

Abstract
DS is the most frequent genetic cause of intellectual disability characterized by the anomalous presence of three copies of chromosome 21. One of the peculiar features of DS is the onset of Alzheimer’s disease neuropathology after the age of 40 years characterized by deposition of senile plaques and neurofibrillary tangles. Growing studies demonstrated that increased oxidative damage, accumulation of unfolded/damaged protein aggregates and dysfunction of intracellular degradative system are key players in neurodegenerative processes. In this study, redox proteomics approach was used to analyze the frontal cortex from DS subjects under the age of 40 compared with age-matched controls, and proteins found to be increasingly carbonylated were identified. Interestingly, our results showed that oxidative damage targets specifically different components of the intracellular quality control system such as GRP78, UCH-L1, V0-ATPase, cathepsin D and GFAP that couples with decreased activity of the proteasome and autophagosome formation observed. We also reported a slight but consistent increase of Aβ 1–42 SDS- and PBS-soluble form and tau phosphorylation in DS versus CTR. We suggest that disturbance in the proteostasis network could contribute to the accumulation of protein aggregates, such as amyloid deposits and NFTs, which occur very early in DS. It is likely that a sub-optimal functioning of degradative systems occur in DS neurons, which in turn provide the basis for further accumulation of toxic protein aggregates. The results of this study suggest that oxidation of protein members of the proteostatis network is an early event in DS and might contribute to neurodegenerative phenomena.
Keywords: Down syndrome, Oxidative stress, Autophagy, Proteasome, Alzheimer disease, Trisomy 21
1. Introduction
Down syndrome (DS) is the most frequent chromosomal abnormality that causes intellectual disability, resulting primarily from trisomy of the distal part of chromosome 21 [1]. The neuropathology of DS is complex and includes development of Alzheimer disease (AD) neuropathology by 40 years of age, altered free radical metabolism and impaired mitochondrial function that lead to neuronal degeneration [2–6]. Although by the fourth decade of life, individuals with DS display many of the neuropathological features of AD, including senile plaques (SPs) and neurofibrillary tangles (NFTs), individuals may not develop dementia but not until after 50 years of age [7–9]. The accumulation of amyloid beta (Aβ)-peptide in DS brain can be observed as early as 8–12 years of age, and increases during the lifespan [10,11]. Deposits of Aβ in DS are first seen in the frontal and entorhinal cortex and spread to other cortical regions and layers with increasing age [6].
However, the onset of dementia typically appears in DS adults over the age of 50 years suggesting a quite long prodromal phase where clinical signs are virtually undetectable [12–15]. For these reasons, it has been suggested that DS may serve as a model for the study of pathophysiological events early in the development of AD neuropathology. The precise mechanisms, by which trisomy 21 lead to the early-onset of AD-like neuropathology and cognitive decline remains to be elucidated.
An increasing number of studies have recently shown that oxidative stress (OS) occurs in AD pathogenesis in DS and progression due to a deregulation of gene/protein expression associated with the trisomy21 [16]. Increased production of ROS is also accompanied by mitochondrial dysfunction, both of which are considered to be a trigger for development of AD in DS population [17]. Markers of oxidative damage have been observed in brain tissues from DS [6,18], and as early as in fetal DS brain glycation products were measured [19]. A recent study from our group reported increased levels of oxidative stress (increased protein oxidation, lipid peroxidation, reduction of glutathione and thioredoxin levels) and induction of the heat shock protein (HSP) response in the amniotic fluid from women carrying DS fetuses compared with non-DS controls [20]. In the same study, we identified, by a redox proteomics approach, selective proteins that showed increased oxidation in amniotic fluid from mothers carrying DS fetuses compared with non-DS controls. These proteins are involved in iron homeostasis (ceruloplasmin and transferin), lipid metabolism (zinc-α2-glycoprotein, retinol-binding protein 4 and apolipoprotein A1) and inflammation (complement C9, α-1B-glycoprotein, collagen α-1 V chain) and possibly play a role in the clinical outcome of DS [20].
Taken together, our findings and those from others suggest that oxidative damage is an early event in DS pathology and might contribute to the development of deleterious DS phenotypes, including abnormal development and AD-like neuropathology.
In addition to OS, characteristic features of neurodegenerative processes also include accumulation of dysfunctional/damaged protein aggregates and dysfunction of intracellular degradative systems, such as autophagy and the ubiquitin-proteasome system (UPS). Growing evidence highlights the role of autophagy in several age-dependent neurodegenerative disorders characterized by protein accumulation, including AD. Indeed, the well-documented decrease in autophagy function with age may contribute to the accumulation of damaged/ dysfunctional proteins in the brain [21]. Autophagy is one of the major intracellular proteolytic systems in which components of the cell are degraded in lysosomes/vacuoles and recycled [22] and may be regarded a predominantly protective process. Under physiological conditions, and also as a consequence of increased oxidative stress, aberrant proteins are targeted for degradation by the UPS [23]. The UPS is a complex enzymatic pathway that ligates ubiquitin (UBB) to other cellular proteins and assigns them to degradation. The accumulation of Aβ and hyperphosphorylated tau, as SPs and NFTs respectively, in DS brain, as well as in AD, suggests that alterations in protein quality control mechanisms – UPS and autophagy – may be directly or indirectly involved in neurodegenerative processes [24].
In this study, we present the hypothesis that DS brains, prior to significant AD pathology, may show early disturbance of the proteostasis network possibly linked to increased oxidative stress conditions. Redox proteomics approaches [25] were used to analyze the frontal cortex from DS brain compared with age-matched controls to identify proteins with increased carbonylation.
2. Materials and methods
2.1. Subjects
DS and young control cases (8 for each group) were obtained from the University of California-Irvine Alzheimer’s Disease Research Center Brain Tissue Repository (supported by NIH NIA Grant #P50 AG16573.), the Eunice Kennedy Shriver NICHD Brain and Tissue Bank for Developmental Disorders (University of Maryland, Baltimore, MD, contract HHSN275200900011C, Ref. No. N01-HD-9-0011). Table 1 shows the characteristics and demographic data of the subjects included in the study. The mean post mortem interval (PMI) is 12.1 ± 4.7 h for CTR subjects and 13.3 ± 2.1 h for DS subjects.
Table 1.
Patients demographic data; AA = African American, Ca = Caucasian, In = Indian.
| Subjects | PMI | Age | Sex | Race | Cause of death |
|---|---|---|---|---|---|
| Control Y 1 | 5.8 | 39 | Female | Unknown | Unknown |
| Control Y 2 | 12 | 22.8 | Male | African American | Arrhythmia due to hypertrophy cardiomyopathy |
| Control Y 3 | 14 | 19.8 | Male | Caucasian | Multiple injuries |
| Control Y 4 | 8 | 33.1 | Male | Caucasian | Cardiac arrhythmia |
| Control Y 5 | 10 | 24.4 | Male | Caucasian | Multiple injuries |
| Control Y 6 | 10 | 10.8 | Female | Caucasian | Asthma |
| Control Y 7 | 17 | 39 | Female | Caucasian | Chest and abdominal injuries |
| Control Y 8 | 20 | 9 | Female | African American | Asthma |
| CTR Y | 12.1 ± 4.7 | 24.7 ± 11.6 | 4F, 4M | 2AA, 5Ca | /////// |
| DS 1 | 13 | 44.5 | Female | Caucasian | Cardiac arrhythmia |
| DS 2 | 14 | 19.9 | Male | Indian | Cardiopulmonary arrest: congenital heart disease |
| DS 3 | 12 | 2.0 | Male | Caucasian | Unknown |
| DS 4 | 14 | 15.5 | Male | Caucasian | Chromosome disorder, trisomy 21 |
| DS 5 | 10 | 40.6 | Male | African American | HCVD (hypertensive cardiovascular disease) |
| DS 6 | 12 | 39.2 | Female | Caucasian | Cancer |
| DS 7 | 15 | 23 | Male | African American | Pneumonia |
| DS 8 | 17 | 2 | Male | African American | Cardiac arrhythmia |
| DS | 13.3 ± 2.1 | 23.1 ± 16.8 | 2F, 6M | 3AA, 4Ca, 1In | //////// |
2.2. Sample preparation
Frozen brain tissues samples (frontal cortex) from control and DS subjects were sonicated in Media 1 lysis buffer (pH 7.4) containing 320 mM Sucrose, 1% of 990 mM Tris–HCl (pH = 8.8), 0.098 mM MgCl2, 0.076 mM EDTA, the proteinase inhibitors leupeptin (0.5 mg/mL), pepstatin (0.7 μg/mL), aprotinin (0.5 mg/mL), and PMSF (40 μg/mL) and phosphatase inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA). Homogenates were centrifuged at 14,000 ×g for 10 min to remove debris. Protein concentration in the supernatant was determined by the Bradford assay (Pierce, Rockford, IL, USA).
