

Abstract
A high-potential nonhaem(μ-oxo)diiron(IV) complex was found to oxidize water to hydroxyl radical via PCET, instead of forming an O–O bond. The rate-determining step requires a second water molecule, proposed to act as a base to promote proton transfer. This work shows that additional factors besides a high redox potential are required to effect O–O bond formation.
The involvement of high-valent metal-oxo species is a common mechanistic feature of biological catalysts that have evolved to cleave strong C–H bonds and to convert water into O2 in photosynthesis. Cytochrome P450 and soluble methane monooxygenase represent iron enzymes of the first group for which [FeIV(O)(porphyrincation radical)]+ and [FeIV2O2] moieties have respectively been characterized as the oxidants responsible for the cleavage of the strong C–H bonds of the substrate.1, 2 The oxygen evolving complex, the only member of the second group, has been shown to have a Mn4CaOx cluster at which water oxidation takes place.3, 4 Because of these biological precedents, there has been an increase in efforts to synthesize biomimetic analogs that carry out these difficult oxidative transformations. Of particular interest is the recent work of Costas and Lloret identifying nonheme iron complexes that can catalyze water oxidation5, 6 as well as the hydroxylation of cyclohexane7, 8 with Ce(IV) as oxidant, where an FeV(O)(OH) species is proposed as the active species common to both reactions. These results suggest that oxidative systems that perform one type of reaction may also be effective in carrying out the other reaction.
Recently we reported the high-yield bulk-electrolytic generation of a (μ-oxo)diiron(IV) complex 1 supported by a pentadentate ligand (shown in Fig. 1) and its characterization by Mössbauer and X-ray absorption spectroscopies.9 Complex 1 has an E1/2 of +1.50 V vs. Fc+/0 in CH3CN at −40 °C, the highest FeIV,IV/FeIII,IV potential determined thus far for a diiron(IV) complex.10–12 As a consequence of the high potential, 1 has been shown to cleave C–H bonds as strong as those of cyclohexane (DC–H = 99.3 kcal mol−1) at rates that are 2~3 orders of magnitude faster than related mononuclear oxoiron(IV) complexes,13 thus appearing to be a powerful oxidant among members of the growing family of synthetic nonhaemoxoiron(IV) complexes.14 Strikingly, 1 can also selectively activate the O–H bonds of MeOH and t-BuOH via PCET, representing the first examples of such O–H bond oxidations by an iron complex. The unique ability of 1 to carry out the oxidation of O–H bonds stimulated us to explore its reaction with H2O, which we report in this communication. This reaction cleaves the O–H bond of water to generate hydroxyl radical via a PCET mechanism, but does not lead to the formation of an O–O bond. Our experimental findings provide mechanistic insights into what is required for the O–O bond formation step.
Fig. 1.
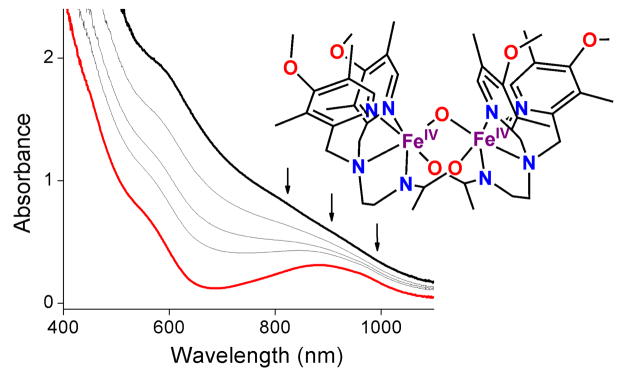
Reduction of 0.3 mM 1 (black line) to 2 (red line) by 0.28 M H2O at 10 °C in CD3CN over the course of 40 sec. Arrows indicate the spectral changes during the reaction. Inset: Schematic drawing of 1.
The introduction of excess H2O to a MeCN solution of 1 at 10 °C causes its rapid conversion to 2, its 1-e− reduced derivative with an FeIII–O–FeIV core (Fig. 1). This reaction is exponential, with the observed pseudo-first-order rate constant (kobs) showing a linear dependence not on the concentration of H2O added into the reaction medium but instead on the square of the H2O concentration (Fig. 2). Taken together, these results indicate that the rate-determining step involves the reaction of 1 with two equivalents of H2O. This termolecular behavior is in contrast to the bimolecular kinetics previously observed in reactions of 1 with hydrocarbons and aliphatic alcohols,9 where linear correlations are observed in all cases between kobs and the substrate concentration. The slope of the linear correlation shown in Fig. 2 affords a third-order rate constant (k3) of 0.73(3) M−2 s−1 for the reaction between 1 and H2O. Interestingly, replacing H2O to D2O as the substrate results in a rate of 0.32(1) M−2 s−1. This kinetic isotope effect (KIE) of 2.3 shows that O–H bond cleavage is an essential component of the rate-determining step.
