

Abstract
Congenital Long QT syndrome (LQTS) is a genetically heterogeneous collection of heritable disorders of myocardial repolarization linked by their shared clinical phenotype of QT prolongation on electrocardiogram and an increased risk of potentially life-threatening cardiac arrhythmias. At the molecular level, mutations in 15 distinct LQTS-susceptibility genes that encode ion channel pore-forming α-subunits and accessory/auxiliary subunits central to the electromechanical function of the heart have been implicated in its pathogenesis. Over the past two decades, our evolving understanding of the electrophysiological mechanisms by which specific genetic substrates perturb the cardiac action potential has translated into vastly improved approaches to the diagnosis, risk stratification, and treatment of patients with LQTS. In this Review, we detail how our understanding of the molecular underpinnings of LQTS has yielded numerous clinically meaningful genotype-phenotype correlations and how these insights have translated into genotype- and phenotype-guided approaches to the clinical management of LQTS.
INTRODUCTION
Over the last two decades, advances at the bench and bedside have broadened our understanding of the pathogenesis and clinical management of congenital long QT syndrome (LQTS), a potentially lethal genetic disorder of cardiac repolarization that represents a leading cause of sudden cardiac death (SCD), particularly autopsy negative SCD, in the young. Clinically, LQTS is characterized clinically by a prolonged heart rate-corrected QT interval (QTc) on electrocardiogram (ECG) and a predilection for LQTS-triggered cardiac events including syncope, seizures, and or sudden cardiac arrest, often during times of emotional or physical duress. [1, 2]
Classically, LQTS follows two distinct patterns of inheritance: the autosomal dominant Romano-Ward syndrome (RWS) [3, 4] with an estimated prevalence between 1:2,000 and 1:5,000 individuals [5] that presents with an isolated cardiac phenotype and the autosomal recessive Jervell and Lange-Nielsen syndrome (JLNS) [6, 7] with an estimated prevalence between 1:1,000,000 and 1:4,000,000 that presents with bilateral sensorineural deafness in addition to a malignant LQTS cardiac phenotype. In reality, LQTS represents a genetically and phenotypically heterogeneous collection of disorders, that also includes rare multisystem disorders, such as Timothy syndrome (TS), characterized by a host of physical and or developmental abnormalities in addition to the classic phenotype of QT prolongation and an increased risk of SCD. [8] Furthermore, as our understanding of the genetic basis of LQTS continues to expand, it has become clear that LQTS, like many monogenic disorders, is subject to the genetic phenomena of incomplete penentrance and variable expressivity, whereby genotype-positive family members display a spectrum of clinical phenotypes ranging from a lifelong asymptomatic state to sudden death in infancy. [9] As such, the interplay between genotype and phenotype in LQTS is likely far more complex than previously envisioned.
While only a small minority of the >250,000 annual sudden deaths in the United States are attributable to LQTS and other heritable arrhythmia syndromes [10, 11], for several reasons it remains important for all practicing cardiologists to develop/maintain a working knowledge of the pathogenic basis, diagnostic approaches, and phenotype- and genotype-guided clinical management of patients with LQTS. First and foremost, LQTS represents a potentially life-threatening, yet highly treatable genetic disorder. Given the marked reduction in mortality observed with proper treatment, there is simply no excuse for clearly symptomatic patients to go undiagnosed, untreated, or improperly managed. Secondly, the level of effort and scrutiny dedicated to the elucidation of genotype-phenotype correlations in LQTS is virtually unrivaled within the realm of cardiovascular disease. As such, the translation of our understanding of the molecular mechanisms underling LQTS pathogenesis to the development of novel and clinically meaningful genotype- and phenotype-specific approaches to LQTS diagnosis and treatment serves as a prototype or paradigm that could be broadly applicable to the study of other inherited and acquired forms of SCD-predisposing cardiovascular disorders in the post-genomic era.
In this review, we describe our current understanding of the electrophysiologic and genetic basis of LQTS, the standard diagnostic approaches used to gleam important genotypic and phenotypic information, and lastly how our growing mechanistic understanding of LQTS pathogenesis has led to the development of clinically meaningful approaches to the genotype- and phenotype-guided clinical management of LQTS.
GENETIC AND ELECTROPHSYIOLOGIC BASIS OF LONG QT SYNDROME
The electromechanical function of the heart, which is reflected by electrocardiographic parameters such as the QT interval, is dependent on the coordinated activation and inactivation of inward depolarizing and outward repolarization currents that underlie the major phases of the cardiac action potential (Figure 1). [12, 13] Genetic defects in the ion channel’s pore-forming (α) and auxiliary subunits responsible for conducting these currents that enhance depolarizing Na+ and Ca2+ currents (INa and ICa,L) or diminish repolarizing potassium currents (IKs, IKr, and IK1) can prolong the ventricular cardiac action potential (Figure 1a), resulting in prolongation of the heart rate-corrected QT interval (QTc) on surface ECG. Although by no means equivalent to a diagnosis of LQTS, a guideline’s based definition of “prolonged QTc” is satisfied by males with a QTc > 450 ms and by females with a QTc > 460 ms (Figure 1a and 1c). [9, 14] In the setting of QT prolongation, increased cardiomyocyte refractoriness and enhancement of the Na+/Ca2+ current leads to the abnormal spontaneous activation of the L-type Ca2+ channel, which may provide the pathogenic substrate for early afterdepolarization-triggered torsades de pointes (TdP), the hallmark and sudden death-predisposing form of polymorphic ventricular fibrillation observed in LQTS. [15]
Figure 1. Electrophysiological basis of long QT syndrome.

a. Tracings of the normal cardiac ventricular action potential (blue) observed in health and prolonged cardiac ventricular action potential (green) observed in long-QT syndrome. b. Schematic representation of a normal ECG (blue) and QT interval prolongation (green). c. Schematic depiction of normal (blue) and prolonged (green) QT intervals. Abbreviations: LQTS, long QT syndrome; ICa,L, L-type calcium current; IK1, inwardly rectifying current; IKr, rapid component of the delayed rectifier potassium current; IKs, slow-component of the delayed rectifier potassium current; INa, cardiac sodium current.
Over the past two decades, 15 distinct LQTS-susceptibility genes, each encoding a critical pore forming α- or auxiliary subunit of key cardiac ion channels, have been identified through a combination of classical linkage analysis and/or mutational analysis of biologically plausible, candidate genes (Figure 2). Following the identification of the 3 major LQTS-susceptibility responsible for the vast majority of LQTS cases [16–18], 12 minor LQTS-susceptibility genes were described subsequently (Table 1). The following paragraphs briefly review the genetic basis of LQTS, including pertinent aspects of the major, minor, and multisystem LQTS genotypes as well as so-called “modifier” genetic loci associated with the modulation of LQTS disease severity.
Figure 2. Current-centric classification of long QT syndrome (LQTS)-susceptibility genes.

Perturbation of ventricular cardiac action potential depolarization (purple), repolarization (orange), or adapter/signaling proteins that influence depolarization/repolarization (maroon) by mutations in long QT syndrome-susceptibility genes are grouped according to the specific current perturbed by the underlying genetic defect. Blue circles represent loss-of-function mutations to the specified current, whereas green circles represent a gain-of-function. Solid lines indicate those disorders that are autosomal dominant, whereas dashed lines indicate those disorders that are autosomal recessive. Abbreviations: ABS, ankyrin-B syndrome; ATS, Andersen-Tawil syndrome; LQT, long-QT syndrome; ICa,L, L-type calcium current; IK1, inwardly rectifying current; IKr, rapid component of the delayed rectifier potassium current; IKs, slow-component of the delayed rectifier potassium current; INa, cardiac sodium current; and TS, Timothy syndrome. Adapted from Giudicessi, J.R., and Ackerman, M.J. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Translational Research 161(1), 1–14 (2013) with permission from Elsevier.
Table 1.
Genetic basis of long-QT syndrome and multisystem syndromes associated with QT prolongation.
| Gene (Genotype) | Locus | Protein | Functional Effect | Mode of Inheritance | Frequency | Ref. |
|---|---|---|---|---|---|---|
| Long QT Syndrome (Major) | ||||||
| KCNQ1 (LQT1) | 11p15.5 | Kv7.1 | Reduced IKs | AD; AR | 30–35% | [16, 25] |
| KCNH2 (LQT2) | 7q35-46 | Kv11.1 | Reduced IKr | AD | 25–30% | [17] |
| SCN5A (LQT3) | 3p21-p24 | Nav1.5 | Increased INa | AD | 5–10% | [18] |
| Long QT Syndrome (Minor) | ||||||
| AKAP9 (AKAP9-LQTS) | 7q21-q22 | Yotiao | Reduced IKs | AD | <1% | [38] |
| CACNA1C (CACNA1C-LQTS) | 12p13.3 | Cav1.2 | Increased ICa,L | AD | <1% | [132] |
| CAV3 (CAV3-LQTS) | 3p25 | Caveolin 3 | Increased INa | AD | <1% | [45] |
| KCNE1 (KCNE1-LQTS) | 21q22.1 | MinK | Reduced IKs | AD | <1% | [37] |
| KCNE2 (KCNE2-LQTS) | 21q22.1 | MiRP1 | Reduced IKr | AD | <1% | [40] |
| KCNJ5 (KCNJ5-LQTS) | 11q24 | Kir3.4 | Reduced IK,ACh | AD | <1% | [48] |
| SCN4B (SCN4B-LQTS) | 11q23. 3 | Nav1.5 β4-subunit | Increased INa | AD | <1% | [46] |
| SNTA1 (SNTA-LQTS) | 20q11.2 | Syntrophin-α1 | Increased INa | AD | <1% | [47] |
| Jervell and Lange-Nielson Syndrome | ||||||
| KCNQ1 (JLNS1) | 11p15.5 | Kv7.1 | Reduced IKs | AR | Very rare | [24] |
| KCNE1 (JLNS2) | 21q22.1 | MinK | Reduced IKs | AR | Very rare | [39] |
| Ankyrin-B Syndrome | ||||||
| ANKB (ABS) | 4q25-q27 | Ankyrin B | Aberrant ion channel/transporter localization | AD | <1% | [51] |
| Andersen-Tawil Syndrome | ||||||
| KCNJ2 (ATS) | 17q23 | Kir2.1 | Reduced IK1 | AD | <1% | [56] |
| Timothy Syndrome | ||||||
| CACNA1C (TS) | 12p13.3 | Cav1.2 | Increased ICa,L | Sporadic | Very rare | [8] |
| Recurrent Infantile Cardiac Arrest Syndrome | ||||||
| CALM1 | 14q24-q31 | Calmodulin 1 | Dysfunctional Ca2+ signaling | Sporadic | <1% | [63] |
| CALM2 | 2p21 | Calmodulin 2 | Dysfunctional Ca2+ signaling | Sporadic | <1% | [63] |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; ABS, Ankyrin-B syndrome; ATS, Andersen Tawil syndrome; LQTS, long-QT syndrome; and TS, Timothy syndrome.