2.3. 2D electrophoresis
Brain sample proteins (200 μg) were precipitated in 15% final concentration of trichloroacetic acid for 10 min in ice. Each individual sample (8 per group) was then spun down at 10 000 g for 5 min and precipitates were washed in ice-cold ethanol-ethyl acetate 1:1 solution four times. The final pellet was dissolved in 200 μl rehydration buffer (8 M urea, 20 mM dithiothreitol (DTT), 2.0% (w/v) Chaps, 0.2% Bio-Lyte, 2 M thiourea, and bromophenol blue). Isoelectric focusing was performed with ReadyStrip IPG Strips (11 cm, pH 3–10; Bio-Rad, Hercules, CA, USA) at 300 V for 2 h linearly, 500 V for 2 h linearly, 1000 V for 2 h linearly, 8000 V for 8 h linearly, and 8000 V for 10 h rapidly. All the above processes were carried out at room temperature. After the first-dimension run the strips were equilibrated two times, first for 10 min in 50 mM Tris–HCl (pH 6.8) containing 6 M urea, 1% (w/v) sodium dodecyl sulfate (SDS), 30% (v/v) glycerol, and 0.5% DTT and again for another 10 min in the same buffer containing 4.5% iodoacetamide in place of DTT. The second dimension was performed using 12% precast Criterion gels (Bio-Rad). The gels were incubated in fixing solution (7% acetic acid, 10% methanol) for 20 min and then stained for 1 h in Bio-Safe Coomassie gel stain (Bio-Rad, Hercules, CA, USA) and destained overnight in deionized water. The Coomassie gels were scanned using a GS 800 densitometer (Bio-Rad, Hercules, CA, USA).
2.4. 2D oxyblot
For 2D OxyBlot, 2D gels (200 μg of proteins) were blotted onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) and 2,4-dinitrophenylhydrazine (DNPH) derivatization was performed. Briefly, membranes were equilibrated in 20% methanol (5 min), then incubated in 2N HCl (5 min), and finally derivatized in 0.5 mM DNPH solution (5 min). After derivatization, three washes using 2 N HCl solution and five washes using methanol 50% were performed (5 min each). Finally the membranes were blocked with 3% albumin in T-TBS and incubated with the primary Rabbit anti-DNP antibody (1:100; Millipore, Billerica, MA, USA) and the secondary antibody alkaline phosphatase-conjugated anti-rabbit IgG (1:5000; Sigma-Aldrich, St Louis, MO, USA). The colorimetric reaction was obtained using 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium solution.
2.5. Image analysis
2D gels and 2D blots were analyzed by PDQuest 2D Analysis (7.2.0 version; Bio-Rad, Hercules, CA, USA). PD-Quest spot-detection software allows the comparison of 2D gels as well as 2D blots from different groups. Powerful auto-matching algorithms quickly and accurately match gels or blots and sophisticated statistical analysis tools identify experimentally significant spots. The intensity value for each spot from an individual gel is normalized using the average mode of background subtraction. This intensity is afterward compared between groups using statistical analysis. Statistical significance was assessed using a two-tailed Student t-test. p values < 0.05 were considered significant for comparison between control and experimental data (CTR vs. DS). PD-Quest software allows normalization of a carbonylated spot intensity on the blot for expression level of the same spot on the gel. One-dimensional blots were analyzed with Quantity One software (4.6.9 version; Bio-Rad, Hercules, CA, USA).
2.6. Trypsin digestion and protein identification by mass spectrometry
Protein spots identified statistically different from controls after PD-Quest analysis were digested in-gel by trypsin. Briefly, spots of interest were excised and then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at room temperature for 15 min. Acetonitrile was added and incubated at room temperature for 15 min. This solvent mixture was then removed and gel pieces were dried. The protein spots were then incubated with 20 ml of 20 mM DTT in 0.1 M NH4HCO3 at 56 °C for 45 min. The DTT solution was removed and replaced with 20 ml of 55 mM iodoacetamide in 0.1 M NH4HCO3. The solution was then incubated at room temperature for 30 min. The iodoacetamide was removed and replaced with 0.2 ml of 50 mM NH4HCO3 at room temperature for 15 min. Acetonitrile (200 ml) was added. After 15 min incubation, the solvent was removed, and the gel spots were dried for 30 min. The gel pieces were rehydrated with 20 ng/ml modified trypsin (Promega, Madison, WI, USA) in 50 mM 67 NH4HCO3 with the minimal volume necessary to cover the gel pieces. The gel pieces were incubated overnight at 37 °C in a shaking incubator. Protein spots of interest were excised and subjected to in-gel trypsin digestion, and the resulting tryptic peptides were analyzed with MALDI ToF and automated nanospray Nanomate Orbitrap XL MS/MS platform [26]. MALDI-ToF MS analyses were performed in a Voyager-DE STR instrument (Applied Biosystems, Framingham, MA, USA) equipped with a 337 nm nitrogen laser and operating in reflector mode. Mass data were obtained by accumulating several spectra from laser shots with an accelerating voltage of 20 kV. Two tryptic autolytic peptides were used for the internal calibration (m/z 842.5100 and 2807.3145). Data were analysed by MoverZ program (v. 2002, http://bioinformatics.genomicsolutions.com), according to default parameters. Identification by peptide mass fingerprint (PMF), with the mono-isotopic mass list, after exclusion of expected contaminant mass values by Peak Erazor program (http://www.protein.sdu.dk/gpmaw/Help/PeakErazor/peakerazor.html), was performed using the Mascot search engine (v. 2.3) against human SwissProt database [(SwissProt 2011_08 (531473 sequences; 188463640 residues)]. Up to one missed cleavage, 50 ppm measurement tolerance, oxidation at methionine (variable modification) and carbamidomethylation at cysteine (fixed modification) were considered. Identifications were validated when the probability-based Mowse protein score was significant according to Mascot. The Orbitrap MS was operated in a data-dependent mode whereby the eight most intense parent ions measured in the FT at 60,000 resolution were selected for ion trap fragmentation with the following conditions: injection time 50 ms, 35% collision energy. MS/MS spectra were measured in the FT at 7500 resolution, and dynamic exclusion was set for 120 s. Each sample was acquired for a total of 2.5 min. MS/MS spectra were searched against the IPI Worms Database using SEQUEST with the following criteria: Xcorr > 2.0, 2.5, 3.0 for +2, +3, and +4 charge states, respectively, and p value (protein and peptide) < 0.01. IPI accession numbers were cross-correlated with SwissProt accession numbers for final protein identification.