Fig. 2.

Plots of the substrate concentration square dependence of the pseudo-first-order rate constants for the oxidation of (■) H2O and (●) D2O by 1 in CD3CN at 10 °C. The red lines represent the best linear fits. Inset: time course of the decay of 0.3 mM 1 (black squares) monitored at 680 nm with the added H2O concentration of (a) 0.28 M, (b) 0.56 M and (c) 1.10 M in CD3CN at 10 °C. The red traces represent the fits according to the pseudo-first-order mode.
Analysis of the H2O oxidation product shows no evidence for the formation of an O–O bond, as neither H2O2 nor O2 was detected. Instead, GC analysis shows the formation of acetamide as the reaction product in a yield of 60±10% relative to 1. This outcome indicates the formation of a hydroxyl radical (•OH) as the H2O oxidation product that was trapped by a solvent CD3CN molecule and accounting for 60% of the oxidizing equivalent used. It has been demonstrated by previous studies that •OH can undergo electrophilic addition to the –C≡N group of nitriles15, 16 or the –C≡C– triple bond of hydrocarbons17 to form respectively amide18 or aldehyde.17 The residual oxidizing equivalent unaccounted for may be ascribed to another decay pathway of •OH in the presence of CD3CN, i.e., hydrogen atom abstraction from the CD3 group.19 However, the desired product formyl cyanide (HC(O)CN) was not identified in this study, presumably due to its instability under current experimental conditions.20
Thus, 1 represents the first example of a well characterized (μ-oxo)diiron(IV) oxidant capable of attacking the very strong O–H bond of H2O (DO–H = 118.8 kcal mol−1). The ability of 1 to cleave strong O–H bonds has also been demonstrated in the reactions of 1 with aliphatic alcohols MeOH and t-BuOH to form the corresponding alkoxyl radicals.9 A PCET mechanism that involves electron transfer (ET) from one of the nonbonding oxygen orbitals and proton transfer (PT) from the O–H group was proposed to rationalize the observed reaction with an O–H/O–D KIE of 1.5. In the case of H2O oxidation, we propose that 1 attacks the O–H bond of H2O also via PCET, given the comparable O–H/O–D KIE of 2.4. However, the termolecular nature of H2O oxidation, as determined by the kinetic measurements, shows a unique rate-determining step that differs from the O–H activation of MeOH and t-BuOH.
Complex 1 oxidizes both H2O and t-BuOH by O–H bond activation with a k3 value of 0.73(3) M−2 s−1 for H2O and a k2 value of 0.30(2) M−1 s−1 for t-BuOH. Thus at substrate concentrations below 0.41 M, t-BuOH is more readily oxidized than H2O, but H2O is more reactive than t-BuOH at substrate concentrations above 0.41 M even though H2O has a much higher ionization energy (12.62 eV) than t-BuOH (9.90 eV). These two substrates also exhibit distinct kinetic behavior in their reactions with 1, namely termolecular for H2O and bimolecular fort-BuOH. Further insight into this kinetic difference was obtained by an Eyring analysis to obtain activation parameters. As shown in Fig. 3, the H2O oxidation has an activation enthalpy that is 2.7 kcal mol−1 smaller than the one for t-BuOH oxidation by 1, despite the fact that H2O has a 2.7-eV higher ionization energy. This result presumably reflects the role of PT in decreasing the kinetic barrier for ET. In H2O oxidation, PT is facilitated by the incorporation of the second H2O molecule as a base, while this feature is apparently absent in the case of t-BuOH. Thus it appears that for the termolecular PCET reaction the kinetic barrier of ET could be dramatically lowered by the PT step induced by the proton acceptor to compensate for the deficiency in thermodynamic driving force.
Fig. 3.

Eyring plots for the reactions of 1 with (■) H2O and (●) t-BuOH in CD3CN with obtained activation parameters listed.
Scheme 1A shows our postulated mechanism for H2O oxidation by 1, which is inspired by the corresponding mechanism proposed for mononuclear RuV=O complexes,21 for which the rate-determining O–O bond formation step involves one H2O molecule that is oxidized and a second H2O molecule that acts as the base to hydrate the released proton (Scheme 1B). This hypothesis was supported by experimental observations from Meyer and co-workers, who found that the H2O oxidation rate can be accelerated by buffering the reaction medium with inorganic bases of different pKa, thereby demonstrating that PT is essentially involved in the rate-determining step.21 The O–H/O–D KIEs in the RuV=O system were determined to fall into the range of 2–7. In line with the above investigations, we speculate that H2O oxidation by 1 proceeds via a similar PCET mechanism, with the first H2O molecule transferring one electron to the diiron(IV) oxidant 1 and the second H2O molecule acting as a proton acceptor to facilitate PT. For both H2O molecules to be involved in the rate-determining step, ET and PT must occur in a concerted manner. The O–H/O–D KIE of 2.4 is consistent with the ones observed in the RuV=O system.21 Unfortunately we have not been able to identify an appropriate organic base that is not oxidized by 1 nor a purely inorganic base that is soluble enough in CD3CN to investigate the effect of base on the kinetic behavior of H2O oxidation by 1.