Major Long QT Syndrome-Susceptibility Genes
Mutations in KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) represent the most common causes of LQTS and collectively account for an estimated 60–75% of genotype-positive LQTS cases (Table 1). [19, 20] KCNQ1 encodes the Kv7.1 pore-forming α-subunit that generates the slowly activating component of the delayed rectifier potassium current (IKs) essential for maintaining the physiologic QT shortening observed with increased sympathetic tone/heart rates [21] and endocochlear potassium cycling required for normal hearing. [22] Heterozygous loss-of-function mutations in KCNQ1 cause autosomal dominant (AD) type 1 LQTS (LQT1), the most prevalent LQTS subtype, and create an arrhythmogenic substrate that predisposes affected individuals to suffering cardiac events during times of physical and emotional duress due to the inability of the defective IKs current to adequately adapt to β-adrenergic stimulation. [16, 19, 20, 23] Classically, homozygous or compound heterozygous mutations in KCNQ1 cause the extremely rare autosomal recessive (AR) JLNS (JLNS1) characterized by extreme QT prolongation, high risk of cardiac events, and bilateral sensorineural hearing loss/deafness secondary to the near abolishment of IKs function in the heart and inner ear. [6, 24] However, emerging evidence suggests that malignant LQT1 cardiac manifestations, akin to those observed in JLNS, without any discernible evidence of sensorineural deafness/hearing loss (so called AR LQT1) may be a more commonly observed phenotype in individuals homozygous/compound heterozygous for mutations in KCNQ1 than JLNS1, at least in countries such as the United States with relatively genetically heterogeneous populations. [25]
The second most prevalent LQTS subtype (LQT2) is caused by heterozygous loss-of-function mutations in the KCNH2-encoded human-ether-a-go-go-related gene potassium channel 1 (hERG1 or Kv11.1) that conducts the rapidly activating component of the delayed rectifier potassium current (IKr), that along with IKs are responsible for phase 3 repolarization of the cardiac action potential (Figure 1a). [13, 17, 19, 20] While the majority of LQT1-causative KCNQ1 mutations appear to form functional Kv7.1 α-subunits capable of co-assembling with wild-type Kv7.1 α-subunits and thereby exerting a dominant-negative effect on the IKs current (≥ 50% reduction), the majority of LQT2-causative KCNH2 mutations produce mutant Kv11.1 α-subunits that are improperly folded, retained in the endoplasmic reticulum, or otherwise fail to make it to the cell surface resulting in haploinsufficiency and ≤ 50% reduction in the IKr current. Similar to LQT1, a small percentage of LQT2 cases may harbor homozygous/compound heterozygous mutations in KCNH2 (or another major LQTS-susceptibility gene) and not surprisingly are associated typically with a more severe cardiac phenotype. [26–28] In addition to causing LQT2, the unique structural features of the tetrameric hERG/Kv11.1 channel make it particularly susceptible to blockade by an array of pharmacologic agents resulting in acquired/drug-induced LQTS, which has been reviewed in detail elsewhere. [29]
Melvin Scheinman: One other clinical situation that draws attention to the possibility of the presence of a forme fruste of LQTS is the presence of a drug-induced torsades. Virtually all such drugs involve block in the Ikr channel, thus mimicking LQTS2. A number of drugs have been removed because of this complication (ie Seldane, Cisapride, Grepafloxacin) while a wide variety of drugs have been implicated including antiarrhythmic agents (sotatol, dofetilide) antibiotics (Erythromycin, floxacim) antipsychotics (Thorazine). Torsades may be due to drug-drug interaction which interfere with drug metabolism (Seldane and ketaconozole CYP450 3A4 substrates) or due to “concealed LQTS” brought out by exposure to QT-prolonging agents. Studies by Roden, et al show that patients with drug-induced torsades will frequently have an underlying genetic mutation associated with LQTS.
Lastly, heterozygous gain-of-function mutations in the SCN5A-encoded Nav1.5 cardiac sodium channel, that conducts the inward sodium current (INa) responsible for phase 0 depolarization, cause LQT3, the third most common cause of congenital LQTS (Figure 1a). [18–20] Mechanistically, LQT3-causative mutations in SCN5A prolong the QT interval via small net increases in the INa current, commonly secondary to an abnormal persistent/sustained late sodium current or impaired Nav1.5 inactivation, observed across the entire voltage range and time course of action potential plateau that perturb the delicate balance between inward and outward currents. [30, 31] Unlike LQT1 (and to an extent LQT2), the QT interval of LQT3 patients adequately shortens at higher heart rates, but tends to prolong excessively at slower heart rates. As a result, LQT3 patients are at greater risk suffering an LQTS-triggered cardiac event at rest, particularly during sleep. [23, 32] While the precise electrophysiological mechanism underlying this phenomenon are still poorly understood, it is has been long speculated [33], and recently demonstrated in a murine model [34], that diurnal variation in cardiac repolarization patterns may underlie the time-dependent vulnerability to ventricular arrhythmias observed in LQT3 and other heritable cardiac arrhythmia syndromes.
Minor Long QT Syndrome-Susceptibility Genes
Following the discovery of the three major LQTS-susceptibility genes, at least 10 additional minor LQTS-susceptibility genes collectively accounting for <5% of LQTS have been described. Given that ion channel pore-forming α-subunits typically function in concert with a number of accessory subunits, it is not surprising that the majority of minor LQTS-susceptibility genes are components of the ion channel macromolecular complexes that function to conduct the INa, IKr, and IKs currents in vivo. As such, an easy way to recall both the genetic and electrophysiologic basis of the minor LQTS-susceptibility genes is through the use of a current-centric model similar to that depicted in Figure 2. The seven minor genes that present with a “pure” LQTS phenotype are described briefly below using a current-centric model, whereas the minor genes that present with QT prolongation in the setting of prominent extracardiac manifestations are covered in the ensuing section.
At minimum, the in vitro recapitulation of the native IKs current requires the assembly of Kv7.1 with the KCNE1-encoded minK β-subunit. [21, 35] Furthermore, minK and the AKAP9-encoded A kinase anchor protein 9/yotiao mediate critical Kv7.1 phosphorylation events required for the physiologic enhancement of the IKs current during times of β-adrenergic stimulation. [36] Not surprisingly, loss-of-function mutations in both KCNE1 (KCNE1-LQT2) [37] and AKAP9 (AKAP9-LQTS) [38] generate defective IKs currents that fail to adequately respond to β-adrenergic stimulation and represent rare causes of IKs-mediated LQTS (Figure 2). Lastly, as minK also functions as a primary molecular constituent of IKs in the heart and the inner ear, homozygous/compound mutations in KCNE1 or digenic compound heterozygous mutations in KCNQ1 and KCNE1 represent a rare cause of JLNS (Table 1). [39]
Although the in vivo role of the KCNE2-encoded MiRP1 β-subunit in the recapitulation of the native IKr current conducted by hERG/Kv11.1 remains at the center of much debate [40–43], loss-of-function mutations in KCNE2 have been associated with both congenital (KCNE2-LQTS) and acquired forms of LQTS. [40, 44] At present KCNE2/MiRP1 remains the only hERG-interacting protein linked to LQTS (Figure 2).
To date, mutations in three β-subunit (SCNB4-encoded β4 subunit/SCNB4-LQTS) or accessory (CAV3-encoded caveolin 3/CAV3-LQTS and SNTA1-encoded syntrophin α1/SNTA1-LQTS) proteins that comprise the larger Nav1.5 macromolecular complex are rare causes of LQTS via the induction of an abnormal persistent/sustained late sodium current that mimics the electrophysiological perturbations associated with many LQT3-causative SCN5A mutations (Figure 2). [45–47] Details of the precise molecular roles of these and other Nav1.5-interacting proteins are reviewed in detail elsewhere. [31]
Lastly, in 2010, Yang et al identified a single loss-of-function mutation in the KCNJ5-encoded Kir3.4 pore-forming α-subunit that conducts the G-protein coupled inwardly rectifying protein current (IKAch) that co-segregated with a LQTS phenotype in a large multigenerational pedigree (Figure 1). [48] While the IKr, IKs, and IK1 currents are responsible primarily for ventricular repolarization, at least in murine models, there is emerging evidence that IKAch is active, but masked by the constitutively active IK1 current, during repolarization phases of the cardiac action potential. [49] Although no additional mutations in KCNJ5 have been described to date, the IKAch current does appear to play a limited, but biologically plausible role in the pathogenesis of LQTS.
Genetics of Multisystem Long-QT Syndrome: Ankyrin-B, Anderson-Tawil, Timothy, and Recurrent Infantile Cardiac Arrest Syndromes
In addition to JLNS, four other LQTS genotypes/subtypes characterized by QT prolongation and an increased risk of syncope, seizures and sudden cardiac death in the setting of a variety of extracardiac manifestations have been described in the literature and thus are best described as “multisystem” forms of LQTS. The genetic and electrophysiological basis of these multisystem forms of LQTS are summarized in chronological order within Table 1 and discussed briefly in the paragraphs below.
