane and shuttles to the outer membrane (9). The shuttle model has recently been ruled out (10). Other models include the propeller model, the pulling model, and the periplasmic binding protein (PBP)-assisted model. The propeller model was suggested based on the homology of ExbBD to flagellar motor proteins and proposes that ExbBD use the PMF to generate a rotatory motion of TonB, which, through association with the TonB box of an outer membrane receptor, causes translocation of its substrate into the periplasm (11). This model faces several challenges, including the lack of anchors to prevent ExbBD from rotating along with TonB (12) and the fact that TonB is most probably a monomer in vivo (13). The pulling model proposes that TonB causes the plug domain of the outer membrane receptor to partially unfold, which in turn allows for translocation of its substrate (14). Finally, the PBP-assisted model was proposed when it was shown that TonB and PBPs could associate (15, 16). TonB was proposed to act as a scaffold to present the PBP FhuD to the outer membrane receptor FhuA to facilitate entry of its siderophore. Subsequently, TonB would transfer FhuD to its inner membrane transporter, FhuBC, to carry the siderophore into the cytoplasm (15). These mechanisms still lack in vivo experimental data to support them.
Most bacteria require micromolar concentrations of iron to grow, because iron serves as a cofactor for numerous biochemical processes. Because lack of iron arrests bacterial growth and functional assimilation pathways are required for host colonization, iron acquisition is an attractive target for antibacterial strategies. Bacterial iron acquisition is already a natural target of the host’s immune system, including a hypoferremic response generated by enhanced iron storage in transport proteins and siderophore binding by siderocalin and lipocalin (17). Thus far, only a limited number of strategies for iron-dependent pathogen control have been explored, including the use of nonmetabolizable chelators or antagonists to compete with siderophores (18). Another strategy involves inhibition of different steps in siderophore biosynthesis or assimilation. Most of the work has focused on enzymes involved in biosynthesis and incorporation of salicyl and dihydroxybenzoate moieties, since such functionalities are absent from human metabolism. A recent review of iron assimilation as a target (19) highlights successful identification of nanomolar affinity inhibitors of siderophore biosynthesis enzymes in Acinetobacter baumannii, Pseudomonas aeruginosa, Yersinia pestis, and Mycobacterium tuberculosis. In all of these cases, the inhibitors identified targeted an enzyme in a single biosynthetic pathway, and that was sufficient to negatively affect growth of these microorganisms under iron-limiting conditions.
In UPEC, however, deletion of single siderophore synthesis pathways does not generally affect growth under low-iron conditions (see Fig. S1 in the supplemental material) and does not impair the ability to colonize the murine urinary tract, although strains carrying mutations in certain receptors lose fitness in vivo (20). A strain with multiple mutations rendering it unable to synthesize siderophores, on the other hand, displays colonization defects in bladder and kidney (20). These results highlight the requirement for effective iron acquisition in pathogenesis but at the same time imply that trying to find inhibitors for specific enzymes in UPEC siderophore synthesis pathways would be an ineffective approach. Multiplicity and redundancy of systems make it impossible to develop assays for any single pathway.
For these reasons, we decided to develop and carry out a growth-based cell assay to test for iron acquisition as a whole, under conditions in which all iron assimilation pathways are upregulated. The vast majority of iron acquisition systems are under transcriptional control of the ferric uptake regulator (Fur), which ensures expression only under iron-limiting conditions (21). Our approach did the following: (i) confirmed that iron acquisition can be a target for small-molecule inhibitors in UPEC, (ii) identified 16 compounds that arrest growth of UPEC under iron-limiting conditions, and (iii) identified the TonB system as the likely target for two compounds.
RESULTS
The goal of this study was to validate iron acquisition systems as a potential target for antimicrobial strategies and to identify small molecules that inhibit iron acquisition in UPEC. We designed and implemented a cell-based high-throughput screen (HTS) to identify such compounds. The assays involved the following: (i) primary HTS hit identification (single point), (ii) hit confirmation in triplicate, (iii) two counterscreens in triplicate to identify compounds that chelate iron or that arrest growth through means other than inhibition of iron acquisition, (iv) determination of the half-maximal activity concentration (AC50) values in low and high iron through 8-point dose-response curves (DRC) in duplicate, and finally (v) purchase and retest of fresh powder compounds in DRC in quadruplicate. Additionally, we tested compounds for activity against wild-type E. coli CFT073 and for proton motive force inhibition. We have also initiated target identification efforts involving a loss-of-target strategy and have identified the potential target for two compounds thus far.
Development of primary assay.
A ΔtolC permeability mutant of E. coli CFT073 that lacks a major efflux pump was constructed for use in the screening assay. Since TolC is involved in efflux of several structurally diverse antibiotics (22), we decided to test using a ΔtolC mutant to maximize the chances of reaching valid intracellular targets. We determined growth curves in commercially available morpholinepropanesulfonic acid (MOPS) minimal medium with no added iron (MOPS no iron) containing 0.2% glucose as the sole carbon source. As illustrated in Fig. 2A, wild-type (WT) E. coli CFT073 and its ΔtolC mutant grow at similar rates in MOPS no iron and in MOPS no iron supplemented with 3 µM FeSO4. Since the growth rate of both strains increases with addition of iron, we concluded that iron was a limiting nutrient in this medium. Growth was abolished if the iron chelator 2′,2′-dipyridyl (DIP) was added, indicating that MOPS no iron still contains traces of the metal ion (Fig. 2A). At this concentration, DIP had no additional toxic effects, since growth was restored by addition of excess iron both in rich medium (LB) and in MOPS no iron (see Fig. S2 in the supplemental material). This also demonstrated that a growth assay in iron-limiting medium could be used to test for compounds that inhibit iron acquisition, since the lack of available iron arrests growth. We then determined growth curves for the ΔtolC mutant in MOPS minimal medium supplemented with different concentrations of FeSO4 as the sole iron source (see Fig. S3). Cultures reached a maximum density in MOPS plus 1 µM FeSO4, demonstrating that in MOPS no iron medium, this metal ion is present at a growth-limiting concentration. Standard MOPS minimal medium contains 10 µM FeSO4. To test growth in a 384-well format, cultures of E. coli CFT073 ΔtolC were incubated in LB medium at 37°C overnight, harvested by centrifugation, and washed three times in MOPS no iron. CFT073 ΔtolC bacteria were used to inoculate 40-µl cultures in 384-well plates to test different inoculum sizes and DIP concentrations. Plates were incubated statically overnight at 37°C, and the OD600 was recorded. For the remaining experiments, an initial OD600 of 0.01 and a DIP concentration of 200 μM as the positive control were selected. Negative controls contained 0.5% dimethyl sulfoxide (DMSO).
FIG 2 .