2.6.1. Aβ levels
Frozen cortical samples were extracted sequentially in ice cold phosphate buffered saline (PBS, pH 7.4) with a complete protease inhibitor cocktail (PIC) (Amresco, Solon, OH), and centrifuged at 20,800 ×g for 30 mins at 4 °C. Following centrifugation, the supernatant was collected and the pellets were sonicated (10 × 0.5 sec pulses at 100W, Fisher Sonic Dismembrator) in 2% SDS with PIC followed by centrifugation (as above, at 14 °C). The supernatant was again collected, and the remaining pellets were sonicated in 70% formic acid (FA) followed by centrifugation at 20,800 ×g for 1 h at 4 °C. Aβ was measured in tissue samples using a standard, well-characterized, two-site sandwich ELISA as described previously [6]. Briefly, an Immulon 4HBX plate was coated with 0.5 ug/well of antibody, incubated overnight at 4 C, then blocked with a solution of Synblock (Serotec), as per the manufacturer’s instructions. Antigen capture was performed using monoclonal antibody Ab9 (against Human Aβ1–16). Antigen detection was performed using biotinylated antibodies 13.1.1. (end specific for Aβ40), and 12F4 (end specific for Aβ42; Covance, Princeton, NJ).
Formic acid extracted material was initially neutralized by a 1:20 dilution in TP buffer (1 M Tris base, 0.5 M Na2HPO4), followed by a further dilution as needed (1:100 to 1:400) in Antigen Capture buffer (AC) (20 mM Na3PO4, 0.4% Block Ace (AbD Serotec, Raleigh, NC), 0.05% NaN3, 2 mM EDTA, 0.4 M NaCl, 0.2% BSA, 0.05% CHAPS, pH 7). SDS soluble fractions were diluted (1:20) in AC buffer alone. PBS fractions were diluted 1:4 in AC buffer alone. A peptide standard curve of Aβ was run on the same plate for comparison, and standards and samples were run at least in duplicate; Aβ values were determined by interpolation relative to the standard curve. Plates were washed between steps with standard PBS containing 0.05% Tween-20 (2–4×) followed by PBS (2–4×). Plates were developed with TMB reagent (Kirkegaard & Perry Laboratories), stopped with 6% o-phosphoric acid, and read at 450 nm using a BioTek multiwell plate reader.
2.7. Western blot
For Western blot, 30 μg of proteins (CTR and DS) were separated by 12% SDS– PAGE and blotted onto a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 3% bovine serum albumin in T-TBS and incubated for 1 h and 30 min at room temperature with primary anti-LC3 antibody (1:100; Novus Biologicals, Littleton, CO, USA), anti proteasome 20S core subunit (1:1000; ENZO Life Science, Farmingdale, NY, USA) and 19S 10B (1:500; Novus Biologicals, Littleton, CO, USA), anti-pTau (ser404) and anti-Tau (1:500; Santa Cruz Biotechnology, Dallas, TX, USA) and for 1 h at room temperature with secondary antibody horseradish peroxidase-conjugated anti-rabbit IgG (1:5000; Sigma–Aldrich, St Louis, MO, USA). Membranes were developed with the Super Signal West Pico chemiluminescent substrate (Thermo Scientific, Waltham, MA, USA).
2.8. Activity assays
2.8.1. Proteasome
Chymotrypsin-like, trypsin-like and caspase-like activity was assayed in young CTR and DS samples according to Keller et al. [27]. Briefly brain tissues were homogenized in proteolysis activity buffer (Tris–HCl 10 mM, pH 7.4, DTT 0.5 mM, ATP 5 mM, 0.035%SDS and MgCl2 5 mM) at 4 °C. Protein determination was performed with Coomassie Protein Assay (Pierce, Rockford, IL, USA). Protein (250 μg) were incubated 30 min at 37 °C with the fluorogenic chymotrypsin-like substrate, succinyl-LLVY-4-methylcoumaryl-7-amide, with the trypsin-like substrate Z-ARR-4-methylcoumaryl-7-amide and with the caspase-like substrate Z-LLE-4-methylcoumaryl-7-amide (Sigma-Aldrich, St Louis, MO, USA), to a final concentration of 50 μM. The reaction was blocked in ice and fluorescence measures were obtained at 380/460 nm excitation and emission, respectively. The background fluorescence was obtained by incubating homogenized samples with MG132 (Sigma-Aldrich, St Louis, MO, USA) a potent, reversible and cell-permeable proteasome inhibitor, to a final concentration of 60 μM. The incubation with MG 132 was performed 30 min before addition of the proteasome substrate.
2.8.2. Cathepsin D
CatD activity was measured according to Grimm et al. [28] by measuring the changes in fluorescence of DS and CTR brain samples incubated with the fluorogenic substrate, Mca-Gly-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys (Dnp) -D-Arg-NH2 (ENZO Life Science, Farmingdale, NY, USA). Brain tissues, 8 CTR and 8 DS, were homogenized on ice, in 100 mM sodium acetate buffer pH 3.5, 4 mM EDTA and 0.2% Triton X100. Homogenated tissues were incubated with substrate 20 μM in 50 mM sodium acetate buffer pH 4.0 at 40 °C for 1 h. The reaction was blocked by adding TCA 5% and fluorescence was measured at 328/393 Ex/Em respectively.
2.8.3. UCH-L1
The activity of UCH-L1 was measured by determining the rate of conversion of ubiquitin-C-terminal 7-amido-4-methylcoumarin (Ub-AMC) (ENZO Life Science, Farmingdale, NY, USA) to ubiquitin and free AMC [29]. In the assay, buffer (50 mM Hepes, pH 7.0, 10 mM DTT, and 0.1 mg/ml ovalbumin) was mixed with 200 μg of frontal cortex homogenate of 8 samples per group for 1 h. Then 10 μl of 200 nM Ub-AMC was added to the enzyme solution, and cleavage of AMC from Ub-AMC was monitored at 380/460 nm excitation and emission respectively.
2.9. Statistical analysis
All statistical analyses were performed using Student’s t-test. Significance was accepted if the p value < 0.05.
3. Results
3.1. Carbonylation results in DS vs. CTR frontal cortex
The redox proteomics analyses showed increased specific carbonylation levels (normalized to expression levels) of six proteins in frontal cortex of DS subjects compared with non-DS cases (Fig. 2 and Table 3). These proteins identified by MS/MS analysis were: ubiquitin carboxyl-terminal hydrolase 1 (UCH-L1) with 2.12-fold increase, cathepsin D (CatD) with 2.11-fold increase, 78 kDa glucose-regulated protein (GRP78) with 12.9-fold increase, V0-type proton ATPase subunit B, brain isoform (V0-ATPase) with 3.9-fold increase, glial fibrillary acidic protein (GFAP) with 8-fold increase and succinyl-CoA:3-ketoacid-coenzyme A transferase 1 mitochondrial (SCOT-1) with 7.9-fold increase (Fig. 3 and Table 3). The expression levels of the above oxidatively modified proteins revealed a slight increase, between 1.15- to 1.27-fold, for UCH-L1, CatD, GRP78 and GFAP, while no alterations for V0-ATPase and a decrease of about 2-fold (0.48) for SCOT-1 were found (Fig. 3 and Table 3).
Fig. 2.

Representative 2D gel and blot from DS and CTR samples. The spots showing significantly increased carbonyl levels are labeled. The spot numbers indicated on the maps are the same as those listed in Table 2.
Table 3.