Scheme 1.
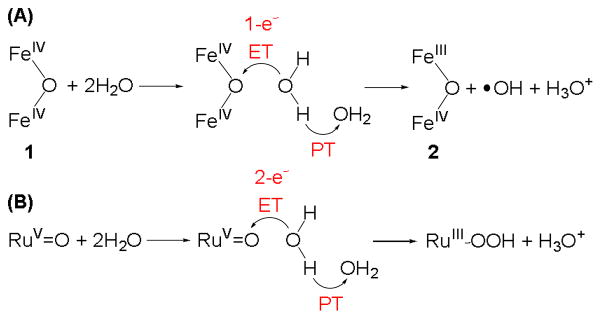
Proposed mechanisms for H2O oxidation by (A) diiron(IV) complex 1 and (B) RuV=O oxidant adapted from ref 21. The red portions highlight the ET and PT processes in both systems.
A significant difference between 1 and the RuV=O oxidant in carrying out H2O oxidation is in the nature of the reaction product, namely •OH and O2, respectively, indicating that 1 cannot promote O–O bond formation. This outcome arises from differences in structural and electrochemical properties between 1 and the RuV=O oxidant. The first difference is the nature of the reactive oxo group, namely bridging oxo in 1 and terminal oxo in RuV=O. O–O bond formation on a bridging oxo would yield a μ-1,1-bridged hydroperoxo species, which DFT calculations suggest to be unstable.22–24 To date, there are only two available examples of such dinuclear metal species with μ-1,1-peroxo bridges, and neither is a diiron complex.25, 26 Secondly, the FeIV,IV/FeIII,IV potential of 1 (+1.50 V vs. Fc+/0, or +2.14 V vs. NHE in CH3CN) is slightly higher than the 1-e− potential of H2O oxidation to •OH (+2.13 V vs. NHE in CH3CN under neutral conditions27), therefore making the 1-e− oxidation of H2O thermodynamically possible. By comparison, the RuV/IV potential for RuV=O is ~+1.60 V vs. NHE, too low to trigger the 1-e− oxidation of H2O (+2.31 V vs. NHE in H2O at pH 727). Thirdly, forming the O–O bond is essentially a 2-e− process to the metal center. However, the potential difference between the 1-e− couples (FeIV,IV/FeIII,IV vs. FeIII,IV/FeIII,III) for 1 is ~0.8 V,9 much larger than the ~0.3 V difference determined between the RuV/IV and RuIV/III couples.21 Therefore, the RuV=O complex has two oxidizing equivalents with more comparable electron affinity than the diiron(IV) complex 1. As the result of the above factors, 1-e− oxidation of H2O is favorable for 1, while 2-e− oxidation of H2O is the favored pathway for the RuV=O complex.
The above notions also apply to the H2O oxidation mechanism proposed for nonhaem iron catalysts of Lloret-Fillol and Costas, which has been explored by DFT calculations.6 It is postulated that the transient FeV(O)(OH) species formed upon CeIV oxidation of the iron catalyst abstracts an H-atom from H2O to generate a hydroxyl radical, which then rapidly reacts with the nascent terminal FeIV=O moiety to form the O–O bond. Therefore, the major reason for the different outcomes between the two iron systems in terms of water oxidation products is likely to be the different binding oxomodes, bridging in 1 and terminal in the other. Similarly, a significant difference in C–H bond cleavage reactivity has also been found for two closely related diiron(IV) complexes with bridging and terminal oxos.28
In summary, we have demonstrated that a nonhaemdiiron(IV) complex 1 is capable of oxidizing H2O via a PCET mechanism. Interestingly, the rate-determining step involves one oxidant and two substrate molecules, a strategy that facilitates PT and lowers the activation barrier of ET. Complex 1 appears to be a powerful 1-e− oxidant, but is incapable of carrying out the 2-e− oxidation of H2O to form an O–O bond. The latter is presumably because of the bridging nature of the oxo ligand and the large difference in redox potential between the FeIV,IV/FeIII,IV and FeIII,IV/FeIII,III couples. Our intriguing results thus provide valuable insights into what is required for H2O oxidation and O–O bond formation and set the stage for future efforts aimed at designing efficient H2O oxidation catalysts.