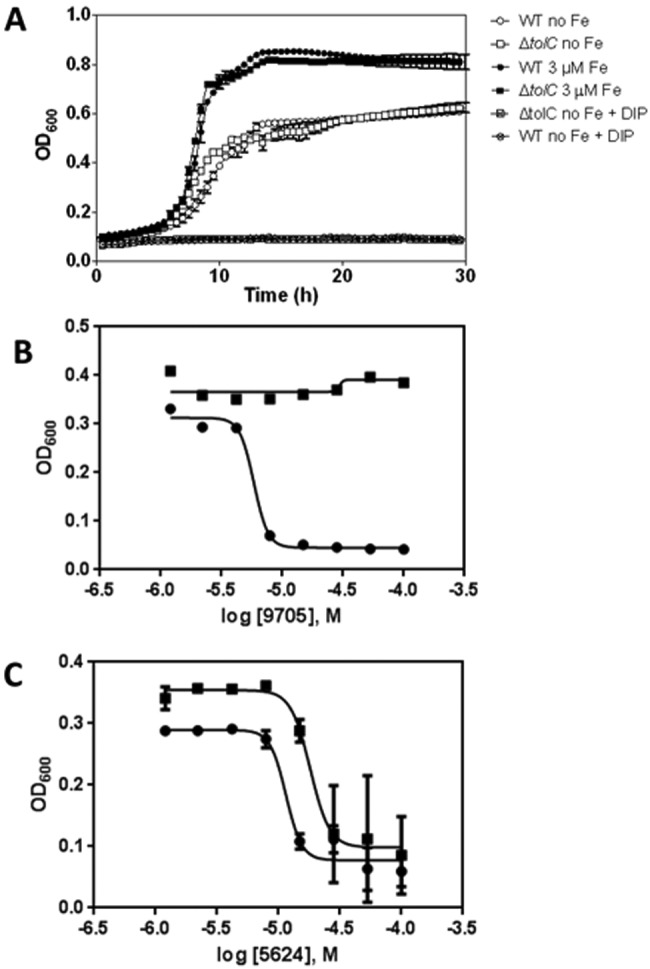
Growth curves of strain CFT073 ΔtolC::kan and representative dose-response curves of compounds confirmed by secondary assays. (A) Growth curves of E. coli CFT073 wild type and the ΔtolC mutant under different iron conditions. MOPS no iron was inoculated as described. “DIP” indicates addition of 200 µM dipyridyl to MOPS no iron. CFT073 ΔtolC::kan grows similarly to the WT under all iron conditions tested. Curves are means ± SD for 5 replicates. (B and C) Representative plots of dose-response curves (377 compounds tested; see attrition rates in Fig. S5 in the supplemental material) using strain CFT073 ΔtolC::kan. Plots were fit by nonlinear regression for sigmoidal kinetics. Each compound was confirmed in replicates (n = 4). The assay readout was optical density at 600 nm after a 16-h incubation in MOPS no iron (circles) or MOPS plus 3 µM FeSO4 (squares). (B) Representative plots of a compound for which pAC50 (MOPS no iron) minus pAC50 (MOPS with 3 µM FeSO4) is >1. Compounds in this category were selected for fresh powder purchase and retested in quadruplicate. (C) Representative plot of a compound for which pAC50 (MOPS no iron) minus pAC50 (MOPS 3 µM FeSO4) is >1. Compounds in this category were removed from further consideration.
Primary screen validation.
We used the primary screen to test two small collections of structurally diverse and biologically active molecules. The MS Spectrum 2000 compounds are represented by 50% drugs, 30% natural products, and 20% biologically active molecules, with 95% purity. The BioFocus NIH Clinical Collection (NCC) library includes 446 small molecules with a history of use in human clinical trials. We included 32 positive-control (200 µM DIP) and 32 negative-control (0.5% DMSO) wells on every plate. The average signal-to-background (S/B) ratio of the pilot assay was 6, with a coefficient of variation (CV) of 6%. The overall Z′ factor was 0.735, whereas the Z′ factor per plate averaged 0.91. Our hit rate for these two libraries combined was 5.4%. Overall, our pilot screen showed a robust assay with excellent reproducibility across plates.
Primary screen and counterscreens.
We ran the primary screen across the entire small-molecule library available at CCG (149,243 compounds) (see Fig. S4 in the supplemental material). The Z′ factor per plate averaged 0.86. Our hit rate was 1.6%, which amounted to 2,467 compounds. After confirmation using the primary assay in triplicate, the 1,728 remaining compounds (1.2%) were subjected to two counterscreens. The first counterscreen was a growth assay similar to the primary assay but with the addition of excess iron (40 µM). The second counterscreen was an iron chelation assay based on the chrome azurol S (CAS) shuttle assay (23). The compounds that displayed >20% activity of the positive controls in either counterscreen were removed from further consideration.
Dose-response analysis.
Triaged compounds with confirmed activity and successful counterscreening (377 compounds) were assayed in dose-response studies according to the screening protocol under two different iron concentrations: no added iron and 3 µM FeSO4. The concentration of small molecules was varied in 2-fold dilutions ranging from 100 to 0.39 µM. Data were plotted as a function of the concentration (µM) of small molecule (pAC) versus percent growth inhibition. Data from compounds that exhibited a sigmoidal dose-response curve were fit by nonlinear regression, and pAC50 values were calculated. Of these, we prioritized compounds for which the difference in pAC50 between medium with no iron and medium with 3 µM iron was higher than 1. From this assay, 202 compounds were confirmed by dose-response analysis, with AC50 values ranging from 0.8 to 80 µM, and 76 were prioritized for further study. Representative data from dose-response analyses are shown in Fig. 2B and C. Fresh powder could be obtained for 58 compounds and retested in dose-response analysis in duplicate. Sixteen compounds were confirmed as active and were studied further as described below. Structures of the 16 compounds and their AC50 are summarized in Fig. 3. A summary of attrition rates from screen to hit is depicted in Fig. S5 in the supplemental material and structures of the compounds.
FIG 3 .

Screening of 149,243 compounds for iron acquisition inhibition. Sixteen hits were identified and confirmed through determining the DRC for fresh powders. AC50 values represent the concentration required to inhibit 50% growth of CFT073 ΔtolC::kan in MOPS no iron. The four compounds in the black-line box are also active against CFT073 WT. The two compounds in the dotted-line box were identified as inhibitors of the TonB system. AC50 values are means ± SD for three to four replicates.
Inhibitors of iron acquisition are bacteriostatic.
We would expect inhibitors of iron acquisition identified through this screen to be bacteriostatic. To test this, we established growth curves in the presence of each of the compounds. For every molecule, we first confirmed that the addition of additional iron relieved inhibition, by culturing CFT073 ΔtolC in the presence of the compounds with or without added iron. All compounds failed to inhibit growth in the presence of 10 µM iron (data not shown). A representative plot of a growth curve for CFT073 ΔtolC in the presence of inhibitors of iron acquisition is shown in Fig. 4A. After 20 h of incubation at 37°C, 10 µM FeS04 was added to the cultures. This resulted in resumed growth for every individual compound tested, demonstrating that the 16 compounds identified through this screen are bacteriostatic.