Protein with increased carbonylation in DS samples vs. CTR identified by proteomics analyses.
| Spot | Protein | Uniprot number | % sequence coverage | Theoretical MW/PI | p value Ox/Exp | Exp fold DS/CTR | Ox fold DS/CTR | Ox/expr Fold DS/CTR |
|---|---|---|---|---|---|---|---|---|
| 1 | Ubiquitin carboxyl-terminal hydrolase 1 (UCLH-1) | P09936 | 40 | 25151/5.33 | <0.05 | 1.19 | 2.52 * | 2.12 ↑ |
| 2 | Cathepsin D (CatD) | P07339 | 16 | 45037/6.10 | <0.05 | 1.15 | 2.42 * | 2.11 ↑ |
| 3 | 78 kDa glucose-regulated protein (GRP78) | P11021 | 40 | 72402/5.07 | <0.05 | 1.27 * | 16.38 * | 12.9 ↑ |
| 4 | V0-type proton ATPase subunit B, brain isoform (V0-ATPase) | P21281 | 30 | 56807/5.57 | <0.05 | 0.99 | 3.86 * | 3.9 ↑ |
| 5 | Glial fibrillary acidic protein (GFAP) | Q96KS4 | 28 | 49907/5.42 | <0.05 | 1.19 | 9.52 * | 8.0 ↑ |
| 6 | Succinyl-CoA:3-ketoacid-coenzyme A transferase 1, mitochondrial (SCOT-1) | P55809 | 23 | 56578/7.14 | <0.05 | 0.48 * | 3.87 * | 7.9 ↑ |
p < 0.05.
Fig. 3.

Bar graph of identified proteins expression, carbonylation and carbonylation/expression values in CTR and DS samples.
3.2. Aβ levels and tau phosphorylation
We analyzed Aβ 1–40/42 PBS-soluble, SDS-soluble and FA extracted levels in DS compared to CTR subjects. No significant increase in Aβ 1–40 forms but significant slight increases for Aβ 1–42 PBS-soluble, SDS-soluble and FA extracted levels were found in DS compared to CTR. Increased phosphorylation (ser404) in DS frontal cortex compared to CTR was observed, consistent with the notion that trisomy of Chr 21 leads to tau hyperphosphorylation (Fig. 1).
Fig. 1.
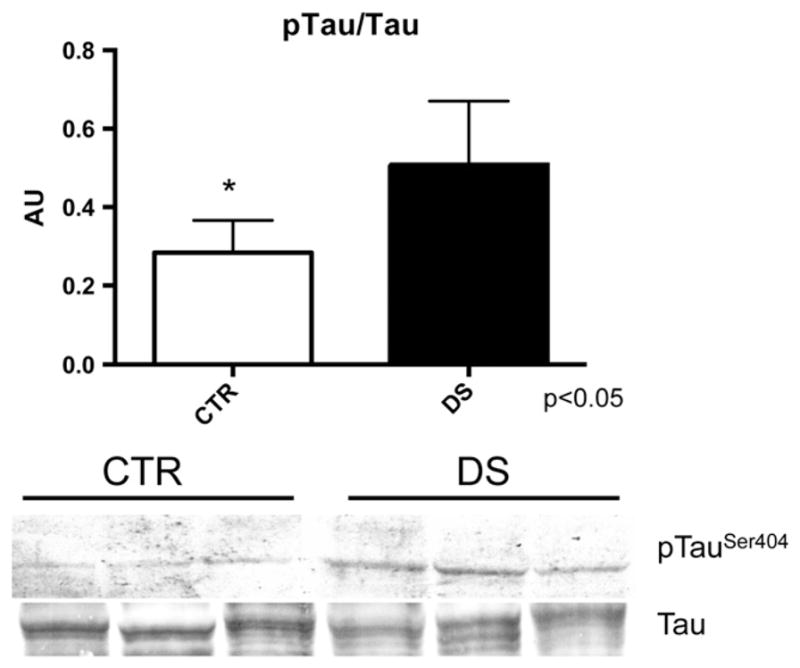
Bar graph and WB image of tau phosphorylation (Ser404) and expression levels in CTR and DS samples.
3.3. Proteasome subunits levels and activity
In order to investigate the functionality of UPS we analyzed proteasome levels (20S and 19S) by WB analysis and proteasome activity by specific assay. The expression levels data show a higher expression of 20S subunit than the 19S subunit of about 80% in CTR subjects, while in DS subject the expression difference of the two subunits is only about 10% (Fig. 4, panel A). The chymotrypsin-like proteasome activity assay demonstrated a significant decrease of proteasome functionality (about 20%) in DS subjects compared with CTR as well as trypsin-like and caspase-like activities that demonstrates about 35 and 40% decrease respectively (Fig. 4, panel B).
Fig. 4.

Panel A: Bar graph of proteasome subunits (20S and 19S) levels in CTR and DS samples measured by WB analysis. Panel B: Proteasome functionality measured by enzymatic assay of chymotrypsin-like, trypsin-like and caspase-like activity in CTR and DS samples. *p < 0.01.
3.4. Cathepsin D and UCH-L1 activity
To analyze if the increased carbonylation of CatD may affect protein function, we measured CatD enzyme activity in the two groups of brain samples. A significant (p < 0.05) slight increase of CatD activity in DS samples, of about 10% compared to CTR samples, was demonstrated (Fig. 5A). As well, the activity of UCH-L1 was measured in order to determine any alteration on its functionality.
Fig. 5.
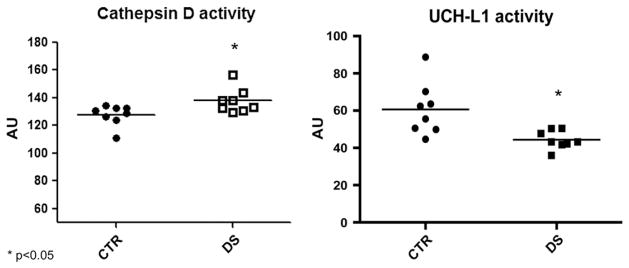
Panel A: Cathepsin D activity in CTR ad DS samples measured by fluorescent assay at 328/393 Ex/Em. Panel B: UCH-L1 activity assay in CTR and DS samples measured by fluorescent assay at 380/460 Ex/Em n = 8; *p < 0.05.
Interestingly, UCH-L1 enzyme activity present a decrease of about 30% in DS compared to CTR.
3.5. LC3 II/I levels
Since our redox proteomics results suggest the impairment of the autophagic pathway, we analyzed by Western blot the expression levels of LC3 II/I, a broadly recognized marker of autophagy activity. Our results shown in Fig. 6 demonstrate about a 40% decrease (from 100 to 60%) of LC3 II/I levels in DS compared to CTR frontal cortex samples.
Fig. 6.

LC3 II/I expression levels in CTR and DS samples. Panel A: Bar graph of the ratio of LC3 II and I values compared to β actin loading control. Panel B: Representative bands of LC3 I and II immunodetection in WB analysis. n = 8; *p < 0.05.
4. Discussion
Growing evidence supports the occurrence of chronic oxidative injury in the brain that could imply a risk factor for subsequent neurodegeneration in aged DS patients [6,30]. The present study aimed to shed light on molecular pathways, perturbed by oxidative stress, which may play a key role in the neurodegenerative phenomena occurring in DS. By applying redox proteomics approaches to analyze the extent of oxidative damage in the cortex of DS brain compared with age-matched controls, we were able to identify specific oxidatively modified proteins. Our findings are discussed in light of the well-established effect of oxidative damage on protein functionality, which results in altered, reduced or toxic gain of function [25].
Interestingly, the majority of these proteins are members of the intracellular Quality Control (QC) system including GRP78, UCH-L1, Cat D, V0-ATPase and GFAP. Our findings suggest that chronic exposure to OS may participate to impair the proteostasis network, which in addition to maintaining correct protein homeostasis is also fundamental to counteract accumulation of Aβ and NFTs [31].