In 1995, Schott et al identified a novel genetic locus (4q25-27) that segregated in large French kindred with the unique clinical phenotype of QT prolongation, sinus node dysfunction, and episodic atrial fibrillation. [50] Nearly a decade later, the causal gene within the 4q25-27 locus was determined to be ANK2-encoded ankyrin-B, a specialized adaptor protein required for the establishment of membrane microdomains, and loss-of-function mutations in ANK2 caused a multisystem form of LQTS now termed sick sinus syndrome with bradycardia or simply ankyrin-B syndrome (ABS). [51, 52] Functionally, ABS arises secondary to the disruption of cellular microdomains involving and a number of cardiac ion channels and transporters including the Na+/K+ ATPase, Na+/Ca2+ exchanger, and the inositol-3-phosphate receptor and the generation of aberrant cytoplasmic Ca2+ release. [53]
Initially described clinically in 1971, Andersen-Tawil syndrome (ATS) is a rare multisystem form of LQTS characterized by the clinical triad of dysmorphic physical features (low-set ears, micrognathia, and clinodactyly), periodic paralysis, and nonsustained ventricular arrhythmia. [54, 55] At the molecular level, heterozygous loss-of-function mutations in the KCNJ2-encoded Kir2.1 inward rectifier potassium channel result in a reduction of the IK1 current that contributes to phase 3 repolarization and prolongation of action potential duration that generates the substrate for re-entrant arrhythmias in ATS. [56, 57] In comparison to the “classical” forms of LQTS (e.g. LQT1–3), ATS patients typically exhibit milder QT prolongation, presence of characteristic broad, high-amplitude U-waves, and decreased risk of life-threatening EAD-triggered ventricular arrhythmias. [58]
Timothy syndrome (TS) is an extremely rare multisystem form of LQTS caused by gain-of-function mutations that impair the voltage-dependent inactivation of the CACNA1C-encoded Cav1.2 channel resulting in an increased L-type Ca2+ current (ICa,L) during the plateau phase of the cardiac action potential (Figure 1) leading to a complex phenotype that includes variable degrees of autism spectrum disorder, syndactyly, and severe cardiac arrhythmias. [8, 59] Unlike most forms of LQTS, which by and large follow Mendelian inheritance patterns, the two TS-causative mutations identified to date, G402S and G406R, in the mutually exclusive exon 8 and exon 8a CACNA1C splice variants, respectively are inherited invariably in a sporadic fashion. [8, 60] As such, it appears that either de novo mutagenesis or parental mosaicism are the primary inheritance patterns of TS. [61, 62]
Most recently, the exome sequencing of two parent-child trios revealed that heterozygous sporadic/de novo mutations in two of the three genes that collectively encode calmodulin (CALM1 and CALM2), a ubiquitous Ca2+ binding protein responsible for a plethora of intracellular signaling processes, cause a multisystem disorder with features of severe LQTS (QTc > 600 ms, 2:1 atrioventricular block, and macroscopic T-wave alternans) characterized by neurodevelopmental delays, seizures, and recurrent cardiac arrest during early infancy. [63] While the precise electrophysiological mechanism(s) by which mutant calmodulin severely disrupts myocardial repolarization in this newly described recurrent infantile cardiac arrest syndrome remains unknown, this intriguing discovery hints at a greater role for intracellular Ca2+ signaling in the pathogenesis of LQTS and possibly sudden infant death syndrome (SIDS).
Genetic Modifiers of Long-QT Syndrome Disease Severity
Interestingly, most of the LQTS subtypes described above are subjected to incomplete penentrance and variable expressivity, that is to say affected individuals within the same multigenerational pedigree, who harbor the same LQTS-causative mutation, paradoxically display variable degrees of disease expression causing them to assume vastly different clinical courses. [9] It is now understood that a complex combination of genetic and environmental factors modulates symptom onset, degree of QTc prolongation, and risk of suffering LQTS-triggered cardiac events that collectively encompass objective measures of LQTS disease severity. Here, we summarize several recently discovered genetic determinants of LQTS disease severity, commonly referred to as modifier genes, which may modulate the phenotype of patients with a primary LQTS-causative mutation.
To date, the bulk of LQTS genetic modifiers described in the literature represent common genetic variants within known LQTS-susceptibility genes that impart a modest, but discernible functional phenotype or effect (Table 2). For example, common aminoacid altering single nucleotide polymorphisms (SNPs) in KCNE1 (D85N) [64], KCNH2 (K897T) [65], and SCN5A (H558R) [66, 67] exert modest electrophysiologic effects that can modulate the in vivo or in vitro phenotypic expression of certain LQT1-, LQT2-, and LQT3-causative mutations, respectively. Furthermore, recent studies have also identified a role for non-coding SNPs within critical genomic regions, such as the promoter or 3′ untranslated regions, known to regulate the expression of established LQTS-susceptibility genes (Table 2). [68, 69] Since SNPs in the promoter or 3′ untranslated region can theoretically enhance or diminish the expression of either wild-type or mutant alleles through the differential binding of critical transcription factors or microRNAs, respectively, these SNPs are referred to as allele-specific modifiers and add an additional layer of complexity to our understanding of the genetic architecture of LQTS.
Table 2.
Common variants shown to modify long QT syndrome disease severity
| Locus | SNP ID | MAF | Amino Acid Change* | Function | QTc/Adrenergic Effect | Reference |
|---|---|---|---|---|---|---|
| Coding variants within LQTS- susceptibility genes | ||||||
| 21q KCNE1 | rs1805128 | 0.01 | D85N | MinK β-subunit (IKs) | ↑ QTc | [64] |
| 7q KCNH2 | rs1805123 | 0.24 | K897T | Kv11.1 α-subunit (IKr) | ↑ QTc | [65] |
| 3p SCN5A | rs1805124 | 0.18 | H558R | Nav1.5 α-subunit (INa) | ↑ QTc | [66, 67] |
| Non-coding variants within LQTS- susceptibility genes | ||||||
| 11p KCNQ1 | rs2519184 | 0.09 | None | 3′UTR of Kv7.1 α- subunit (IKs) | Allele-specific QTc effect | [68] |
| rs8234 | 0.49 | None | 3′UTR of Kv7.1 α- subunit (IKs) | Allele-specific QTc effect | [68] | |
| rs10798 | 0.49 | None | 3′UTR of Kv7.1 α- subunit (IKs) | Allele-specific QTc effect | [68] | |
| Variants outside of LQTS- susceptibility genes | ||||||
| 4p ADRA2C | rs61767072 | 0.06 | Del322–325 | α2 adrenergic receptor | ↑ Adrenergic response* | [73] |
| 10q ADRB1 | rs1801253 | 0.3 | G389R | β1 adrenergic receptor | ↑ Adrenergic response* | [72, 73] |
| 1q NOS1AP | rs16857031 | 0.14 | None | Nitric oxide synthetase 1 adaptor protein (nNOS signaling) | ↑ QTc | [74, 75] |
| rs4657139 | 0.41 | None | Nitric oxide synthetase 1 adaptor protein (nNOS signaling) | ↑ QTc | [74, 75] |
Increased adrenergic response as assessed by higher baroreflex sensitivity values has associated with an increased risk of cardiac events in LQT1, but does not appear to be mediated by QTc interval.
Abbreviations: 3′UTR, 3′ untranslated region; LQTS, long-QT syndrome; and nNOS, neuronal nitric oxide synthetase
In addition to SNPs within established LQTS-susceptibility genes, several studies have illustrated that SNPs within genes that modulate cardiac ion channel function through post-translation- or transcription-level events can serve as genetic modifiers of LQTS disease severity (Table 2). First, LQT1 disease severity/expressivity can be modified by common amino-acid altering SNPs in the genes encoding the α2 and β1 adrenergic receptors secondary to a loss of α2 autoinhibitory feedback/increased presynaptic epinephrine release (ADRA2C-del322-325) [70] or enhanced β1 activity secondary to improved coupling to adenylyl cyclase (ADRB1-G389R) [71] resulting in increased baroreceptor/autonomic responsiveness. [72, 73] Second, common non-coding SNPs (rs4657139 and rs16847548) in NOS1AP-encoded nitric oxide synthetase 1 adaptor protein, previously associated with QT interval duration in the general population, have been associated with modest QT prolongation and risk of SCD in the large South African KCNQ1-A341V LQT1 kindred [74], as well a prospective registry LQTS patients. [75]
CLINICAL PRESENTATION AND DIAGNOSIS OF LONG QT SYNDROME
Prevalence and Clinical Presentation
In 2009, Schwartz et al used population-based ECG and molecular screening of 44,456 Italian infants to place the estimated prevalence of congenital LQTS at ~1 in 2,000 persons. [5] While this study provided the first data-driven estimate of infants with the phenotype of an abnormally long QTc, it did not take into account those individuals who may harbor a disease-causative mutation but fail to display objective evidence of QTc prolongation. Interestingly, recent analysis of population-scale exome sequencing from the National Heart Lung and Blood Institute Exome Sequencing Project (ESP) database placed the prevalence of a potentially pathogenic LQTS genotype, defined as a variant previously shown to co-segregate with disease and or that features a functionally perturbed electrophysiological phenotype, at ~1 in 80. [76] While incomplete penetrance and variable expressivity certainly contribute to the discordance observed between the population-based estimates of “pathogenic” LQTS genotype (1:80) and an expressed QTc clinical phenotype (1:2000) prevalence, the precise mechanism(s) that underlie this discordance are not fully understood and worthy of future investigations.
Phenotypically, LQTS is characterized objectively by the presence of QTc prolongation on 12-lead ECG (with QTc values > 470 ms for males and > 480 ms for females representing approximate 99th percentile values) in the absence of structural heart disease or secondary causes of a QTc prolongation and an increased risk of syncope, seizures, and tragically sudden death secondary to torsades de pointes (TdP), the characteristic form of polymorphic ventricular tachycardia observed in LQTS (Figure 3). [14] However, just as QTc values beyond the 99th percentile do not equal necessarily a diagnosis of LQTS, normal QTc values do not exclude LQTS. In fact, an estimated 10–40% of genotype-positive individuals do not display any objective evidence of a QT abnormality and are classified as “normal QT interval” LQTS or “concealed” LQTS. [77, 78]
Figure 3. Electrocardiographic and clinical hallmarks of long QT syndrome.