FIG 4 .
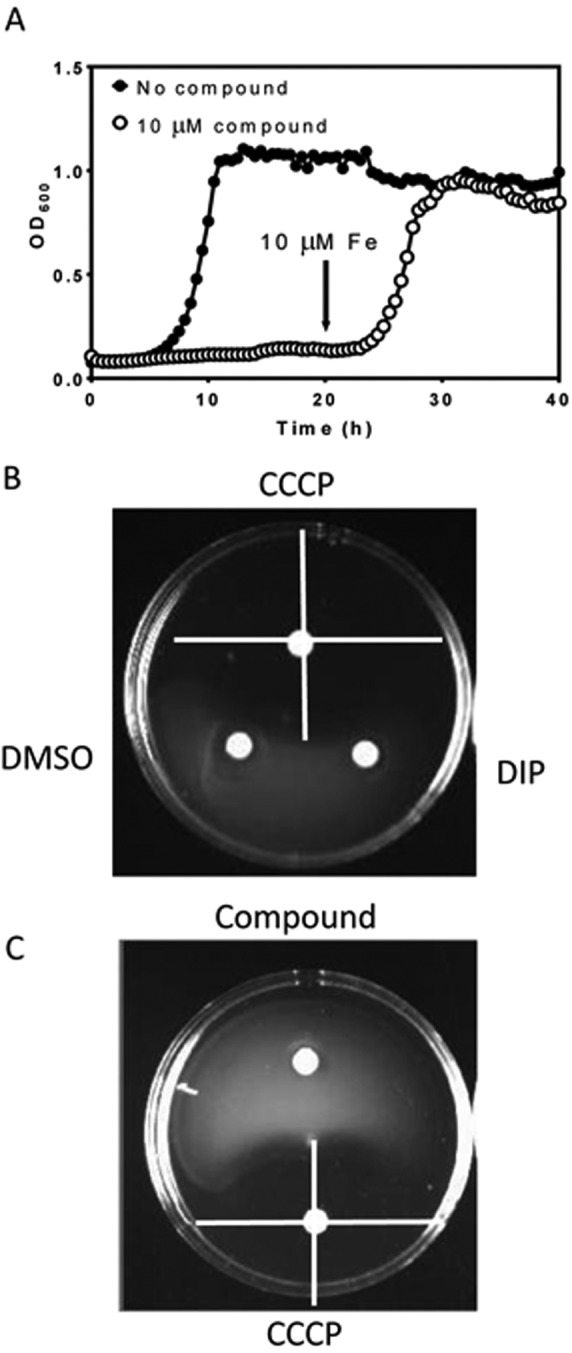
Iron acquisition inhibitors are bacteriostatic and do not inhibit the proton motive force. (A) Representative growth curves of E. coli CFT073 ΔtolC in the presence of iron acquisition inhibitors. Cultures in MOPS with (closed circles) or without (open circles) additional iron were allowed to grow for 40 h in the presence of each of the compounds. For all compounds, growth was not inhibited when excess iron was added to the medium. At 20 h, 10 µM FeSO4 was added. Addition of excess iron to E. coli CFT073 ΔtolC cells in the presence of inhibitory compounds restores growth. Curves are means ± SD for 5 independent replicates. (B) Swimming motility controls. Neither DMSO (10 µl) nor 10 µM dipyridyl (10 µl) inhibited swimming motility of CFT073 ΔtolC. CCCP (10 µl of a 10 µM solution in DMSO) inhibits swimming motility. Inhibition of the PMF is seen as a halo of swimming inhibition surrounding the paper disk. (C) Iron acquisition inhibitors do not inhibit the PMF. Swimming agar plates were inoculated with CFT073 ΔtolC as described. Compounds were tested by pipetting 10 µl of a 10 µM solution in DMSO into paper disks and placing them between the inoculation point and the edge of the plate. The positive control was CCCP (10 µl of a 10 µM solution in DMSO).
Activity toward wild-type E. coli CFT073.
Compounds were tested against wild type E. coli CFT073 in dose-response experiments (data not shown). Of 16 compounds, 4 were able to inhibit the wild-type strain as well as the ΔtolC mutant (Fig. 3), implying that these compounds are either not subject to efflux by this pump or act on outer membrane targets.
Assay for PMF inhibition.
Compounds were tested for their ability to inhibit the proton motive force (PMF). The PMF provides the necessary energy for internalization of siderophores and heme, so a PMF inhibitor would prevent growth in iron-limiting medium. We employed swimming motility as a test for PMF inhibition, since it is a phenotype dependent on PMF but not dependent on iron. The negative controls used were dipyridyl and DMSO, both of which showed no effect on motility, and as a positive control we used the proton ionophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) (Fig. 4B). None of the 16 compounds inhibited swimming motility (Fig. 4C).
Loss-of-target strategy for target identification.
To pinpoint the cellular target of the iron acquisition inhibitors identified through this screen, we applied a loss-of-target strategy. This consists in generating a panel of deletion mutants of genes involved in iron acquisition in CFT073. This is an ongoing strategy, since targets for all compounds have not been identified yet. Mutants generated and tested in a first step include single outer membrane receptor mutants and mutants lacking the ent operon (defective in catecholate siderophore synthesis), iuc genes (defective in aerobactin synthesis), iro genes (defective in salmochelin synthesis), or tonB (see Table S1 in the supplemental material for a complete list of mutants tested).
For each mutant generated, we tested growth in MOPS no iron and in MOPS with various iron concentrations to establish minimal iron requirements. For the majority of mutants, growth in MOPS no iron was not affected. CFT073 ΔtonB::kan and CFT073 ΔtonB ΔtolC::kan were unable to grow in MOPS no iron and required addition of 1 µM FeSO4 to grow to levels similar to those reached by the wild-type strain in MOPS no iron (data not shown). For the remainder of the experiments, these two strains were grown in MOPS 1 µM FeSO4.
When tested in DRC analysis, most compounds continued to inhibit the ΔtonB ΔtolC::kan strain to the same level to which they inhibited the ΔtolC::kan strain (data not shown), but the double mutant was resistant to two compounds, 120304 and 175472 (Fig. 5A and B). We pursued confirmation of the target by overexpressing the putative target TonB in pBAD. Figure 5A and B show that inhibition by 120304 and 175472 is relieved by overexpression of TonB. To further test inhibition of TonB function independently from iron acquisition, we conducted a reversible adsorption test with bacteriophage λh80 (having bacteriophage φ80 specificity), which recognizes the TonB-dependent receptor FhuA and requires a functional TonB system to enter the cell. E. coli CFT073 is not susceptible to bacteriophage φ80, so testing was conducted using an E. coli MG1655 strain and corresponding mutant strains. When either WT (not shown) or ΔtolC::kan cells are incubated with phage, the number of unadsorbed phage particles in the supernatant decreases over time, reflecting an increasing number of phage particles irreversibly adsorbing to the cells in a TonB-dependent process (Fig. 5C). In a strain lacking TonB function, phage cannot irreversibly adsorb, and the number of phage particles recovered from the supernatant remains constant over time. Compound 120304 was able to prevent irreversible adsorption of φ80 to E. coli ΔtolC::kan cells (Fig. 5C), further confirming that the TonB system is the target of this molecule. A single similar experiment with compound 175472 yielded similar results (data not shown). However, due to the unavailability from the vendor, this compound was not tested further during this work. Incubation of E. coli MG1655 ΔtolC::kan cells with compound 120304 did not affect adsorption of bacteriophage λ, which does not rely on TonB for entry (Fig. 5B).