We observed a slight increase of Aβ 1–42 PBS-soluble, SDS-soluble and FA extracted levels in DS frontal cortex compared to CTR and also increased levels of tau phosphorylation. Our results are, consistent with reported increased Aβ levels early in DS, prior to extracellular Aβ deposits [32]. Subsequently high molecular weight aggregates of Aβ may accumulate, enhancing the deposition of plaques and, through Aβ oligomers, ROS production [14,33]. In addition aberrantly phosphorylated tau conceivably might accumulate as a consequence of impaired activity of both proteasome and autophagy systems thus contributing to neurodegeneration in DS.
Intracellular Aβ oligomers may accumulate in the endoplasmic reticulum (ER) and activate the unfolded protein response (UPR) [34,35]. UPR is a primary line of defense of the cell against toxic accumulation of misfolded proteins [24,36] and UPR occurs mainly in three steps: 1) upregulation of molecular chaperones to assist in protein refolding; 2) arrest of further protein synthesis; and 3) ubiquitination of misfolded proteins and their translocation to proteasomes for degradation (i.e., ER-associated degradation, ERAD) [36].
As the first step, the protective mechanism used by cells to adapt to stress of the ER is the induction of members of the glucose-regulated protein (Grp) family, mainly GRP78. The induction of mammalian Grp proteins in response to ER stress involves a complex network of regulators and novel mechanisms. Our data demonstrate a relationship between OS and protein misfolding in DS brain, showing that GRP78 can be a target of OS in vivo. Oxidative damage may result in GRP78 dysfunction (inability to bind to the misfolded proteins), which provides a conceivable mechanistic link between deficits in molecular chaperones, accumulation of misfolded proteins, and risk of cognitive decline. According to our previous studies [20,37], increased levels of GRP78 indicate the induction of the UPS system, as results of the presence of unfolded/misfolded proteins; however, the pro-oxidant environment in DS appears to result in GRP78 oxidation that might lead to dysfunction of the UPR system.
Upon activation of ER-stress response, ERAD mediates ubiquitination of misfolded proteins, which occur in the cytoplasm [38]. Here, the proteasome system involves the activity of different enzymes, ubiquitinating and deubiquitinating enzymes, leading to degradations of cytsolic proteins as well as proteins that fail to pass the QC in the ER. Protein ubiquitination/deubiquitination has emerged as an important mechanism for regulating a variety of cellular processes, including protein degradation, synaptic function, and neuronal apoptosis [39]. We observed that UCH-L1 is a target of oxidative damage in DS brains, and its oxidative modification likely leads to a decreased function in DS brain as demonstrated by activity assay. UCH-L1 has an important role in recycling of ubiquitin through hydrolysis of peptide-ubiquitin bonds and processing of ubiquitin precursors, but it also possesses ubiquitin ligase activity UCH-L1 possesses a well-characterized de-ubiquitinating activity that catalyzes the hydrolysis of carboxyl-terminal esters and amides of ubiquitin to generate monomeric ubiquitin [40]. Oxidative modifications causing irreversible alteration in the conformation and/ or enzymatic activity of UCH-L1 result in deleterious effects on neuronal function and survival. Aberrant ubiquitin hydrolase and/or ligase activity for the identified oxidative modifications of UCH-L1 might lead to dysfunction of the neuronal ubiquitination/de-ubiquitination machinery, causing synaptic deterioration and neuronal degeneration in DS as well as demonstrated in AD brains [41–45]. Previous studies from our laboratory and others demonstrated that the levels of UCH-L1 are decreased in AD brain and also that this enzyme is oxidatively modified [44]. One of the major consequences of aberrant UCH-L1 activity is an impaired proteasome proteolytic system, which will lead to accumulation of damaged proteins and formation of protein aggregates. The proteolytic core of the proteasome or “20S proteasome” is involved in the degradation of oxidatively modified proteins [46–48]. Oxidatively modified proteins can undergo chemical fragmentation or form aggregates because of covalent cross-linking reactions and increased surface hydrophobicity [49]. The recognition of hydrophobic amino acid residues of oxidized proteins and their subsequent degradation by the 20S proteasome could be a selective mechanism to remove oxidatively damaged proteins from the cell [50]. Although we found increased levels of the 20S subunit, the activities of trypsin-like, chymotrypsin-like and caspase-like of the proteasome were decreased in DS brain compared with age-matched controls. It is likely that proteasome activation may be a signal to enhance protein degradation in affected neurons to prevent the formation of toxic aggregates, but at the same time the enzyme is directly or indirectly damaged by ROS and its functionality is affected.
Another major degradation system reported to be activated by ER stress in vitro is autophagy [51–53]. During autophagy, a double membrane is wrapped around an organelle or protein aggregate and this autophagosome subsequently fuses with a lysosome, forming an autophagolysosome where degradation takes place. Healthy neurons only rarely show autophagic structures, whereas many autophagic vacuoles can be observed in AD neuronal cell bodies and dystrophic neuritis [54].
Autophagy is considered as a basal cellular mechanism to control homeostasis of intracellular proteins and organelles. However, autophagy can be also induced to protect cells under various physiological stresses such as nutrient depletion, genotoxic agents, aggregated proteins, or cytokines. In particular, if the first defense mechanism by non-enzymatic molecules and enzymatic scavengers is overwhelmed under oxidative stress, autophagy plays a crucial role in cell protection from harmful oxidized materials by effectively removing them [55]. Unfortunately, these defense mechanisms are not always enough to overcome the oxidative stress, resulting in autophagic cell death (type II programmed cell death) that is distinct from apoptosis (type I programmed cell death) [56]. However, the mechanistic basis coordinating autophagic processes under oxidative stress is still not clear and further studies are needed.
We hypothesize that the oxidation of V0-ATPase pump could lead to autophagy dysfunction. This proton pump is essential for acidic lysosomal pH. Mutation of lysosomal ATPase genes is a well-recognized risk factor for autophagy related neurodegenerative diseases [57,58]. It is reasonable to speculate that once oxidized V0-ATPase has altered ability to regulate intracellular pH thus affecting proper lysosome functionality and autophagy, as was previously seen in a PD model [59]. In addition, a recent report showed that V0-ATPase is necessary for amino acids to activate mTORC1, thus suggesting that V0-ATPase is an active component of the mTOR pathway [60]. Recently, it has been demonstrated that PS1 directly regulates the acidification of lysosomes by controlling the targeting of V03ATPase to these vesicles. AD-linked PS1 mutations or neurons lacking PS1 result in defective lysosomal proteolysis, having a detrimental impact on protein homeostasis and neuronal function. Restoration of lysosomal function by genetic intervention was able to prevent neuropathology and cognitive deficits of an AD mouse model [61,62].
A characteristic hallmark of autophagic activation is the formation of cellular autophagosome punctae containing LC3 II. A decreased ratio of LC3II/I is an index of decreased autophagosome formation, as we show in our results, which suggest a decrease of autophagic flux occurring early in DS brain. Recent studies reported that insulin-like growth factor binding protein-3 (IGFBP-3) enhances autophagy via binding to GRP78 [63], consistent with previous studies that showed that GRP78 is required for stress-induced autophagy [64]. In this context, the oxidation of GRP78, as previously mentioned, could also contribute to autophagy dysfunction.
A newly recognized member of the autophagy machinery is also GFAP. GFAP is expressed in activated astrocytes, where it is thought to help maintain mechanical strength, as well as the shape of cells [65]. The exact function of GFAP remains an enigma, despite the huge number of studies using it as a marker for astrocyte activation. Increased expression of GFAP is a characteristic feature of reactive astrocytes during chronic inflammation that could be associated with the presence of tangles, neuritic plaques and Aβ pathology, and is closely associated with the maturation of the astrocyte [66]. The levels of GFAP dynamically respond to aging as well as to neurodegenerative lesions, [67,68], and many studies have shown that the amount of GFAP generally increases in neurodegenerative diseases such as AD [69]. A recent study showed for the first time that GFAP is an important regulator for chaperone-mediated autophagy (CMA), a degradative mechanism for cytosolic proteins in lysosomes [70]. GFAP was proposed to interact at the lysosomal membrane either with the lysosome-associated membrane protein type 2A (LAMP-2A), an important component of the translocation complex, or with the elongation factor 1a (ef1a). Since CMA is activated as part of the cellular response to oxidative stress required for targeting oxidized proteins to lysosomes, oxidation of GFAP might contribute to disrupt the complex network involved in autophagy processes [71]. Previous in vitro studies showing the carbonylation of GFAP by Aβ (1–42) are consistent with our hypothesis [72].