Acknowledgments
We gratefully acknowledge National Institutes of Health grant GM-38767 (to L.Q.) for support of this work.
Notes and references
- 1.Groves JT. J Inorg Biochem. 2006;100:434. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 3.Barber J. Chem Soc Rev. 2009;38:185. doi: 10.1039/b802262n. [DOI] [PubMed] [Google Scholar]
- 4.McEvoy JP, Brudvig GW. Chem Rev. 2006;106:4455. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 5.Fillol JL, Codola Z, Garcia-Bosch I, Gomez L, Pla JJ, Costas M. Nat Chem. 2011;3:807. doi: 10.1038/nchem.1140. [DOI] [PubMed] [Google Scholar]
- 6.Codola Z, Garcia-Bosch I, Acuna-Pares F, Prat I, Luis JM, Costas M, Lloret-Fillol J. Chem Eur J. 2013;19:8042. doi: 10.1002/chem.201301112. [DOI] [PubMed] [Google Scholar]
- 7.Prat I, Mathieson JS, Guell M, Ribas X, Luis JM, Cronin L, Costas M. Nat Chem. 2011;3:788. doi: 10.1038/nchem.1132. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Bosch I, Codola Z, Prat I, Ribas X, Lloret-Fillol J, Costas M. Chem Eur J. 2012;18:13269. doi: 10.1002/chem.201202147. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Farquhar ER, Stubna A, Münck E, Que L., Jr Nat Chem. 2009;1:145. doi: 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue G, Fiedler AT, Martinho M, Münck E, Que L., Jr Proc Natl Acad Sci USA. 2008;105:20615. [Google Scholar]
- 11.Xue G, Wang D, De Hont R, Fiedler AT, Shan X, Münck E, Que L., Jr Proc Natl Acad Sci USA. 2007;104:20713. doi: 10.1073/pnas.0708516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh A, Oliveira FTd, Yano T, Nishioka T, Beach ES, Kinoshita I, Münck E, Ryabov AD, Horwitz CP, Collins TJ. J Am Chem Soc. 2005;127:2505. doi: 10.1021/ja0460458. [DOI] [PubMed] [Google Scholar]
- 13.Kaizer J, Klinker EJ, Oh NY, Rohde JU, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L., Jr J Am Chem Soc. 2004;126:472. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- 14.Que L., Jr Acc Chem Res. 2007;40:493. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 15.Draganic I, Draganic Z, Petkovic L, Nikolic A. J Am Chem Soc. 1973;95:7193. [Google Scholar]
- 16.Murata M, Ohnishi S, Kawanishi S. Chem Res Toxicol. 2001;14:1421. doi: 10.1021/tx010081s. [DOI] [PubMed] [Google Scholar]
- 17.Walling C, El-Taliawi G. J Am Chem Soc. 1973;95:848. [Google Scholar]
- 18.Kim TS, Kim J, Bae S, Choi YY, Kim S. Ind Eng Chem Res. 2007;46:4799. [Google Scholar]
- 19.Tyndall GS, Orlando JJ, Wallington TJ, Hurley MD. J Phys Chem A. 2001;105:5380. [Google Scholar]
- 20.Lewis-Bevan W, Gaston RD, Tyrrell J, Stork WD, Salmon GL. J Am Chem Soc. 1992;114:1933. [Google Scholar]
- 21.Chen Z, Concepcion JJ, Hu X, Yang W, Hoertz PG, Meyer TJ. Proc Natl Acad Sci USA. 2010;107:7225. doi: 10.1073/pnas.1001132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 23.Torrent M, Musaev DG, Morokuma K. J Phys Chem B. 2001;105:4453. [Google Scholar]
- 24.Skulan AJ, Brunold TC, Baldwin J, Saleh L, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2004;126:8842. doi: 10.1021/ja049106a. [DOI] [PubMed] [Google Scholar]
- 25.Mahroof-Tahir M, Muthy NN, Karlin KD, Blackbum NJ, Shaik SN, Zubieta J. Inorg Chem. 1992;31:3001. [Google Scholar]
- 26.Suzuki M. Acc Chem Res. 2007;40:609. doi: 10.1021/ar600048g. [DOI] [PubMed] [Google Scholar]
- 27.Sawyer DT, Sobkowiak A, Roberts JL., Jr . In: Electrochemistry for Chemists. Sawyer DT, Sobkowiak A, Roberts JL Jr, editors. Ch. 9. John Wiley & Sons, Inc; New York: 1995. p. 358. [Google Scholar]
- 28.Xue G, De Hont R, Münck E, Que L., Jr Nat Chem. 2010;2:400. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]