FIG 5 .

TonB is identified as a putative target in a loss-of-target strategy. (A and B) Dose-response curves for compounds 120304 and 175472 were conducted using CFT073 WT and mutant strains in MOPS no iron supplemented with FeSO4 to match their respective minimal iron concentration and the appropriate antibiotics. No arabinose was added for the ΔtolC/pBAD-TonB strain. The ΔtolC (squares), ΔtolC ΔtonB (circles), and ΔtolC/pBAD-TonB (triangles) strains are shown. (C) Compound 120304 inhibits irreversible adsorption of bacteriophage φ80. Cells were incubated with TonB-dependent bacteriophage φ80, and unadsorbed phage particles in the supernatant were counted at different time points. The higher the unadsorbed fraction, the lower the relative level of TonB function. Each point represents the mean ± SD for three independent assays. *, P < 0.05 by two-way analysis of variance (ANOVA). (D) Compound 120304 does not affect adsorption of bacteriophage λ to cells. Cells were incubated with TonB-independent bacteriophage λ, and phage particles in the supernatant were counted at different time points as described for φ80. Each point represents the mean ± SD of data from three independent assays.
The remaining mutants in outer membrane receptors and in siderophore synthesis operons displayed no difference in DRC for any of the compounds tested (data not shown). This suggests that the deleted genes or operons do not encode the target(s) of these compounds. Additional experiments are under way to identify the cellular targets of the 14 remaining compounds.
DISCUSSION
Urinary tract infections place a significant burden on our health system. These infections affect a large proportion of the population, causing an estimated annual cost in the United States alone of $3.5 billion (24). The dramatic increase in resistance to known antibiotics in recent years threatens to make UTI treatment especially challenging, forcing physicians to prescribe more-expensive and often less-effective drugs. In this context, we set out to uncover novel targets for antibacterial strategies within the iron acquisition systems of UPEC.
Iron acquisition is an attractive target for antibacterial strategies against UPEC for several reasons. First, E. coli requires significant amounts of iron for growth, averaging 105 to 106 Fe atoms per cell (25). Second, there are no equivalent systems in eukaryotes, which makes this strategy specific against bacteria. Third, iron acquisition systems of UPEC are necessary for full virulence in the mouse model of ascending UTI (20) and are highly expressed during infection in humans (26). Last, a strategy based on iron acquisition would conceivably be effective only within the urinary tract, preserving the commensal strains of E. coli present in the gut, where iron is presumably less restricted.
To identify small molecules with the potential to arrest growth of UPEC under iron-restricted conditions, we developed and performed an HTS assay. The main difficulties in dealing with iron acquisition in UPEC are the multiplicity and redundancy of iron acquisition systems. In our approach, we targeted the whole cell under iron-limiting conditions, in which all iron acquisition systems are expected to be expressed based on our own previous transcriptional data (26, 27). Since this project was at the same time a proof of concept of the druggability of iron acquisition in a complex system like UPEC and an exploration of possible targets, we conducted the screen with a strain isogenic with E. coli CFT073 except for the deletion of the gene encoding the major efflux pump, TolC. The TolC-AcrAB system is well studied and has been shown to participate in efflux of a variety of antimicrobial compounds (22). Of 16 compounds identified in our screen as targeting iron acquisition, 12 are active only in the ΔtolC background, suggesting that their macromolecular target is intracellular. The remaining 4 are equally active against WT CFT073, which could indicate either an outer membrane target or a lack of recognition of the compounds by efflux systems.
The initial hit rate of our screen is high (1.6%), as expected for a nonspecific cellular assay with off-target effects. The primary screen identified any compound that inhibits growth of UPEC in minimal medium under low-iron conditions. That includes not only all antibiotics and toxic compounds but also any compounds that increase the stress for bacterial cells already under the stress of growing in minimal medium with severe iron limitation. By removing from further consideration all compounds for which growth could not be rescued by addition of extra iron, we rejected antibiotics, toxic compounds, and any compound that inhibited growth by means other than iron acquisition. The only class of compounds that could not be discarded by the high-iron counterscreen was that encompassing molecules capable of chelating iron, hence the development of a second counterscreen to identify iron chelators.
In the pilot assay, the CAS assay successfully identified known iron chelators that were hits in the primary assay, such as EDTA and deferoxamine mesylate. However, it is possible that due to differences in the pH at which these two assays are performed (4.5 for CAS and 7.4 for primary assay), some compounds could fail to chelate iron when protonated at the lower pH but function as chelators at a higher pH. Nevertheless, the simplicity and reproducibility of the CAS assay made it the best choice for high throughput. Moreover, compounds that inhibit growth due to iron chelation, such as deferoxamine mesylate, are active both against CFT073 WT and the ΔtolC mutant, whereas most of the compounds identified by this screen are not active against the WT. This suggests that even if they do chelate iron to a certain extent, this is not the main mode by which they arrest growth under iron-limiting conditions.
The inhibitors identified through this assay are of moderate potency, with AC50 values ranging from high nanomolar to midmicromolar. These values, however, reflect the potency in vivo against growth of the bacterial cell and not the affinity for their target.
Since entry of iron through the majority of UPEC systems is ultimately driven by the PMF, we tested our compounds for PMF inhibition. While a strong PMF inhibitor like CCCP would also prevent growth in high iron and should be removed by the counterscreen in high iron, we nonetheless tested the compounds for PMF inhibition. Swimming motility is a PMF-dependent process with an easy readout and independent from iron acquisition. None of the 16 compounds identified in this work inhibited swimming motility, suggesting that they do not affect the PMF.
Whereas whole-cell approaches like the one we carried out have the distinct advantage of inherently selecting compounds that act against the cell, the nature of the target remains to be solved. This is a major complication when screening for general antibacterial compounds against an organism. We have an advantage in that only a small subset of the bacterial genome encodes proteins related to acquisition of iron or regulation of iron homeostasis. This allows us to pursue strategies such as loss of target and overexpression of target in a delimited set of genes. This combination of loss of target and inhibition relief by overexpression allowed us to identify the cellular target of two compounds as the TonB system. This system is required for entry of multiple iron sources, and it is one of the few evident global targets in iron acquisition. In CFT073, TonB is required for fitness and virulence in the mouse model of urinary tract infection (20). Two of the sixteen compounds lost inhibitory activity when TonB was not present and also when TonB was overexpressed (Fig. 5A and B), strongly suggesting that this is their cellular target. We further confirmed this with a bacteriophage adsorption assay.