a. Schematic ECGs displaying the broad-based T wave pattern associated with long-QT syndrome type 1 (LQT1), notched T wave pattern associated with long-QT syndrome type 2 (LQT2), and long isoelectric segment with normal symmetrical T wave pattern associated with long-QT syndrome type 3 (LQT3) and QT prolongation observed in LQT1–LQT3 compared to normal. b. Triggers such as strenuous exercise (LQT1), sudden noises (LQT2), or sleep/rest (LQT3) can cause the stable, but prolonged myocardial repolarization to degenerate into torsades de pointes (TdP), the characteristic form of polymorphic ventricular fibrillation observed in long QT syndrome, depicted in this schematic ECG. c. The clinical manifestations of TdP in long QT syndrome are dependent on whether (syncope/seizures) or not (sudden death) order is restored to cardiac rhythm, either spontaneously or by a defibrillator.
While QT interval prolongation serves as the key electrocardiographic hallmark of LQTS, careful analysis of T-wave morphology can also provide useful diagnostic information. For instance, specific ST-T wave patterns correlate with each of the major LQTS genotypes (broad-based T-waves in LQT1, low-amplitude notched T-waves in LQT2, and late-onset peaked/biphasic T-waves in LQT3; Figure 3A) providing the astute clinician with the ability to anticipate the possible genotype prior to the initiation of genetic testing. [79, 80] Furthermore, T-wave alternans, in either polarity or amplitude, is as a marker of cardiac electrical instability which identifies a higher risk subset. [81]
Standard Diagnostic Approaches
Despite the plethora of advanced imaging, diagnostic, and genetic tests available today, the most important factor needed to establish a diagnosis of LQTS still remains the patient’s overall clinical picture. In fact, attempts to interpret a patient’s 12-lead ECG, commercial genetic testing, or other adjunct test results without first obtaining a meticulous personal and family history (e.g. insufficient evidence) represent some of the most common diagnostic miscues leading to the premature/incorrect diagnosis of LQTS. [82]
Accordingly, the first step towards establishing a diagnosis of LQTS should always be to carefully assess the patient’s overall clinical picture by obtaining a meticulous personal and family history. Here, the primary goal is to ascertain if the patient has suffered any LQTS-triggered episodes of syncope, seizure, or aborted sudden cardiac arrest him/herself and if a history of similar LQTS-triggered cardiac events, sudden unexplained deaths/accidents/drownings, or long-standing diagnosis of a seizure disorder is present amongst first, second, and third degree relatives. Given that the rate of reflex vasovagal syncope is similar between patients with LQTS and the general population [83], any personal or family history of fainting merits further scrutiny before the initiation of additional testing (e.g. 12-lead ECG) as the misinterpretation of vasovagal symptoms in the setting of a borderline QTc is the most common cause of a premature/incorrect LQTS diagnosis. [82] While syncope upon standing or preceding nausea are suggestive of a vasovagal origin, syncope while supine (e.g. rest or sleep), during times of emotional or physical duress, or preceding palpitations or auditory stimuli are reported more frequently by LQTS patients than otherwise healthy individuals and therefore should increase suspicion for arrhythmic syncope. [84]
As a rule of thumb, any individual with a personal or family history suspicious for LQTS should undergo a thorough cardiac evaluation including but not limited to a 12-lead ECG. As mentioned previously, the primary diagnostic characteristic of LQTS is a prolonged QTc on 12-lead ECG (Figure 3), defined by the latest AHA/ACC/HRS guidelines as a QTc > 450 ms in men and > 460 ms in women. [85] However, the use of these ~95th percentile values, without corroborating clinical data that raises the index of clinical suspicion, will result in an unacceptable number of diagnostic mishaps given the known overlap in QTc values between LQTS patients and healthy individuals (Figure 4). [82] Thus, employing the 99th percentile of the QTc value distribution (> 470 in adult males and >480 in adult females) will improve the positive predictive value for LQTS, especially when a clinical picture suggestive of LQTS accompanies a QTc that exceeds the 99th percentile for sex and age.
Figure 4. QTc distribution in health and disease.

The distribution of QTc values in health was derived from nearly 80,000 healthy adult males and females. [81] The distribution of QTc values in LQTS were derived from all patients with genetically proven LQTS evaluated in the Mayo Clinic’s Long QT Syndrome Clinic. Permission obtained from the American Heart Association © Taggart, N.W. et al. Diagnostic miscues in congenital long-QT syndrome. Circulation 115(20), 2613–2620 (2007).
At this point in the evaluation, the “Schwartz score”, a diagnostic scorecard that takes into account elements of the ECG, personal history, family history, can be helpful to quantitatively assess the clinical probability of LQTS for a given index case (Table 3). [86, 87] While not a strict cut-off value, a Schwartz score > 3.5 (high probability of LQTS) is a useful metric for determining which patients and families would benefit most from further assessment in order to solidify the diagnosis of LQTS, namely the judicious use of provocation/stress tests and/or genetic tests as described below.
Table 3.
Diagnostic criteria and score for LQTS
| Points* | |
|---|---|
| Electrocardiographic findings# | |
| QTc§ interval | |
| ≥ 480 ms | 3 |
| 460–479 ms | 2 |
| 450–459 ms (men) | 1 |
| QTc§ ≥ 480 ms during 2nd – 4th minute of recovery from exercise stress test | 1 |
| Documented torsades de pointes¥ | 2 |
| T-wave alternans | 1 |
| Notched T wave in 3 leads | 1 |
| Resting heart rate below second percentile for age | 0.5 |
| Clinical history | |
| Syncope¥ | |
| With stress | 2 |
| Without stress | 1 |
| Congenital deafness | 0.5 |
| Family history | |
| Relatives with clinically definitive LQTS† | 1 |
| Unexplained sudden cardiac death in immediate relative < 30 years of age† | 0.5 |
Total score indicates probability of LQTS: ≤ 1 point (low), 2–3 points (intermediate), ≥ 3.5 points (high).
In the absence of medications, electrolyte abnormalities, or disorders known to influence these electrocardiographic parameters.
QTccalculated using Bazett’s formula (QTc = QT/√RR)
Mutually exclusive
Same family member cannot be counted twice
Abbreviations: LQTS, long QT syndrome; QTc, heart-rate corrected QT interval
Provocation/Stress Tests and Genetic Testing
While provocation/stress tests and clinical genetic testing are not needed to establish the diagnosis of LQTS in the setting of a robust clinical phenotype, unmasking or identifying a specific LQTS genotype (e.g. LQT1, LQT2, LQT3, etc.) has assumed an increasingly important role when it comes to assessing the risk of SCD, selecting appropriate therapeutic interventions, and identifying potentially at-risk relatives. [88]
By and large, the usefulness of catecholamine provocation and exercise stress tests in the diagnosis of LQTS is confined to unmasking a LQT1 genotype. Paradoxical QTc prolongation during the recovery phase of the Bruce treadmill stress test protocol (i.e. > 470 ms at 2–4 minutes of recovery) or paradoxical lengthening of the absolute QT interval by > 30 ms following low-dose epinephrine administration (< 0.1 mcg/kg/min) can be indicative of the blunted physiologic response of the defective IKs current to β-adrenergic stimulation seen in LQT1. [79, 89, 90] While the presence of a positive treadmill stress and epinephrine provocation tests certainly increases the pre-test probability of LQT1, paradoxical QT lengthening does not equal a diagnosis of LQT1 or LQTS in general, nor can the presence of normal QTc shortening rule out other types of LQTS. [32]
Melvin Scheinman: The authors point out the importance of provocative stress testing in order to better define the cause of the LQTS. In addition to standard exercise stress testing and epinephrene infusions, one other simple test (proposed by Viskin) is of value. This test involves measurement of the QTc both supine and in the erect position. Standing will result in mild increases in heart rate and abnormal prolongation of the QTc in affected individuals. It should be emphasized that QTc measurement during exercise may be misleading when using the Bazzet correction since the QT interval will shorten much less than the exercise heart rate, hence resulting in over correction. The most meaningful measurements are made 4–5 minutes into recovery. In addition, while the epinephrine challenge is an important provocateur of abnormal QTc, this medication results in augmentation of the U waves in normals and hence care must be taken to measure the QT interval alone without the confounding influence of the U wave. According to Lepeshkin, the interval from peak of T wave to peak of the U wave is > 150ms. This is helpful in distinguishing notched T waves from T-U waves.
Finally, some important considerations regarding genetic testing in the diagnostic evaluation of patients with suspected LQTS are necessary. Given that approximately 4% of ostensibly healthy white individuals and 6–8% of black individuals harbor a rare, amino acid-altering genetic variant in one of the three major LQTS-susceptibility genes (KCNQ1, KCNH2, or SCN5A) [91], beginning any LQTS evaluation with genetic testing is flat-out dangerous and represents a fundamental failure to recognize the probabilistic, rather than deterministic/binary, nature of clinical genetic testing. That said, current Heart Rhythm Society (HRS)/European Heart Rhythm Association (EHRA) guidelines recommend the judicious use of comprehensive (major and minor LQTS genes listed in Table 1) or targeted (major LQTS genes listed in Table 1) LQTS genetic testing for 1) any individual with a strong clinical suspicion of LQTS based on clinical/family history and electrocardiographic phenotype, 2) any asymptomatic individual with unexplained QTc prolongation (> 480 ms before puberty and > 500 ms after puberty), and 3) appropriate relatives, regardless of clinical/electrocardiographic phenotype, when a bona fide LQTS-causative mutation has been identified in the index case. [92]
As with any clinical test, the proper interpretation of LQTS genetic testing results requires a firm understanding of all potential sources of false positive (e.g. frequency of rare, but innocuous genetic variants within a particular gene in health) and false negative (e.g. prevalence of a concealed LQTS phenotype secondary to incomplete penetrance/variable expressivity) results that contribute to the test’s collective “signal-to-noise” ratio. [93] Importantly, whenever ordering or attempting to interpret genetic test results, it is paramount to remember that as the strength of LQTS clinical phenotype decreases (i.e. the pre-test probability of disease), the possibility of a false positive genetic test result increases significantly. Unfortunately, even when LQTS genetic testing is used in an appropriate and judicious manner, rare “Variants of Uncertain Significance” (VUS) alterations in the normal sequence of a gene whose association with disease risk is uncertain due to insufficient or inconclusive evidence (commonly used criteria are listed in Table 4) to confidently label the variant as “pathogenic/disease-causative”, can still be encountered in LQTS-susceptibility genes.