Intriguingly, we found that CatD is oxidized in DS compared with non-DS controls resulting in a slight increase of enzyme activity. CatD is normally localized within lysosomes and participates in the degradation of proteins, downstream autophagy, and processing of precursor proteins [73,74]. Evidence exists that CatD is involved in AD pathogenesis [75–77] and CatD has been found within neuritic plaques in AD brains [78,79]. Indeed, it has been reported that CatD possess beta-secretase activity and this function may play a key role in Aβ processing [80]. The hypothesis of a CatD role in amyloidogenesis initially came from the first evidence by Nixon and co-workers in 1990 [79]. The authors observed in sections from AD brains, stained by antisera to both cathepsin D and B, high levels of inmunoreactivity in senile plaques. However, APP intracellular processing to generate Aβ has remained controversial. It has been proposed that APP cleavage may occur in different organelles along the endolysosomal pathway including the ER, the Golgi apparatus, the plasma membrane, early, late, or recycling endosomes and the lysosome [81]. It is likely that a crosstalk between these different organelles may occur and the exact location of APP processing would depend on the active traffic of the precursor and the secretase enzymes. Within this frame, we suggest that increased oxidation of CatD results in a “gain of its function” as already demonstrated for other proteins, such as SOD-1 in the case of amyotrophic lateral sclerosis [29,82]. The activation of CatD, resulting either from its increased expression or enzyme activity, may be a compensatory response to partially disturbed autophagy and/or alternatively be involved in APP processing thus resulting in increased Aβ production, as occurs early in DS pathology [77,80].
Recent in vitro data suggest that activation of lysosomal cathepsin, either D and B forms, may be reflective of early compensatory processes that keep protein accumulation events partially in check and contribute to the progressive nature of AD pathology [83].
All the above-mentioned processes rely, to varying extents, on ATP consumption and are closely related to mitochondrial integrity. Indeed, the AD brain has altered mitochondrial functions and altered glucose metabolism, which ultimately culminates in decreased ATP synthesis [84]. Alteration in the brain metabolic profile of AD is associated with a concomitant metabolism of ketone bodies to compensate for the decline in glucose-driven ATP generation. It has been demonstrated that ketone bodies and fatty acids can replace glucose as the primary energy source for brain under pathophysiological conditions [85]. We found in the current study that succinyl-CoA:3-ketoacid-coenzyme A transferase 1 (SCOT) mitochondrial, is more oxidized in DS. This homodimeric mitochondrial matrix enzyme plays a central role in extrahepatic ketone body catabolism by catalyzing the reversible transfer of coenzyme A from succinyl-CoA to acetoacetate (SCOT). Thus, oxidation of SCOT might indicate a partial failure of compensatory responses to generate an alternative fuel source for a reduction in glucose utilization.
5. Conclusions
Our study suggests that DS may be included in the family of “protein deposition disorders”, where inappropriate protein deposits arise when the normally efficient protein folding quality control system is overwhelmed [86]. The correct balance between folding and degradation of misfolded proteins is critical for cell viability [87]. Taken together, our findings highlight that oxidative damage targets mainly proteasome and autophagy systems in DS subjects and contribute to disturbance of the proteostasis network. The cross-talk between different members of the QC machinery is essential to avoid dangerous accumulation of misfolded/aggregated proteins [21].
We suggest that accumulation of Aβ and NFT in DS may result both from its increased production and from the disruption of cellular degradative systems (e.g. UPS and autophagy). Indeed studies from Nixon’s group demonstrated alterations in function of the endosomal-autophagic-lysosomal system in DS [88]. In this scenario chronic oxidative stress occurs in DS brain and contributes to Aβ and NFT accumulation, fostering the question: how do neuronal cells try to survive?
ER stress elicits the unfolded protein response, including upregulation of molecular chaperones and induction of ERAD. In ERAD, the Aβ oligomers are translocated from the ER into the cytoplasm to be degraded by proteasomes. Damaged ER and undergraded Aβ oligomers in the cytoplasm are engulfed by autophagosomes and sorted to lysosomes to be digested. Oxidation of GRP78, UCH-L1, CatD, V0-ATPase and GFAP demonstrate modification and relative dysfunction of proteins involved at different steps of the proteostasis network. This dysfunction is exacerbated by the decreased activities of the proteasome and the potential impairment of autophagy.
In parallel, hyperphosphorylated tau might be able to escape dysfunctional quality control mechanisms represented by ER, proteasome and autophagy activities, leading to its aggregation and NFT formation (Scheme 1).
Scheme 1.

Putative scenario of QC protein oxidation and Aβ and hpTau degradation/accumulation in DS brain.
Though observational and comparative, it is tempting to speculate that DS neurons are characterized by increased oxidative stress combined with defective degradative systems which in turn results in further accumulation of toxic protein aggregates. Since DS may be considered a prodromal human model of AD, our study suggests that the impairment of proteostatis network represents an early event in neurodegenerative phenomena. Future studies directed to investigate whether activation/modulation of the QC system may be a therapeutic target are underway.
Table 2.
Amyloid β 1–40 and 1–42 levels in frontal cortex of CTR and DS subjects.
| Aβ 1–42 PBS-soluble (pmol/mg) | Aβ 1–42 SDS-soluble (pmol/mg) | Aβ 1–42 FA-extracted (pmol/mg) | Aβ 1–40 PBS-soluble (pmol/mg) | Aβ 1–40 SDS-soluble (pmol/mg) | Aβ 1–40 FA-extracted(pmol/mg) | |
|---|---|---|---|---|---|---|
| CTR (Avg) | 52.9 | 233.6 | 1502.7 | 3.4 | 198.2* | 1409.2 |
| DS (Avg) | 147.4* | 4315.3* | 3100.9* | 1.3 | 6.5 | 1794.3 |
Significant differences.
Acknowledgments
This work is partially supported by PRIN2009 to C.C. and C.M. and NIH [AG-05119] to D.A.B. Brain tissue was acquired by EH under funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institute on Aging Grant #NIH 1RO1HD064993-01.