Whereas the TonB system is required for entry of ferrisiderophore complexes, growth of a tonB mutant can be achieved provided there is at least 1 µM iron in the growth medium (Fig. 5). It is likely that in the absence of an active TonB system, bacteria utilize lower-affinity iron acquisition systems, such as the ferrous iron (FeoABC) uptake system (28). This system relies on ferrous iron accessing the periplasm through yet-unidentified porins and being transported across the cell membrane by the transmembrane protein FeoB through an ATP-driven active transport process. FeoC is thought to act as a transcriptional regulator, whereas the role of FeoA is not yet clear, though experiments suggest that it interacts with FeoB (29). Other TonB-independent systems for iron uptake have been described, including the SitABCD system encoded by CFT073, which has been involved in iron and manganese transport (30, 31).
It would seem obvious that if deletion of a gene or operon does not alter the ability of CFT073 to grow on limited-iron medium, that gene product is not the target of an iron acquisition inhibitor. This would make testing of any mutant capable of growing in MOPS no iron, such as receptors and synthesis mutants, irrelevant. However, recent data have suggested that the roles of gene products in iron acquisition pathways are not as straightforward as once thought. Work with an avian extraintestinal pathogenic E. coli strain closely related to CFT073 has shown that catecholate siderophore synthesis mutants retain full virulence whereas catecholate siderophore export mutants show decreased fitness in a cochallenge infection model (32). Recently, genes involved in catecholate siderophore synthesis have been shown to protect against reactive oxygen species in Salmonella (33). Without knowing the full extent of the interplay between iron acquisition systems, we decided to pursue a comprehensive approach and generate mutants in all genes that allegedly participate in iron acquisition. Our results so far seem to substantiate the obvious, that is, the identified target for two compounds is the only one of the tested mutants that failed to grow in MOPS no iron.
The approach described in this article deals only with free iron acquisition by UPEC. However, heme acquisition in particular has been shown to be important at different steps in the mouse model of ascending UTI (5, 20). The druggability of heme acquisition has been underexplored, with only a few reported efforts targeting the key enzyme in heme catabolism, specifically heme monooxygenase (34, 35). We are currently developing an HTS similar to the one described in this work to look for inhibitors of heme acquisition by UPEC.
The majority of current therapeutic strategies aim at the same group of targets, mainly nucleic acid synthesis, bacterial ribosome, and cell wall assembly (36). Given the high diversity in the prokaryotic evolutionary tree, it is possible that common targets have already been exploited and the era of broad-spectrum antibiotics has come to an end. Most industrial efforts have been focused on molecules acting on highly conserved molecular processes, but future studies might focus on strain- and disease-specific therapeutics. Limited spectral coverage has its own unique advantages. As we begin to understand the human microbiome, drugs that engender less perturbation of the natural gut and vaginal ecology than traditional antibiotics might reduce the incidence of opportunistic secondary infections (such as Clostridium difficile or vaginal yeast infections). Strategies such as the one we are pursuing, in which the target is present across a broad spectrum of organisms but its importance is site specific, might represent the answer for future antibacterial drugs.
In summary, we have developed a simple and robust cell-based growth assay for testing inhibition of iron acquisition in large libraries. Our assay addresses the problem of redundancy and multiplicity of iron acquisition pathways to be targeted by utilizing whole cells grown in culture under conditions where genes related to iron acquisition are upregulated. We found 16 compounds validated through dose-response curves with fresh powders that arrest bacterial growth through inhibition of iron acquisition. These compounds do not function primarily as iron chelators, are bacteriostatic, and act only under low-iron conditions. We are applying a combination of strategies, including loss of target and overexpression/underexpression arrays, to identify the cellular targets of these compounds. So far, we have identified the TonB system as the likely target of two of the compounds and confirmed it through a TonB-dependent bacteriophage adsorption assay. Further experiments to identify the cellular target of the remaining compounds, as well as the mechanism of inhibition of TonB function for compounds 120304 and 175472, are under way.
MATERIALS AND METHODS
Reagents.
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). Morpholinepropanesulfonic acid (MOPS) with no added iron (MOPS no iron) was acquired from Teknova. It should be noted that while MOPS no iron has no added iron, traces of the metal are still present. All water used during this study was Milli-Q quality and was run through Chelex resin (Bio-Rad) to chelate any metal ion present. Fresh powder compounds were purchased from the original library vendors.
Bacterial strains and culture conditions.
The prototypical uropathogenic E. coli strain CFT073, a human pyelonephritis isolate (37), was used. Wild-type and mutant strains were routinely cultured in LB broth at 37°C.
Mutant strain construction.
E. coli CFT073 (ΔtolC::kan ΔtonB::kan) and E. coli MG1655 (ΔtolC::kan) mutants were constructed using the Lambda Red recombinase system (38). Primers containing 50-nucleotide (nt) sequences homologous to the 5′ and 3′ ends of the tolC or tonB sequence were designed and used to amplify the kanamycin resistance cassette from plasmid pKD4. E. coli CFT073 or MG1655 cells carrying plasmid pKD46 encoding the Lambda Red recombinase were transformed with the resulting PCR product. Primers homologous to flanking regions of the tolC or tonB gene were used to confirm recombination in Kanr colonies. MG1655 ΔtonB was provided by David Friedman.
Plasmid construction.
The tonB locus was cloned into pBAD under the control of an arabinose-inducible promoter using the primers 5′ ATGTCACCATGGTTGCATTTAAAATCGGGGCC 3′ (NcoI site is underlined) and ATCTCAAAGCTTTTACTGAATTTCGGTAGTGCCG-3′ (HindIII site is underlined, stop codon is in boldface).
Growth curves.
Bacteria were cultured in 5 ml LB broth overnight at 37°C with shaking, collected by centrifugation, and washed 3× with 5 ml MOPS no iron. Cultures were inoculated in appropriate medium at an initial optical density at 600 nm (OD600) of 0.01. Growth curves were generated using a Bioscreen C automated growth curve analysis system (Growth Curves USA) in a final volume of 300 µl per well. Culture plates were incubated at 37°C with continuous shaking for 12 to 48 h, and turbidity was measured at 600 nm every 30 min.
Small-molecule libraries.
The primary assay was validated using the Spectrum Collection 2000 (2,000 compounds; MicroSource, Connecticut) and NCC BioFocus (446 compounds; NIH, USA) libraries. In the primary screen, 149,243 compounds were tested at the Center for Chemical Genomics (CCG) (University of Michigan) using the primary assay. Among the libraries tested were a chemical diversity set (120,000 compounds; ChemDiv, San Diego, CA), a Maybridge HitFinder library (16,000 compounds; Thermo, Fisher Scientific), and a subset of the ChemBridge core diversity library stock (13,000 compounds; ChemBridge Corp., San Diego, CA).