Table 4.
Principles of rare variant interpretation
| Major Pathogenicity Criteria |
| Co-segregation of variant with disease in a multigenerational pedigree |
| Absence/extreme rarity of variant in healthy controls and public exomes/genomes |
| Radical (e.g. nonsense, frameshift, or insertion/deletion) mutation |
| Amino acid-altering variants localizing to key structure-function domain* |
| Minor Pathogenicity Criteria |
| Perturbed electrophysiological phenotype observed during in vitro functional studies |
| Agreement of multiple in silico phenotype prediction tools on variant pathogenicity |
For example the transmembrane and pore-regions of KCNQ1, KCNH2, or SCN5A. Additional key structure-function domains in the major LQTS-susceptibility genes are detailed in Figure 5.
Luckily, studies coupling the established rate of rare and presumably innocuous background genetic variation in health with large compendia of mutations identified in clinically definite LQTS cases have yielded a number of clinically meaningful observations that have improved how an indeterminant variant (i.e. a VUS) in major LQTS-susceptibility genes can be interpreted. [91, 94] Specifically, certain mutation types (e.g. radical/truncating) and missense mutations localizing to particular topological structure-function domains of the Kv11.1/hERG, Kv7.1, and Nav1.5 channels (e.g. pore/transmembrane regions) are associated with a high (>90%) estimated predictive value (EPV). When identified in a case with high clinical probability for LQTS, these variants are more likely to be the disease-causative mutation. [91, 93] Furthermore, coupling ion channel topology with the synergistic use of multiple independent in silico phenotype prediction tools such as “Sorting Intolerant From Tolerant” (SIFT) and “Polymorphism Phenotyping” (PolyPhen) have displayed the ability to enhance the interpretation of genetic variants localizing to regions where the topology-driven EPVs are suboptimal. [94] Collectively, these insights have enabled an algorithm to aid in the probabilistic interpretation of a LQTS genetic testing result (Figure 5).
Figure 5. Evidence-based algorithm designed to aid in the interpretation of a long QT syndrome (LQTS) genetic test result.

Algorithm for interpreting a “positive” LQTS genetic test. Radical mutations that significantly alter/truncate Kv7.1 or Kv11.1 such as insertions/deletions, alteration of intronic/exonic splice site boundaries, and nonsense mutations, are probably LQTS-associated. Those raremissense mutations that localize to Kv7.1 (TM/pore, SA, or C terminal domains), Kv11.1 (C terminal when ≥ 3 tools are in agreement, TM/Pore, PAS/PAC, or cNBD), or Nav1.5 (TM/pore/linker or C terminus) are probably or possibly pathogenic. Variants outside these topological structure-function domains are truly ambiguous variants or variants of uncertain significance (VUS) without the aid of additional evidence (e.g. co-segregation with disease, LQTS-like electrophysiological phenotype etc.). Abbreviations: cNBD, cyclic nucleotide binding domain; EPV, estimated predictive value; IDL, interdomain linker; PAC, per-arnt-sim C-terminal associated; PAS, per-arnt-sim; SA, subunit assembly; TM, transmembrane. Adapted from Giudicessi, J.R. and Ackerman, M.J. Genetic testing in heritable cardiac arrhythmia syndromes; differentiating pathogenic mutations from background genetic noise. Current Opinion in Cardiology 28(1), 63–71 (2013) with permission from Wolters Kluwer Health.
GENOTYPE- AND PHENOTYPE-GUIDED RISK STRATIFICATION AND MANAGEMENT OF LONG QT SYNDROME
Genotype- and Phenotype-Driven Risk Stratification
At present, both genotypic (e.g. LQTS genetic, intragenic, and mutation-specific subtype) and phenotypic (e.g. gender, QTc, and history of cardiac events) characteristics are used to guide the risk stratification, and ultimately the clinical management, of patients with LQTS (Figure 6). Those patients who harbor bona fide LQT1-causative mutations on > 1 KCNQ1 allele (e.g. JLNS and AR-LQT1) [25, 95, 96], have suffered ≥ 10 cardiac events before the age of 18 [97], or have Timothy syndrome are at highest risk (≥ 80%) of suffering from one or more LQTS-associated cardiac events before the age of 40, including a high rate of sudden cardiac arrest/death, and thus require aggressive clinical management, which often involves more invasive approaches such as left cardiac sympathetic denervation (LCSD) and or use of an implantable cardioverterter-defibrillator (ICD).
Figure 6. Genotype- and phenotype-guided risk classification of long QT syndrome patients.
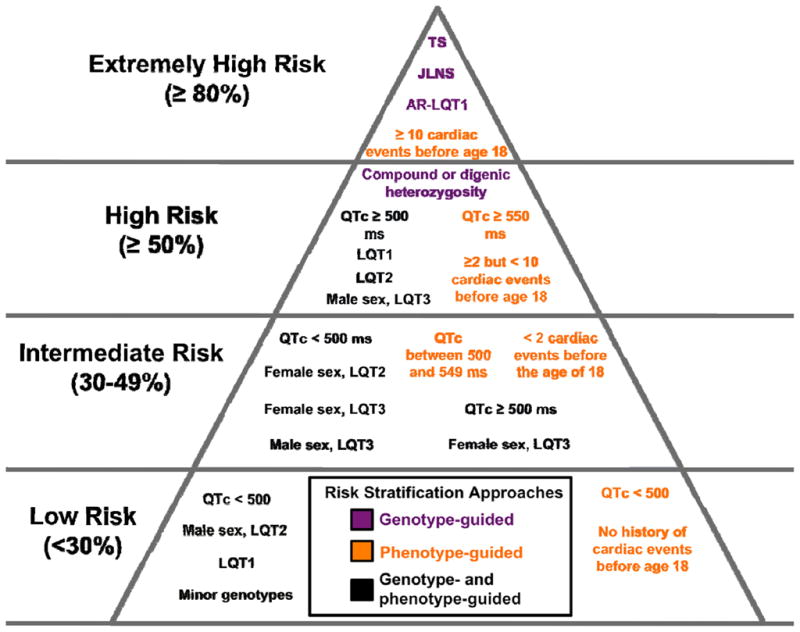
Risk groups have been defined based on the previously published probability of suffering a first or recurrent cardiac event (syncope, seizure, sudden cardiac arrest, or sudden cardiac death) before 40 years of age without appropriate therapeutic interventions. A probability of suffering a first cardiac event > 80% defines the extremely high-risk group, > 50% the high-risk group, between 30% and 49% as the intermediate-risk group, and below 30% as the lowest-risk group. Genotype-guided recommendations are indicated by purple text, phenotype-guided recommendations are indicated by orange text, and a combination of genotype- and phenotype-guided recommendations is indicated by black text within the figure.
Similarly, individuals with a QTc ≥ 550 ms regardless of LQTS genotype [97], a QTc ≥ 500 ms with an LQT1, LQT2, or males with an LQT3 genotype [88], non-JLNS patients with bona fide LQTS-causative mutations on > 1 major LQTS-susceptibility allele (e.g. compound heterozygosity or digenic heterozygosity) [27, 98], or individuals who have suffered ≥ 2 but < 10 cardiac events before the age of 18 [97] are at higher risk (≥ 50%) for suffering an LQTS-associated cardiac event(s) before the age of 40 and often require a combination of medical, surgical, or device-related management.
Those individuals with a QTc between 500 and 549 ms regardless of genotype, females with major genotype-positive LQTS, male LQT3 patients with a QTc < 500 ms, and any individual who has experienced < 2 cardiac events before the age 18 are at intermediate risk for experiencing an LQTS-associated cardiac event(s) before the age of 40 and require some form of treatment, typically β-adrenergic blockers. [88, 97]
All other LQTS patients (e.g. asymptomatic patients with a QTc < 500 ms aside from certain high-risk gender/genotype combinations like postpubertal LQT2 females; Figure 6) are at lower risk and the selection of appropriate therapy, if any, is performed on an individualized basis.
Medical, Surgical, and Device-Related Management
In general, regardless of symptomatic status, all LQTS patients should avoid QT-prolonging medications whenever possible and maintain adequate hydration, and thereby normal electrolyte levels, especially in the setting of emesis, diarrhea, or other medical conditions known to cause hypokalemia. Furthermore, given that sudden cardiac arrest/death can be the sentinel event, appropriate tailored therapeutic interventions should be initiated in most patients with LQTS with the possible exception of some patients with asymptomatic, concealed (QTc < 460 ms) LQTS. [2] Currently, LQTS therapy targets two distinct strategies: 1) reduction in sympathetic/adrenergic tone, and therefore arrhythmia risk, via the use of β-adrenergic receptor antagonists and or LCSD and 2) correction/cessation of life-threatening arrhythmias through the timely delivery of electrical impulses by an ICD. The medical, surgical, and device-related interventions commonly used to fulfill these principles in the clinical management of patients with LQTS are reviewed briefly below.