References
- 1.Ness S, Rafii M, Aisen P, Krams M, Silverman W, Manji H. Down’s syndrome and Alzheimer’s disease: towards secondary prevention. Nat Rev Drug Discov. 2012;11:655–656. doi: 10.1038/nrd3822. [DOI] [PubMed] [Google Scholar]
- 2.Busciglio J, Pelsman A, Helguera P, Ashur-Fabian O, Pinhasov A, Brenneman DE, Gozes I. NAP and ADNF-9 protect normal and Down’s syndrome cortical neurons from oxidative damage and apoptosis. Curr Pharm Des. 2007;13:1091–1098. doi: 10.2174/138161207780618957. [DOI] [PubMed] [Google Scholar]
- 3.Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33:677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 4.de Haan JB, Susil B, Pritchard M, Kola I. An altered antioxidant balance occurs in Down syndrome fetal organs: implications for the “gene dosage effect” hypothesis. J Neural Transm Suppl. 2003:67–83. doi: 10.1007/978-3-7091-6721-2_6. [DOI] [PubMed] [Google Scholar]
- 5.Lott IT, Head E. Down syndrome and Alzheimer’s disease: a link between development and aging. Ment Retard Dev Disabil Res Rev. 2001;7:172–178. doi: 10.1002/mrdd.1025. [DOI] [PubMed] [Google Scholar]
- 6.Cenini G, Dowling AL, Beckett TL, Barone E, Mancuso C, Murphy MP, Levine H, III, Lott IT, Schmitt FA, Butterfield DA, Head E. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta. 2012;1822:130–138. doi: 10.1016/j.bbadis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mann DM, Yates PO, Marcyniuk B, Ravindra CR. The topography of plaques and tangles in Down’s syndrome patients of different ages. Neuropathol Appl Neurobiol. 1986;12:447–457. doi: 10.1111/j.1365-2990.1986.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 8.Visser FE, Aldenkamp AP, van Huffelen AC, Kuilman M, Overweg J, van Wijk J. Prospective study of the prevalence of Alzheimer-type dementia in institutionalized individuals with Down syndrome. Am J Ment Retard. 1997;101:400–412. [PubMed] [Google Scholar]
- 9.Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM. Alzheimer’s disease in Down’s syndrome: clinicopathologic studies. Neurology. 1985;35:957–961. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- 10.Wegiel J, Wisniewski HM, Dziewiatkowski J, Popovitch ER, Tarnawski M. Differential susceptibility to neurofibrillary pathology among patients with Down syndrome. Dementia. 1996;7:135–141. doi: 10.1159/000106868. [DOI] [PubMed] [Google Scholar]
- 11.Leverenz JB, Raskind MA. Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol. 1998;150:296–304. doi: 10.1006/exnr.1997.6777. [DOI] [PubMed] [Google Scholar]
- 12.Head E, Lott IT, Patterson D, Doran E, Haier RJ. Possible compensatory events in adult Down syndrome brain prior to the development of Alzheimer disease neuropathology: targets for nonpharmacological intervention. J Alzheimers Dis. 2007;11:61–76. doi: 10.3233/jad-2007-11110. [DOI] [PubMed] [Google Scholar]
- 13.Head E, Doran E, Nistor M, Hill M, Schmitt FA, Haier RJ, Lott IT. Plasma amyloid-beta as a function of age, level of intellectual disability, and presence of dementia In down syndrome. J Alzheimers Dis. 2011;23:399–409. doi: 10.3233/JAD-2010-101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Head E, Lott IT. Down syndrome and beta-amyloid deposition. Curr Opin Neurol. 2004;17:95–100. doi: 10.1097/00019052-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Schupf N, Sergievsky GH. Genetic and host factors for dementia in Down’s syndrome. Br J Psychiatry. 2002;180:405–410. doi: 10.1192/bjp.180.5.405. [DOI] [PubMed] [Google Scholar]
- 16.Perluigi M, Butterfield DA. Oxidative stress and Down syndrome: a route toward Alzheimer-like dementia. Curr Gerontol Geriatr Res. 2012;2012:724904. doi: 10.1155/2012/724904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in-vitro. Nature. 1995;378:776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds GP, Cutts AJ. Free radical damage in Down’s syndrome brain. Biochem Soc Trans. 1993;21:221S. doi: 10.1042/bst021221s. [DOI] [PubMed] [Google Scholar]
- 19.Odetti P, Angelini G, Dapino D, Zaccheo D, Garibaldi S, Dagna-Bricarelli F, Piombo G, Perry G, Smith M, Traverso N, Tabaton M. Early glycoxidation damage in brains from Down’s syndrome. Biochem Biophys Res Commun. 1998;243:849–851. doi: 10.1006/bbrc.1998.8186. [DOI] [PubMed] [Google Scholar]
- 20.Perluigi M, di Domenico F, Fiorini A, Cocciolo A, Giorgi A, Foppoli C, Butterfield DA, Giorlandino M, Giorlandino C, Schinina ME, Coccia R. Oxidative stress occurs early in Down syndrome pregnancy: a redox proteomics analysis of amniotic fluid. Proteomics Clin Appl. 2011;5:167–178. doi: 10.1002/prca.201000121. [DOI] [PubMed] [Google Scholar]
- 21.Nijholt DA, De Kimpe L, Elfrink HL, Hoozemans JJ, Scheper W. Removing protein aggregates: the role of proteolysis in neurodegeneration. Curr Med Chem. 2011;18:2459–2476. doi: 10.2174/092986711795843236. [DOI] [PubMed] [Google Scholar]
- 22.Klionsky DJ. An autophagy glossary. Autophagy. 2010;6 [Google Scholar]
- 23.Riederer BM, Leuba G, Vernay A, Riederer IM. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp Biol Med (Maywood) 2011;236:268–276. doi: 10.1258/ebm.2010.010327. [DOI] [PubMed] [Google Scholar]
- 24.Scheper W, Nijholt DA, Hoozemans JJ. The unfolded protein response and proteostasis in Alzheimer disease: preferential activation of autophagy by endoplasmic reticulum stress. Autophagy. 2011;7:910–911. doi: 10.4161/auto.7.8.15761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butterfield DA, Perluigi M, Reed T, Muharib T, Hughes CP, Robinson RA, Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal. 2012;17:1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mangialasche F, Polidori MC, Monastero R, Ercolani S, Camarda C, Cecchetti R, Mecocci P. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res Rev. 2009;8:285–305. doi: 10.1016/j.arr.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 28.Grimm S, Horlacher M, Catalgol B, Hoehn A, Reinheckel T, Grune T. Cathepsins D and L reduce the toxicity of advanced glycation end products. Free Radic Biol Med. 2012;52:1011–1023. doi: 10.1016/j.freeradbiomed.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 29.Poon HF, Hensley K, Thongboonkerd V, Merchant ML, Lynn BC, Pierce WM, Klein JB, Calabrese V, Butterfield DA. Redox proteomics analysis of oxidatively modified proteins in G93A–SOD1 transgenic mice—a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;39:453–462. doi: 10.1016/j.freeradbiomed.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 30.Pagano G, Castello G. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv Exp Med Biol. 2012;724:291–299. doi: 10.1007/978-1-4614-0653-2_22. [DOI] [PubMed] [Google Scholar]
- 31.Gestwicki JE, Garza D. Protein quality control in neurodegenerative disease. Prog Mol Biol Transl Sci. 2012;107:327–353. doi: 10.1016/B978-0-12-385883-2.00003-5. [DOI] [PubMed] [Google Scholar]
- 32.Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25:1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 33.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 34.Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H. Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res. 2011;89:1031–1042. doi: 10.1002/jnr.22640. [DOI] [PubMed] [Google Scholar]
- 35.Chafekar SM, Zwart R, Veerhuis R, Vanderstichele H, Baas F, Scheper W. Increased Abeta1-42 production sensitizes neuroblastoma cells for ER stress toxicity. Curr Alzheimer Res. 2008;5:469–474. doi: 10.2174/156720508785908883. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 37.Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, Butterfield DA. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: implications for progression of AD. J Proteome Res. 2009;8:471–482. doi: 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goder V. Roles of ubiquitin in endoplasmic reticulum-associated protein degradation (ERAD) Curr Protein Pept Sci. 2012;13:425–435. doi: 10.2174/138920312802430572. [DOI] [PubMed] [Google Scholar]
- 40.Gong B, Leznik E. The role of ubiquitin C-terminal hydrolase L1 in neurodegenerative disorders. Drug News Perspect. 2007;20:365–370. doi: 10.1358/dnp.2007.20.6.1138160. [DOI] [PubMed] [Google Scholar]
- 41.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 42.Butterfield DA, Gnjec A, Poon HF, Castegna A, Pierce WM, Klein JB, Martins RN. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer’s disease: an initial assessment. J Alzheimers Dis. 2006;10:391–397. doi: 10.3233/jad-2006-10407. [DOI] [PubMed] [Google Scholar]