Primary HTS assay.
E. coli CFT073 ΔtolC::kan was cultured overnight in LB supplemented with 25 µg/ml kanamycin, and bacteria were collected by centrifugation at 6,000 rpm, washed three times in MOPS no iron to remove the metal, and resuspended in the same medium. Bacteria were inoculated to an initial OD600 of 0.01 in 384-well plates (Corning, catalog number 3680) previously spotted in column numbers 3 to 22 with 0.2-µl library compounds in dimethyl sulfoxide (DMSO) (final concentration, 10 µM) using a Biomek FX laboratory automation workstation with high-density replication (HDR) (pin tool). In each plate, columns 1 and 2 were spotted with DMSO (final concentration, 0.5%), and columns 23 and 24 contained 2,2′-dipyridyl (DIP) in DMSO (final concentration, 200 µM) as negative and positive controls, respectively. Plates were briefly centrifuged and incubated for 16 h at 37°C. Readings were taken using an Envision Multimode plate reader (PerkinElmer). The average of the 16 DMSO and 16 DIP control wells on each individual plate was used to calculate the percent inhibitory activity for each well according to the following equation: % inhibition = {1 − [(OD600 compound-treated well − average OD600 DMSO controls)/(average OD600 DIP controls − average OD600 DMSO controls)]} × 100.
Z’ factors were calculated as previously described (39) for each individual 384-well plate screened.
Data analysis.
Hits were considered actives if they attained >30% growth inhibition. Compounds were removed from the “actives” list if they were a hit against Shigella flexneri in rich medium under high-iron conditions (from other screens run at the CCG), if the molecular weight (MW) was >500, if they were structurally related to known antibiotics, if they were promiscuous (defined as being a hit in >30% of the total number of screens run at CCG, tested a minimum of 12 times), or if they contained structural alerts used to exclude reactive compounds from the NIH MLSMR collection (http://mlsmr.evotec.com/MLSMR_HomePage/pdf/MLSMR_Excluded_Functionality_Filters_200605121510.pdf). A confirmation screen of the actives at a 10 µM final concentration was run in triplicate, and compounds were confirmed as hits if the median of three replicates was >30% inhibition.
Counterscreen in high iron.
Triaged compounds with confirmed activity in triplicate were assayed in an endpoint growth assay in MOPS–0.2% glucose supplemented with 40 µM FeSO4. Plates were inoculated and read as in the primary assay described above. Compounds with more than 20% inhibitory activity at 10 µM in this assay were considered positives.
Chelator assay.
The chrome azurol S (CAS) shuttle assay (23) was used to quantify iron chelation. Briefly, compounds that were hits in the primary assay were spot plated (0.2 µl) using a Mosquito X1 hit-picking liquid handler (TTP Labtech). A CAS shuttle solution containing 0.6 mM hexadecyltrimethylammonium bromide (HDTMA), 0.015 mM FeCl3, 0.15 mM CAS, 0.75 M hydrochloric acid, 0.5 M anhydrous piperazine, and 4 mM 5-sulfosalicylic acid was added to the plates (40 µl) and incubated for 30 min at room temperature, and absorbance at 630 nm was measured in an Envision Multimode plate reader (PerkinElmer). Positive controls for iron chelation were EDTA, deferoxamine mesylate, and DIP dissolved in DMSO and were spotted at the same final concentration as the library compounds (10 µM). Compounds with more than 20% activity of EDTA were considered positives in the chelator assay.
Dose-response analysis.
Compounds that passed the triage, confirmation, and counterscreening (377 small molecules) were assayed in a dose-response study according to the screening protocol. Compounds were picked from library plates using a Mosquito X1 hit-picking liquid handler (TTP Labtech) and tested in duplicate in concentrations ranging from 100 to 0.39 µM. Data were plotted as a function of the concentration of small molecule (pAC) versus percent inhibition. For those compounds that exhibited a sigmoidal dose-response curve (DRC), data were fit by nonlinear regression to the following equation using the software program GraphPad Prism (GraphPad Software, Inc.):
where B is the bottom plateau of the curve, and is T is the top plateau of the curve.
Compounds for which the difference in pAC50 between no-iron and 3 µM FeSO4 media was lower than 1 were deprioritized from this study.
Fresh powder compounds.
Selected compounds that were available from vendors (58 out of 76) were purchased as fresh powders. Compounds were dissolved in DMSO at a final concentration of 20 mM and tested in DRC in quadruplicate as described for library compounds. DRC for mutants in iron acquisition systems were conducted in a similar way at the lowest iron concentration that provided growth similar to that of the wild type (WT) in MOPS no iron. A minimal iron concentration was achieved in MOPS no iron for all mutants tested (see Table S1 in the supplemental material) except for CFT073 ΔtonB::kan, which was assayed at 1 µM FeSO4.
Swimming diameter assay.
Swimming plates (1 g/liter tryptone, 0.5 g/liter NaCl, and 0.25 g/liter agar) were stabbed in the center with an overnight culture of E. coli strain CFT073 ΔtolC::kan (diluted to an OD600 of 1). Filter disks containing 10 µl of a 10 µM compound solution in DMSO were placed equidistant from the center, and inhibition haloes were measured. Carbonyl cyanide m-chlorophenyl hydrazine (CCCP) at a final concentration of 10 µM and DMSO were used as positive and negative controls.
Bacteriophage adsorption assay.
E. coli MG1655 and corresponding ΔtolC::kan and ΔtonB mutant cells were cultured in LB to an OD600 of 0.4. Aliquots (1 ml) were pelleted and resuspended in 100 µl fresh LB supplemented with 5 mM CaCl2 and 100 µM compound 120304 or the DMSO control. Bacteriophage λh80 (λ with bacteriophage φ80 selectivity) was added to the cells to a multiplicity of infection of 0.5 PFU/cell and incubated statically at 37°C. At different time points, 10-µl aliquots were removed and diluted in 1 ml LB plus 0.5% Cl3CH4, vigorously vortexed to release reversibly bound bacteriophage particles, and centrifuged at 13,000 rpm for 1 min. Supernatants were placed on ice and titrated on E. coli MG1655 to quantify unadsorbed phage. Results are the means ± standard deviations (SD) for three independent experiments.