Since the 1970’s, β-adrenergic receptor antagonists (β-blockers) have been first-line therapy for the prevention of life-threatening arrhythmias in LQTS that are often triggered by sudden increases in sympathetic activity. [99] While the efficacy of β-blockers in the reduction of LQTS-associated cardiac events, particularly in LQT1 and LQT2, is undisputable [100, 101], the observation that 20% to 30% of previously symptomatic LQTS patients suffer breakthrough cardiac events [102, 103] and that different β-blockers have variable effects (e.g. blockade) on the late/sustained cardiac Na+ current (propranolol > nadolol ≫ metoprolol) [104] has led to widespread concern that not all β-blockers share an equivalent level of antiarrhythmic efficacy. [105] To this end, a multicenter study of symptomatic LQTS patients receiving β-blockers recently demonstrated that propranolol and nadolol are significantly more effective than metoprolol at preventing breakthrough cardiac events. [106] Previously, concerns regarding the efficacy of atenolol, were raised based on a smaller observational study. [105] Among the largest LQTS specialty centers throughout the world, propranolol (2 to 4 mg/kg/day; half-life 4–5 hours) and nadolol (1 to 2 mg/kg/day; half-life 14–24 hours) are recommended for the initial treatment of all forms of LQTS. Propranolol may be the preferred beta blocker for LQT3.
Unfortunately, in some cases, particularly those with malignant forms of LQTS such as TS and JLNS, β-blocker monotherapy may not provide adequate protection against life-threatening ventricular arrhythmias resulting in breakthrough events or the dosage needed to achieve adequate protection is poorly tolerated. In these patients, an extrapleural [107] or video-assisted thoracoscopic [108, 109] LCSD, which involves the removal of the lower half the stellate ganglion (T1) and thoracic ganglia (T2–T4) of the left sympathetic chain with preservation of the upper half of the stellate ganglion (T1) to avoid iatrogenic Horner’s syndrome (Figure 7), often provides astrong anti-fibrillatory and QTc-attenuating effect via the localized attenuation of norepinephrine release in the left ventricular myocardium. [110, 111] In a large series of 147 high-risk LQTS patients (average QTc 563 ± 65 ms; 99% symptomatic), Schwartz et al demonstrated a >90% overall reduction in cardiac events postdenervation with a mean follow-up of 8 years. [111] Importantly, the 5 patients in this larger study who underwent LCSD secondary to multiple ICD shocks/electrical storms displayed a 95% reduction in the number shocks during a 4 year follow-up. [111] Similar observations have been seen for patients with medically refractory LQTS and malignant LQTS subtypes such as JLNS. [112, 113] As such, LCSD should be considered for LQTS patients who either 1) experience LQTS-triggered breakthrough cardiac events despite adequate β-blockade, 2) cannot tolerate β-blocker therapy secondary to undesirable side effects or absolute contraindications such as asthma, 3) experience > 1 appropriate ventricular fibrillation-terminating ICD shock(s) or electrical storms, or 4) require a so-called “bridge to ICD” due to young age and particularly malignant/high risk LQTS genotype/phenotype. [109]
Figure 7. Left cardiac sympathetic denervation.

a. Anatomical drawing depicting the extrapleural exposure of the left cardiac sympathetic chain during video assisted thoracic surgery left cardiac sympathetic denervation (VATS-LCSD). The stellate ganglion is located under the superior edge of the incision. The dashed line indicates the resection of the lower half of the left stellate ganglion occurring just above the major lower branches. b. Videoscopic stillframe from a VATS-LCSD depicting the left cardiac sympathetic chain before dissection of the pleura. c. Videoscopic stillframe from a VATS-LCSD depicting the left cardiac sympathetic chain after dissection of the pleura. Permission obtained from Elsevier © Collura et al. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm 6(6), 752–759 (2009).
Melvin Scheinman: The authors emphasize the role of left cardiac sympathetic denervation (LCSD) for high risk LQTS patients and treatment of patients with LQTS refractory to beta-blocker therapy. It should be emphasized that there are no randomized control trials which systematically evaluate the results of this treatment. The current data relative to LCSD comes largely from a small number of centers. In a comprehensive review by Schwartz et al, the incidence of syncope and sudden cardiac death was halved but clearly LCSD can not be used in lieu of ICS insertion in the very high risk LQTS group.
Our own experience is less sanguine. In a long term study of 10 patients who underwent left stellate (one) or cervicothoracic sympathectomy (nine) followed for a mean of 38.6 +/− 19 months, 8 patients experienced recurrent symptoms and 3 experienced cardiac arrest (fatal in one). We also found evidence of re eneration of the left sympathetics in patients who originally developed Horner’s syndrome. The work of Shiuknman and colleagues emphasize that both stelata and T1–T4 activate the heart and clinically for patients with ventricular tachycardia storm bilateral denervation is more effective than unilateral denervation.
Lastly, as the awareness and diagnosis of heritable cardiac arrhythmia syndromes such as LQTS increases, the number of young individuals receiving implantable cardioverter-defibrillators (ICDs) has also increased precipitously. While few would argue against immediate ICD implantation following a documented LQTS-triggered cardiac arrest, the long-term complications and quality of life issues associated with early ICD implantation make the decision to implant an ICD in those who have not suffered a cardiac arrest more tenuous.
Interestingly, an examination of the largest series of LQTS patients with ICDs (n = 233) confirmed the reality that the majority of LQTS patients receiving an ICD had not suffered a cardiac arrest and many had not even failed initial β-blocker therapy suggesting that the practice of “defensive medicine” influences clinical decision making. [114] Furthermore, during an average follow-up of <5 years, 28% of LQTS patients with an ICD received an appropriate shock, whereas 31% of patients suffered at least one adverse event including but not limited to device-related infections, lead-related complications, ICD revisions, and inappropriate shocks. [114] As a result, a clinical scorecard (M-FACT; Table 5), based on simple clinical variables, was developed to identify those patients where ICD implantation may be most appropriate. Based in part on M-FACT criteria (Table 5) and that single center studies indicate that a vast majority of LQTS patients can be treated effectively without an ICD [115], an ICD should be considered for those LQTS patients who 1) survived a cardiac arrest despite adequate β-blockade or LCSD, 2) survived a cardiac arrest off therapy, except when a reversible/preventable cause such as QT-prolonging medications or electrolyte abnormalities are identified, 3) suffer from recurrent LQTS-triggered syncope despite adequate β-blockade when LCSD is not a viable option, 4) suffer from recurrent LQTS-triggered syncope despite adequate β-blockade and LCSD, and 4) in rare extenuating circumstances such as asymptomatic patients with a QTc ≥ 550 ms with overt signs of electrical instability (e.g. T-wave alternans) on ECG and or additional objective evidence of being high risk (e.g. postpubertal LQT2 women) despite adequate β-blockade and LCSD. [2]
Table 5.
M-FACT Risk Score
| −1 Point | 0 Point | 1 Point | 2 Points | |
|---|---|---|---|---|
| Event free on therapy for > 10 years | Yes | |||
| QTc, ms | ≤ 500 | > 500 to ≤ 550 | > 550 | |
| Prior aborted cardiac arrest | No | Yes | ||
| Events on therapy | No | Yes | ||
| Age at ICD implantation, year | < 20 | ≤ 20 |
The acronym M-FACT denotes M for Minus 1 point for being free of cardiac events while on therapy for > 10 years; F for Five hundred and Fifty millisecond QTc; A for age ≤ 20 years at time of ICD implantation; C for Cardiac arrest; and T for events on Therapy.
Genotype-Guided Management
Over the past two decades, the progressive unraveling of the numerous genetic, electrophysiological, and clinical underpinnings of LQTS described above has led to the enumeration of multiple clinically meaningful genotype-phenotype correlations that have unlocked previously unforeseen management strategies and enabled genotype-guided management of LQTS to become a reality. Genotype-specific recommendations for the clinical management of the major LQTS subtypes (LQT1–LQT3), minor LQTS subtypes, and malignant forms of LQTS (TS, JLNS, AR LQT1, etc.) are described below.
Given that LQT1 patients have an increased risk of LQTS-triggered cardiac events in the setting of increased sympathetic tone [23], often secondary to emotional or physical duress, not surprisingly the use of anti-adrenergic interventions (β-blockers and or LCSD) has proven extremely effective. In fact, β-blocker noncompliance and or the concomitant use of QT-prolonging medications are responsible for the vast majority of life-threatening breakthrough cardiac events in single mutation LQT1 patients who have come to clinical attention. [116] While strenuous exercise, particularly swimming [117], has long been recognized as a “trigger” for cardiac events in LQT1 patients, recently our group decided to respectfully re-evaluate the strict competitive sports participation ban previously recommended by the 2005 36th Bethesda Conference [118] and European Society for Cardiology (ESC) [119] guidelines. While the majority of LQT1 (108/182, 59.3%) and LQT1–LQT3 patients in general (223/353, 63.2%) followed at the Mayo Clinic were not involved or elected to discontinue involvement in sports at the time of diagnosis, no difference in mortality or the rate of cardiac events was observed between non-athletes and the 60 LQT1–LQT3 (33 LQT1) patients who elected to continue sports participation in contradiction of both the Bethesda and ESC guidelines. [120, 121] In light of the low rate of LQTS-triggered cardiac events during sports and rising obesity rates in the U.S. pediatric population, the Mayo Clinic LongQT Syndrome Clinic has elected to adopt a patient/family-centered approach that embraces patient/family autonomy when LQTS athletes have been evaluated meticulously, accurately risk stratified, treated robustly, and thoroughly counseled in regards to potential risks/dangers before returning to the playing field.
Melvin Scheinman: The data from the national sports registry conducted by Rachel Lampert supports relaxation in allowing LQTS patients with defibrillators to participate in vigorous sporting activity. This includes the patients with LQTS who were formerly (Bethesdal guidelines) restricted from vigorous activities. My own clinical experience is on accord with the more relaxed approach to exercise in those with defibrillators.
Compared to LQT1, individuals with LQT2 are more susceptible to LQTS-triggered cardiac events when serum potassium levels fall [2], aroused from sleep/rest by sudden noises such as alarm clocks, telephones, or crying babies [23, 122], and during the postpartum period [123, 124]. As such, LQT2-specific management recommendations include 1) careful maintenance of serum potassium levels with a combination of diet, oral potassium supplementation, and if necessary use of potassium-sparring diuretics such as spironolactone, 2) blunting or removal of sudden noise from the bedroom and education of family members and other individuals sharing the home to avoid yelling or otherwise startling the patient, and 3) counseling LQT2 women and their partners on the necessity of β-blocker compliance, adequate rest, and avoidance of QT-prolonging medications during the postpartum period. While β-blockers remain first-line therapy for the treatment of LQT2, given the higher rate of life-threatening breakthrough cardiac events (6–7%), [102] specifically resuscitated SCAs, ultimately many high-risk LQT2 patients will require LCSD or if clinically indicated an ICD.