- 43.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 44.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 45.Hegde AN. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010;17:314–327. doi: 10.1101/lm.1504010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grune T, Reinheckel T, Joshi M, Davies KJA. Proteolysis in cultured liver epithelial-cells during oxidative stress — role of the multicatalytic proteinase complex, proteasome. J Biol Chem. 1995;270:2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- 47.Rivett AJ. The effect of mixed-function oxidation of enzymes on their susceptibility to degradation by a nonlysosomal cysteine proteinase. Arch Biochem Biophys. 1985;243:624–632. doi: 10.1016/0003-9861(85)90540-5. [DOI] [PubMed] [Google Scholar]
- 48.Giulivi C, Pacifici RE, Davies KJA. Exposure of hydrophobic moieties promotes the selective degradation of hydrogen peroxide-modified hemoglobin by the multicatalytic proteinase complex, proteasome. Arch Biochem Biophys. 1994;311:329–341. doi: 10.1006/abbi.1994.1245. [DOI] [PubMed] [Google Scholar]
- 49.Butterfield DA, Stadtman ER. Protein oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- 50.Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11:526–534. [PubMed] [Google Scholar]
- 51.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imaizumi K. Autophagy is activated for cell survival after ER stress. J Pharmacol Sci. 2007;103:45P. [Google Scholar]
- 53.Yorimitsu T, Nair U, Yang ZF, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barnett A, Brewer GJ. Autophagy in aging and Alzheimer’s disease: pathologic or protective? J Alzheimers Dis. 2011;25:385–394. doi: 10.3233/JAD-2011-101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006;27:444–454. doi: 10.1016/j.mam.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci. 2000;113(Pt 7):1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 57.Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, Marini P, Squitieri F, Horstink MW, Montagna P, Libera AD, Stocchi F, Goldwurm S, Ferreira JJ, Meco G, Martignoni E, Lopiano L, Jardim LB, Oostra BA, Barbosa ER, Bonifati V N. Italian Parkinson Genetics. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68:1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08. [DOI] [PubMed] [Google Scholar]
- 58.Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 59.Di Domenico F, Sultana R, Ferree A, Smith K, Barone E, Perluigi M, Coccia R, Pierce W, Cai J, Mancuso C, Squillace R, Wiengele M, Dalle-Donne I, Wolozin B, Butterfield DA. Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: relevance to Parkinson disease. Antioxid Redox Signal. 2012;17:1490–1506. doi: 10.1089/ars.2011.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morawe T, Hiebel C, Kern A, Behl C. Protein homeostasis, aging and Alzheimer’s disease. Mol Neurobiol. 2012;46:41–54. doi: 10.1007/s12035-012-8246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grkovic S, O’Reilly VC, Han S, Hong M, Baxter RC, Firth SM. IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene. 2012 doi: 10.1038/onc.2012.264. [DOI] [PubMed] [Google Scholar]
- 64.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15:1460–1471. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol. 2011;93:421–443. doi: 10.1016/j.pneurobio.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 66.Korolainen MA, Auriola S, Nyman TA, Alafuzoff I, Pirttila T. Proteomic analysis of glial fibrillary acidic protein in Alzheimer’s disease and aging brain. Neurobiol Dis. 2005;20:858–870. doi: 10.1016/j.nbd.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 67.Eddleston M, Mucke L. Molecular profile of reactive astrocytes — implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomes FC, Paulin D, Moura Neto V. Glial fibrillary acidic protein (GFAP): modulation by growth factors and its implication in astrocyte differentiation. Braz J Med Biol Res. 1999;32:619–631. doi: 10.1590/s0100-879x1999000500016. [DOI] [PubMed] [Google Scholar]
- 69.Beach TG, Walker R, McGeer EG. Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia. 1989;2:420–436. doi: 10.1002/glia.440020605. [DOI] [PubMed] [Google Scholar]
- 70.Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell. 2010;39:535–547. doi: 10.1016/j.molcel.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boyd-Kimball D, Castegna A, Sultana R, Poon HF, Petroze R, Lynn BC, Klein JB, Butterfield DA. Proteomic identification of proteins oxidized by Abeta(1–42) in synaptosomes: implications for Alzheimer’s disease. Brain Res. 2005;1044:206–215. doi: 10.1016/j.brainres.2005.02.086. [DOI] [PubMed] [Google Scholar]
- 73.Grimm S, Ernst L, Grotzinger N, Hohn A, Breusing N, Reinheckel T, Grune T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radic Res. 2010;44:1013–1026. doi: 10.3109/10715762.2010.495127. [DOI] [PubMed] [Google Scholar]
- 74.Benes P, Vetvicka V, Fusek M. Cathepsin D—many functions of one aspartic protease. Crit Rev Oncol Hematol. 2008;68:12–28. doi: 10.1016/j.critrevonc.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schuur M, Ikram MA, van Swieten JC, Isaacs A, Vergeer-Drop JM, Hofman A, Oostra BA, Breteler MM, van Duijn CM. Cathepsin D gene and the risk of Alzheimer’s disease: a population-based study and meta-analysis. Neurobiol Aging. 2011;32:1607–1614. doi: 10.1016/j.neurobiolaging.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 76.Aluise CD, Robinson RA, Beckett TL, Murphy MP, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Preclinical Alzheimer disease: brain oxidative stress, Abeta peptide and proteomics. Neurobiol Dis. 2010;39:221–228. doi: 10.1016/j.nbd.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, Nixon RA. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early upregulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 78.Bernstein HG, Bruszis S, Schmidt D, Wiederanders B, Dorn A. Immunodetection of cathepsin-D in neuritic plaques found in brains of patients with dementia of Alzheimer type. J Hirnforsch. 1989;30:613–618. [PubMed] [Google Scholar]
- 79.Cataldo AM, Thayer CY, Bird ED, Wheelock TR, Nixon RA. Lysosomal proteinase antigens are prominently localized within senile plaques of Alzheimer’s disease: evidence for a neuronal origin. Brain Res. 1990;513:181–192. doi: 10.1016/0006-8993(90)90456-l. [DOI] [PubMed] [Google Scholar]
- 80.Hook V, Schechter I, Demuth HU, Hook G. Alternative pathways for production of beta-amyloid peptides of Alzheimer’s disease. Biol Chem. 2008;389:993–1006. doi: 10.1515/BC.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choy RW, Cheng Z, Schekman R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid beta (Abeta) production in the trans-Golgi network. Proc Natl Acad Sci U S A. 2012;109:E2077–E2082. doi: 10.1073/pnas.1208635109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karch CM, Prudencio M, Winkler DD, Hart PJ, Borchelt DR. Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc Natl Acad Sci U S A. 2009;106:7774–7779. doi: 10.1073/pnas.0902505106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Butler D, Hwang J, Estick C, Nishiyama A, Kumar SS, Baveghems C, Young-Oxendine HB, Wisniewski ML, Charalambides A, Bahr BA. Protective effects of positive lysosomal modulation in Alzheimer’s disease transgenic mouse models. PLoS One. 2011;6:e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sultana R, Butterfield DA. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. J Bioenerg Biomembr. 2009;41:441–446. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yao J, Rettberg JR, Klosinski LP, Cadenas E, Brinton RD. Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol Aspects Med. 2011;32:247–257. doi: 10.1016/j.mam.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soto C, Estrada LD. Protein misfolding and neurodegeneration. Arch Neurol. 2008;65:184–189. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- 87.McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: chaperones culling corrupt conformations. Nat Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- 88.Cataldo AM, Mathews PM, Boiteau AB, Hassinger LC, Peterhoff CM, Jiang Y, Mullaney K, Neve RL, Gruenberg J, Nixon RA. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol. 2008;173:370–384. doi: 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]