SUPPLEMENTAL MATERIAL
Growth of CFT073 siderophore synthesis mutants in MOPS no iron. Growth curves were recorded as described for MOPS no iron. Mutants depicted lack the synthesis pathway for catecholate siderophores (ent) or aerobactin (iuc). Curves are means ± SD for 3 independent replicates. Download
Iron is required for CFT073 growth. Growth curves of CFT073 in LB (A) or MOPS no iron (B) are shown. In both cases, addition of the nonmetabolizable iron chelator dipyridyl abolishes growth. Supplementation with additional iron restores growth. Curves are means ± SD for 3 independent replicates. Download
CFT073 ΔtolC growth curves in MOPS supplemented with different iron concentrations. Growth curves were recorded as described for MOPS no iron (circles) or MOPS supplemented with 0.1 µM (squares), 1 µM (upward triangles), or 10 µM (downward triangles) FeSO4. Curves are means ± SD for 3 independent replicates. Download
Assay for iron acquisition inhibition. The plot depicts percent growth inhibition relative to results for DIP controls versus well number. Positive controls (red) are 200 µM dipyridyl and represent 100% inhibition. Negative controls (blue) are 0.5% DMSO and represent 0% inhibition. Green dots represent the activity of tested compounds. The red line marks 3 standard deviations from results for negative controls. Download
Attrition rates from screen to hit. The 149,243 compounds in the primary screening were tested to identify inhibitors of E. coli CFT073 growth in MOPS no iron. Primary triage discarded compounds in the categories described under “Data analysis.” The confirmation screen was run in triplicate and resulted in a 60% confirmation rate. Confirmed compounds were tested in a high-iron counterscreen and a CAS chelation assay. Compounds with >20% activity of controls in either counterscreen were discarded during confirmation triage. Dose-response curve analysis was conducted, and 53.6% of compounds were titrated. Titration triage discarded compounds with <1 difference in pAC50 between MOPS no iron and MOPS 3 µM iron. Fresh powder was purchased for 58 compounds, and dose-response analysis was repeated. A total of 16 fresh compounds were retested, giving a final hit rate of 0.6% of actives. Download
E. coli CFT073 mutants tested in loss-of-target strategy
ACKNOWLEDGMENTS
We thank Hollis Showalter for help evaluating compound structures, Erin Garcia for mutant CFT073 ΔtonB::kan, and David Friedman for phages, strains, and advice.
This work was supported by Public Health Service grants DK097362 and AI043363 from the National Institutes of Health.
Footnotes
Citation Yep A, McQuade T, Kirchhoff P, Larsen M, Mobley HLT. 2014. Inhibitors of TonB function identified by a high-throughput screen for inhibitors of iron acquisition in uropathogenic Escherichia coli CFT073. mBio 5(2):e01089-13. doi:10.1128/mBio.01089-13.
REFERENCES
- 1. Sanchez GV, Master RN, Karlowsky JA, Bordon JM. 2012. In vitro antimicrobial resistance of urinary Escherichia coli isolates among U.S. outpatients from 2000 to 2010. Antimicrob. Agents Chemother. 56:2181–2183. 10.1128/AAC.06060-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schito GC, Naber KG, Botto H, Palou J, Mazzei T, Gualco L, Marchese A. 2009. The ARESC study: an international survey on the antimicrobial resistance of pathogens involved in uncomplicated urinary tract infections. Int. J. Antimicrob. Agents 34:407–413. 10.1016/j.ijantimicag.2009.04.012 [DOI] [PubMed] [Google Scholar]
- 3. Anderson GJ, Vulpe CD. 2009. Mammalian iron transport. Cell. Mol. Life Sci. 66:3241–3261. 10.1007/s00018-009-0051-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Masson PL, Heremans JF, Schonne E. 1969. Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J. Exp. Med. 130:643–658. 10.1084/jem.130.3.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hagan EC, Mobley HL. 2009. Haem acquisition is facilitated by a novel receptor Hma and required by uropathogenic Escherichia coli for kidney infection. Mol. Microbiol. 71:79–91. 10.1111/j.1365-2958.2008.06509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wiener MC. 2005. TonB-dependent outer membrane transport: going for Baroque? Curr. Opin. Struct. Biol. 15:394–400. 10.1016/j.sbi.2005.07.001 [DOI] [PubMed] [Google Scholar]
- 7. Schauer K, Rodionov DA, de Reuse H. 2008. New substrates for TonB-dependent transport: do we only see the “tip of the iceberg”? Trends Biochem. Sci. 33:330–338. 10.1016/j.tibs.2008.04.012 [DOI] [PubMed] [Google Scholar]
- 8. Krewulak KD, Vogel HJ. 2011. TonB or not TonB: is that the question? Biochem. Cell Biol. 89:87–97. 10.1139/O10-141 [DOI] [PubMed] [Google Scholar]
- 9. Larsen RA, Letain TE, Postle K. 2003. In vivo evidence of TonB shuttling between the cytoplasmic and outer membrane in Escherichia coli. Mol. Microbiol. 49:211–218. 10.1046/j.1365-2958.2003.03579.x [DOI] [PubMed] [Google Scholar]
- 10. Gresock MG, Savenkova MI, Larsen RA, Ollis AA, Postle K. 2011. Death of the TonB shuttle hypothesis. Front. Microbiol. 2:00206. 10.3389/fmicb.2011.00206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cascales E, Lloubès R, Sturgis JN. 2001. The TolQ-TolR proteins energize TolA and share homologies with the flagellar motor proteins MotA-MotB. Mol. Microbiol. 42:795–807. 10.1046/j.1365-2958.2001.02673.x [DOI] [PubMed] [Google Scholar]
- 12. De Mot R, Vanderleyden J. 1994. The C-terminal sequence conservation between OmpA-related outer membrane proteins and MotB suggests a common function in both gram-positive and gram-negative bacteria, possibly in the interaction of these domains with peptidoglycan. Mol. Microbiol. 12:333–334. 10.1111/j.1365-2958.1994.tb01021.x [DOI] [PubMed] [Google Scholar]
- 13. Peacock R, Weljie AM, Peter Howard S, Price FD, Vogel HJ. 2005. The solution structure of the C-terminal domain of TonB and interaction studies with TonB box peptides. J. Mol. Biol. 345:1185–1197. 10.1016/j.jmb.2004.11.026 [DOI] [PubMed] [Google Scholar]
- 14. Gumbart J, Wiener MC, Tajkhorshid E. 2007. Mechanics of force propagation in TonB-dependent outer membrane transport. Biophys. J. 93:496–504. 10.1529/biophysj.107.104158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carter DM, Miousse IR, Gagnon JN, Martinez E, Clements A, Lee J, Hancock MA, Gagnon H, Pawelek PD, Coulton JW. 2006. Interactions between TonB from Escherichia coli and the periplasmic protein FhuD. J. Biol. Chem. 281:35413–35424. 10.1074/jbc.M607611200 [DOI] [PubMed] [Google Scholar]
- 16. James KJ, Hancock MA, Gagnon JN, Coulton JW. 2009. TonB interacts with BtuF, the Escherichia coli periplasmic binding protein for cyanocobalamin. Biochemistry 48:9212–9220. 10.1021/bi900722p [DOI] [PubMed] [Google Scholar]