Melvin Scheinman: Another very important genotype-phenotype interaction was described by Barsheshet et al. they studied 860 patients with mutations in the JCNQ1 channel. Patients were divided into those with missense mutations in the membrane – spanning domain (44%), cytoplasmic loops (15%), C/N terminus (20%) or non-missense mutations. The patients were followed from birth to age 40. They recorded 27 aborted and 78 sudden death events. They concluded that missense mutations in the C loops (intracytoplasmic loops) exhibited the highest risk for sudden death and beta-blocker therapy was more beneficial for those with C loop abnormalities and attenuated for the other, finally, expression studies showed impaired regulation of PKA activity as the mechanism of these findings.
Of the major LQTS subtypes, patients with LQT3 tend to experience the highest rate of breakthrough cardiac events while on β-blocker therapy (10–15%). As a result, there has been increasing interest in targeting the pathogenic late sodium current produced by LQT3-causative SCN5A “gain-of-function” mutations with a combination of a β-blocker, preferably propranolol, and an adjuvant sodium channel blocker such as mexiletine or ranolazine as a LQT3-specific management strategy. [125, 126] However, despite the successful use of a combination of propranolol and mexiletine to treat isolated cases of malignant perinatal LQT3 caused by the unique SCN5A-G1631D mutations [127], the effect of mexiletine appears to be largely mutation-specific [126] necessitating a cumbersome drug challenge under continuous ECG monitoring to assess both therapeutic efficacy and the potential of eliciting an unwanted type 1 Brugada syndrome-like ECG pattern (PR prolongation and ST-segment elevation in the right precordial leads) given the pleiotropic nature of some SCN5A mutations. [128] While ranolazine has shown great promise as a direct late sodium current blocker in experimental systems [129] and small case series [130], the widespread clinical efficacy of ranolazine in the treatment of LQT3 remains unknown.
Similar to LQT2, given the higher rate of breakthrough cardiac events on β-blocker therapy, ultimately more LQT3 patients may require LCSD and/or ICD implantation. However, contrary to popular belief, the mere presence of an LQT3-causative mutation should not be viewed as a clinical indication for an ICD. Instead, it should be one factor that is considered in the context of the patient’s entire clinical picture.
Given the rare nature of the minor LQTS subtypes and multisystem forms of LQTS (Table 1), no specific genotype-phenotype correlations exist nor are there any true evidence-based guidelines for the management of these patients. That said, since most of the minor genotypes perturb the IKs (AKAP9-LQTS and KCNE1-LQTS), IKr (KCNE2-LQTS), or INa (CAV3-LQTS, SCN4B-LQTS, and SNTA1-LQTS) currents in an analogous fashion to what is observed LQT1, LQT2, and LQT3, respectively. Practically, minor LQTS subtypes can be managed in the same way as the corresponding major LQTS subtype (e.g. AKAP9-LQTS and KCNE1-LQTS can be managed like LQT1). However, this rule of thumb does not hold for the more malignant/multisystem forms of LQTS, such as TS and JLNS, where β-blocker therapy alone is often insufficient and early initiation of individualized combination therapy consisting of β-blockers, adjunct anti-arrhythmic agents, LCSD, and/or ICD therapy should be considered strongly.
Management of “Concealed/Low Risk” Long QT Syndrome
Owing to incomplete penetrance and variable expressivity, roughly 25% of genotype-positive LQTS patients (relatives ≫> index cases) fail to manifest any overt clinical hallmark of the disease (i.e. asymptomatic with a QTc ≤ 440 ms). Although such individuals with “concealed” LQTS have a markedly reduced risk of sudden cardiac death/aborted cardiac arrest (4%) compared to those with “expressive” phenotypes (15%), they still carry a >10 fold higher relative risk than their genotype-negative/phenotype negative relatives (0.4%). [131] This creates a clinical management conundrum where overtreatment will likely continue as some genotype-positive/phenotype-negative individuals might needprophylactic β-blocker therapy. While individuals with a concealed LQTS phenotype who harbor LQT1- and LQT3-causative missense mutations in the Kv7.1 and Nav1.5 transmembrane domains appear to be at the highest risk for life-threatening cardiac events [131], the inability to more accurately risk stratify genotype-positive/phenotype-negative individuals highlights the need to deepen our understanding of the complex interplay between genetic and environmental determinants that modulate the penetrance/expressivity of the primary LQTS-causative mutation as well as to develop further genotype-, intragenic-, and mutation-specific approaches to risk stratification and eventually treatment of LQTS.
SUMMARY
In summary, over the past two decades, tireless work from bench to bedside has unraveled many of the electrophysiologic and genetic underpinnings of LQTS allowing for the elucidation of meaningful genotype-phenotype correlations that have advanced how individuals with this potentially life-threatening disorder are diagnosed, risk-stratified, and clinically managed. For the visually inclined, the various clinically meaningful genotype-phenotype correlations, diagnostic approaches, risk stratification strategies, and therapeutic interventions discussed in the preceding sections have been synthesized into a single evidence-based algorithm designed to provide practicing cardiologists, who likely only rarely encounter LQTS patients, with a quick reference that outlines the most pertinent aspects of the genotype- and phenotype-guided management of congenital LQTS (Figure 8).
Figure 8. An integrated approach to the diagnosis, risk stratification, and genotype- and phenotype-guided management of patients with long QT syndrome.

Blue boxes denote the recommended steps in assessing the index of clinical suspicion for long QT syndrome based on personal and family history, thorough cardiac evaluation including a 12-lead electrocardiogram, and if appropriate clinical long QT syndrome genetic testing. For those individuals where a bona fide long QT syndrome-causative mutation is identified, purple boxes indicate the recommended genotype-guided management of specific long QT syndrome genetic substrates based in part on established genotype-phenotype correlations and our current understanding of the pathogenesis/electrophysiological mechanisms of these disorders. Lastly, orange boxes indicate the recommended phenotype-guided management of individuals where a specific long QT syndrome genotype remains elusive or cannot be established as well as genotype-positive individuals already receiving gene-specific treatment. Importantly, it should be noted that for genotype-positive individuals that both genotype- and phenotype-guided management strategies are utilizedconcurrently. Abbreviations: ACA, aborted cardiac arrest; AR LQT1, autosomal recessive long-QT syndrome type 1; ICD, implantable cardioverter-defibrillator; JLNS, Jervell and Lange-Nielsen syndrome; LCSD, left cardiac sympathetic denervation; LQTS, long QT syndrome; QTc, heart-rate corrected QT interval; SCD, sudden cardiac death; TS, Timothy syndrome; and VUS, variant of unknown significance.
CONCLUDING REMARKS
It seems that as quickly as one set of puzzles are solved, new and inherently more complex ones always seem to emerge. While this is certainly the case for LQTS in the post-genomic era, the rise of new technologies such as whole exome and genome sequencing and recent generation of patient-specific LQTS induced pluripotent stem cell models seem poised to address complex lingering issues such as 1) the genetic substrates responsible for the ~20% of LQTS cases that remain currently genetically elusive, 2) the novel genetic determinants that contribute to phenotypic expressivity or the lack thereof with concealed LQTS, 3) the discrepancy observed between the public domain prevalence of a possible LQTS genotype (~1:80) and LQTS phenotype (~1:2,000), and 4) the development of therapeutic interventions with less undesirable side effects and better safety profiles than those in use today. Hopefully, insights into the pathophysiological mechanisms, clinical manifestations, and therapeutic responses gained from these efforts will pave the way for the development of refined and novel approaches to the genotype- and phenotype-guided clinical management of patients afflicted by this potentially lethal, yet highly treatable, genetic disorder.
Melvin Scheinman: The authors are to be congratulated on a splendid and authoritative review of the LQTS. They offer a superb review of both the genetics and basic electrophysiology of this syndrome. The authors take advantage of extensive data base from the Mayo Clinic and formulate an eminently reasonable approach to the clinical diagnoses and treatment of these patients. The manuscript is replete with superb figures and is a valuable read not only for the general cardiologist but for cardiac electrophysiologists as well.
Acknowledgments
Funding Sources: This work was supported by the Windland Smith Rice Sudden Comprehensive Sudden Cardiac Death Program (to M.J.A.). J.R.G is supported by a National Heart Lung and Blood Institute Ruth L. Kirschstein National Research Service Award individual pre-doctoral MD/PhD fellowship (F30-HL106993).
ABBREVIATIONS
- ABS
ankyrin-B syndrome
- ATS
Andersen-Tawil syndrome
- ECG
electrocardiogram
- ICa,L
L-type calcium current
- IK1
inward rectifier potassium current
- IKAch
acetylcholine-activated inward rectifier potassium current
- IKr
rapid component of the delayed rectifier potassium current
- IKs
slow component of the delayed rectifier potassium current
- INa
sodium current
- ICD
implantable cardioverter-defibrillator
- JLNS
Jervell and Lange-Nielsen syndrome
- LCSD
left cardiac sympathetic denervation
- LQTS
long QT syndrome
- QTc
heart rate-corrected QT interval
- SCD
sudden cardiac death
- SNPs
single nucleotide polymorphisms
- TS
Timothy syndrome
Biographies
JOHN R. GIUDICESSI
J. R. Giudicessi is an MD/PhD candidate at the Mayo Graduate and Medical Schools, Mayo Clinic, Rochester, MN, USA. After receiving his bachelor’s degree from Lawrence University in Appleton, WI, USA, he entered the Medical Scientist Training Program at the Mayo Clinic where he recently completed his PhD thesis in the Windland Smith Rice Sudden Death Genomics Laboratory of Dr. Michael J. Ackerman.