- 17. Cassat JE, Skaar EP. 2013. Iron in infection and immunity. Cell Host Microb. 13:509–519. 10.1016/j.chom.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brock JH, Licéaga J, Kontoghiorghes GJ. 1988. The effect of synthetic iron chelators on bacterial growth in human serum. FEMS Microbiol. Immunol. 1:55–60 [DOI] [PubMed] [Google Scholar]
- 19. Foley TL, Simeonov A. 2012. Targeting iron assimilation to develop new antibacterials. Expert Opin. Drug Discov. 7:831–847. 10.1517/17460441.2012.708335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Torres AG, Redford P, Welch RA, Payne SM. 2001. TonB-dependent systems of uropathogenic Escherichia coli: aerobactin and heme transport and TonB are required for virulence in the mouse. Infect. Immun. 69:6179–6185. 10.1128/IAI.69.10.6179-6185.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carpenter BM, Whitmire JM, Merrell DS. 2009. This is not your mother’s repressor: the complex role of Fur in pathogenesis. Infect. Immun. 77:2590–2601. 10.1128/IAI.00116-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Routh MD, Zalucki Y, Su CC, Zhang Q, Shafer WM, Yu EW. 2011. Efflux pumps of the resistance-nodulation-division family: a perspective of their structure, function, and regulation in gram-negative bacteria. Adv. Enzymol. Relat. Areas Mol. Biol. 77:109–146. 10.1002/9780470920541.ch3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160:47–56. 10.1016/0003-2697(87)90612-9 [DOI] [PubMed] [Google Scholar]
- 24. Litwin MS, Saigal CS. 2007. Urologic diseases in America. NIH Publication 07-5512:3-7 NIH, Bethesda, MD. [Google Scholar]
- 25. Andrews SC, Robinson AK, Rodríguez-Quiñones F. 2003. Bacterial iron homeostasis. FEMS Microbiol. Rev. 27:215–237. 10.1016/S0168-6445(03)00055-X [DOI] [PubMed] [Google Scholar]
- 26. Hagan EC, Lloyd AL, Rasko DA, Faerber GJ, Mobley HL. 2010. Escherichia coli global gene expression in urine from women with urinary tract infection. PLOS Pathog. 6:e1001187. 10.1371/journal.ppat.1001187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Snyder JA, Haugen BJ, Buckles EL, Lockatell CV, Johnson DE, Donnenberg MS, Welch RA, Mobley HL. 2004. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infect. Immun. 72:6373–6381. 10.1128/IAI.72.11.6373-6381.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kammler M, Schön C, Hantke K. 1993. Characterization of the ferrous iron uptake system of Escherichia coli. J. Bacteriol. 175:6212–6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lau CK, Ishida H, Liu Z, Vogel HJ. 2013. Solution structure of Escherichia coli FeoA and its potential role in bacterial ferrous iron transport. J. Bacteriol. 195:46–55. 10.1128/JB.01121-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kehres DG, Janakiraman A, Slauch JM, Maguire ME. 2002. SitABCD is the alkaline Mn(2+) transporter of Salmonella enterica serovar Typhimurium. J. Bacteriol. 184:3159–3166. 10.1128/JB.184.12.3159-3166.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sabri M, Léveillé S, Dozois CM. 2006. A SitABCD homologue from an avian pathogenic Escherichia coli strain mediates transport of iron and manganese and resistance to hydrogen peroxide. Microbiology 152:745–758. 10.1099/mic.0.28682-0 [DOI] [PubMed] [Google Scholar]
- 32. Caza M, Lépine F, Dozois CM. 2011. Secretion, but not overall synthesis, of catecholate siderophores contributes to virulence of extraintestinal pathogenic Escherichia coli. Mol. Microbiol. 80:266–282. 10.1111/j.1365-2958.2011.07570.x [DOI] [PubMed] [Google Scholar]
- 33. Achard ME, Chen KW, Sweet MJ, Watts RE, Schroder K, Schembri MA, McEwan AG. 2013. An antioxidant role for catecholate siderophores in Salmonella. Biochem. J. 454:28. 10.1042/BJ20121771 [DOI] [PubMed] [Google Scholar]
- 34. Stojiljkovic I, Kumar V, Srinivasan N. 1999. Non-iron metalloporphyrins: potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol. Microbiol. 31:429–442. 10.1046/j.1365-2958.1999.01175.x [DOI] [PubMed] [Google Scholar]
- 35. Hom K, Heinzl GA, Eakanunkul S, Lopes PE, Xue F, MacKerell AD, Wilks A. 2013. Small molecule antivirulents targeting the iron-regulated heme oxygenase (HemO) of P. aeruginosa. J. Med. Chem. 56:2097–2109. 10.1021/jm301819k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walsh C. 2003. Antibiotics: actions, origins, resistance. ASM Press, Washington, DC [Google Scholar]
- 37. Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. 1990. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect. Immun. 58:1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73. 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth of CFT073 siderophore synthesis mutants in MOPS no iron. Growth curves were recorded as described for MOPS no iron. Mutants depicted lack the synthesis pathway for catecholate siderophores (ent) or aerobactin (iuc). Curves are means ± SD for 3 independent replicates. Download
Iron is required for CFT073 growth. Growth curves of CFT073 in LB (A) or MOPS no iron (B) are shown. In both cases, addition of the nonmetabolizable iron chelator dipyridyl abolishes growth. Supplementation with additional iron restores growth. Curves are means ± SD for 3 independent replicates. Download
CFT073 ΔtolC growth curves in MOPS supplemented with different iron concentrations. Growth curves were recorded as described for MOPS no iron (circles) or MOPS supplemented with 0.1 µM (squares), 1 µM (upward triangles), or 10 µM (downward triangles) FeSO4. Curves are means ± SD for 3 independent replicates. Download
Assay for iron acquisition inhibition. The plot depicts percent growth inhibition relative to results for DIP controls versus well number. Positive controls (red) are 200 µM dipyridyl and represent 100% inhibition. Negative controls (blue) are 0.5% DMSO and represent 0% inhibition. Green dots represent the activity of tested compounds. The red line marks 3 standard deviations from results for negative controls. Download
Attrition rates from screen to hit. The 149,243 compounds in the primary screening were tested to identify inhibitors of E. coli CFT073 growth in MOPS no iron. Primary triage discarded compounds in the categories described under “Data analysis.” The confirmation screen was run in triplicate and resulted in a 60% confirmation rate. Confirmed compounds were tested in a high-iron counterscreen and a CAS chelation assay. Compounds with >20% activity of controls in either counterscreen were discarded during confirmation triage. Dose-response curve analysis was conducted, and 53.6% of compounds were titrated. Titration triage discarded compounds with <1 difference in pAC50 between MOPS no iron and MOPS 3 µM iron. Fresh powder was purchased for 58 compounds, and dose-response analysis was repeated. A total of 16 fresh compounds were retested, giving a final hit rate of 0.6% of actives. Download
E. coli CFT073 mutants tested in loss-of-target strategy