MICHAEL J. ACKERMAN
M. J. Ackerman is the Windland Smith Rice Cardiovascular Genomics Research Professor and Professor of Medicine, Pediatrics, and Pharmacology, director of the Windland Smith Rice Sudden Death Genomics Laboratory, and Consultant in the Divisions of Cardiovascular Diseases and Pediatric Cardiology at the Mayo Clinic, Rochester, MN, USA. After receiving his MD and PhD degrees from the Mayo Graduate and Medical Schools, he completed a Pediatric and Adolescent Medicine residency and Pediatric Cardiology fellowship at the Mayo Graduate School of Medicine. He joined Mayo Clinic’s faculty in 2000. Currently, he also serves as the President of the Sudden Arrhythmia Death Syndromes (SADS) Foundation.
Footnotes
Disclosures: M.J.A. is a consultant for Transgenomic. Intellectual property derived from M.J.A.’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals and now Transgenomic). J.R.G. has nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moss AJ. Long QT Syndrome. JAMA. 2003;289:2041–4. doi: 10.1001/jama.289.16.2041. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–77. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romano C, Gemme G, Pongiglione R. Rare Cardiac Arrythmias of the Pediatric Age. Ii. Syncopal Attacks Due to Paroxysmal Ventricular Fibrillation. (Presentation of 1st Case in Italian Pediatric Literature) Clin Pediatr (Bologna) 1963;45:656–83. [PubMed] [Google Scholar]
- 4.Ward OC. A New Familial Cardiac Syndrome in Children. J Ir Med Assoc. 1964;54:103–6. [PubMed] [Google Scholar]
- 5.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59–68. doi: 10.1016/0002-8703(57)90079-0. [DOI] [PubMed] [Google Scholar]
- 7.Fraser GR, Froggatt P, Murphy T. Genetical Aspects of the Cardio-Auditory Syndrome of Jervell and Lange-Nielsen (Congenital Deafness and Electrocardiographic Abnormalities) Ann Hum Genet. 1964;28:133–57. doi: 10.1111/j.1469-1809.1964.tb00469.x. [DOI] [PubMed] [Google Scholar]
- 8.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res. 2013;161:1–14. doi: 10.1016/j.trsl.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104:2158–63. doi: 10.1161/hc4301.098254. [DOI] [PubMed] [Google Scholar]
- 11.Fishman GI, Chugh SS, Dimarco JP, Albert CM, Anderson ME, Bonow RO, et al. Sudden cardiac death prediction and prevention: report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation. 2010;122:2335–48. doi: 10.1161/CIRCULATIONAHA.110.976092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 13.Giudicessi JR, Ackerman MJ. Potassium-channel mutations and cardiac arrhythmias--diagnosis and therapy. Nat Rev Cardiol. 2012;9:319–32. doi: 10.1038/nrcardio.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372:750–63. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 15.Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293:H2024–38. doi: 10.1152/ajpheart.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 17.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–11. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 19.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–85. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 20.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–17. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–3. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 22.Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 24.Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. 1997;336:1562–7. doi: 10.1056/NEJM199705293362204. [DOI] [PubMed] [Google Scholar]
- 25.Giudicessi JR, Ackerman MJ. Prevalence and Potential Genetic Determinants of Sensorineural Deafness in KCNQ1 Homozygosity and Compound Heterozygosity. Circ Cardiovasc Genet. 2013 doi: 10.1161/CIRCGENETICS.112.964684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases?: the intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1120–1. doi: 10.1046/j.1540-8167.2003.03339.x. [DOI] [PubMed] [Google Scholar]
- 27.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–41. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 28.Itoh H, Shimizu W, Hayashi K, Yamagata K, Sakaguchi T, Ohno S, et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: a Japanese multicenter study. Heart Rhythm. 2010;7:1411–8. doi: 10.1016/j.hrthm.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 29.Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev. 2010;62:760–81. doi: 10.1124/pr.110.003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–5. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 31.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–48. doi: 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–6. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 33.Stramba-Badiale M, Priori SG, Napolitano C, Locati EH, Vinolas X, Haverkamp W, et al. Gene-specific differences in the circadian variation of ventricular repolarization in the long QT syndrome: a key to sudden death during sleep? Ital Heart J. 2000;1:323–8. [PubMed] [Google Scholar]
- 34.Jeyaraj D, Haldar SM, Wan X, McCauley MD, Ripperger JA, Hu K, et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature. 2012;483:96–9. doi: 10.1038/nature10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 36.Lo CF, Numann R. Independent and exclusive modulation of cardiac delayed rectifying K+ current by protein kinase C and protein kinase A. Circ Res. 1998;83:995–1002. doi: 10.1161/01.res.83.10.995. [DOI] [PubMed] [Google Scholar]
- 37.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–40. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104:20990–5. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation. 1998;97:142–6. doi: 10.1161/01.cir.97.2.142. [DOI] [PubMed] [Google Scholar]
- 40.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 41.Weerapura M, Nattel S, Chartier D, Caballero R, Hebert TE. A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J Physiol. 2002;540:15–27. doi: 10.1113/jphysiol.2001.013296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pourrier M, Zicha S, Ehrlich J, Han W, Nattel S. Canine ventricular KCNE2 expression resides predominantly in Purkinje fibers. Circ Res. 2003;93:189–91. doi: 10.1161/01.RES.0000084851.60947.B5. [DOI] [PubMed] [Google Scholar]
- 43.Jiang M, Zhang M, Tang DG, Clemo HF, Liu J, Holwitt D, et al. KCNE2 protein is expressed in ventricles of different species, and changes in its expression contribute to electrical remodeling in diseased hearts. Circulation. 2004;109:1783–8. doi: 10.1161/01.CIR.0000124225.43852.50. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y, Mahaut-Smith MP, Huang CL, Vandenberg JI. Mutant MiRP1 subunits modulate HERG K+ channel gating: a mechanism for pro-arrhythmia in long QT syndrome type 6. J Physiol. 2003;551:253–62. doi: 10.1113/jphysiol.2003.046045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–12. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 46.Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–42. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–60. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y, Liang B, Liu J, Li J, Grunnet M, Olesen SP, et al. Identification of a Kir3.4 Mutation in Congenital Long QT Syndrome. Am J Hum Genet. 2010 doi: 10.1016/j.ajhg.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beckmann C, Rinne A, Littwitz C, Mintert E, Bosche LI, Kienitz MC, et al. G protein-activated (GIRK) current in rat ventricular myocytes is masked by constitutive inward rectifier current (I(K1)) Cell Physiol Biochem. 2008;21:259–68. doi: 10.1159/000129381. [DOI] [PubMed] [Google Scholar]
- 50.Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Mapping of a gene for long QT syndrome to chromosome 4q25–27. Am J Hum Genet. 1995;57:1114–22. [PMC free article] [PubMed] [Google Scholar]
- 51.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–9. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 52.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137–42. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohler PJ, Le Scouarnec S, Denjoy I, Lowe JS, Guicheney P, Caron L, et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007;115:432–41. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- 54.Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr Scand. 1971;60:559–64. doi: 10.1111/j.1651-2227.1971.tb06990.x. [DOI] [PubMed] [Google Scholar]
- 55.Tawil R, Ptacek LJ, Pavlakis SG, DeVivo DC, Penn AS, Ozdemir C, et al. Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol. 1994;35:326–30. doi: 10.1002/ana.410350313. [DOI] [PubMed] [Google Scholar]
- 56.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511–9. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 57.Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) J Clin Invest. 2002;110:381–8. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang L, Benson DW, Tristani-Firouzi M, Ptacek LJ, Tawil R, Schwartz PJ, et al. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation. 2005;111:2720–6. doi: 10.1161/CIRCULATIONAHA.104.472498. [DOI] [PubMed] [Google Scholar]
- 59.Barrett CF, Tsien RW. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc Natl Acad Sci U S A. 2008;105:2157–62. doi: 10.1073/pnas.0710501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–96. doi: 10.1073/pnas.0502506102. discussion 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Etheridge SP, Bowles NE, Arrington CB, Pilcher T, Rope A, Wilde AA, et al. Somatic mosaicism contributes to phenotypic variation in Timothy syndrome. Am J Med Genet A. 2011;155A:2578–83. doi: 10.1002/ajmg.a.34223. [DOI] [PubMed] [Google Scholar]
- 62.Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Maternal Mosaicsim Confounds the Neonatal Diagnosis of Type 1 Timothy Syndrome. Pediatrics. 2013 doi: 10.1542/peds.2012-2941. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, et al. Calmodulin Mutations Associated with Recurrent Cardiac Arrest in Infants. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lahtinen AM, Marjamaa A, Swan H, Kontula K. KCNE1 D85N polymorphism--a sex-specific modifier in type 1 long QT syndrome? BMC Med Genet. 2011;12:11. doi: 10.1186/1471-2350-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–8. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 66.Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, et al. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821–8. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- 67.Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J Clin Invest. 2003;111:341–6. doi: 10.1172/JCI16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J. 2012;33:714–23. doi: 10.1093/eurheartj/ehr473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park JK, Martin LJ, Zhang X, Jegga AG, Benson DW. Genetic variants in SCN5A promoter are associated with arrhythmia phenotype severity in patients with heterozygous loss-of-function mutation. Heart Rhythm. 2012;9:1090–6. doi: 10.1016/j.hrthm.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hein L, Altman JD, Kobilka BK. Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature. 1999;402:181–4. doi: 10.1038/46040. [DOI] [PubMed] [Google Scholar]
- 71.Mason DA, Moore JD, Green SA, Liggett SB. A gain-of-function polymorphism in a G-protein coupling domain of the human beta1-adrenergic receptor. J Biol Chem. 1999;274:12670–4. doi: 10.1074/jbc.274.18.12670. [DOI] [PubMed] [Google Scholar]
- 72.Paavonen KJ, Swan H, Piippo K, Laitinen P, Fodstad H, Sarna S, et al. Beta1-adrenergic receptor polymorphisms, QTc interval and occurrence of symptoms in type 1 of long QT syndrome. Int J Cardiol. 2007;118:197–202. doi: 10.1016/j.ijcard.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 73.Schwartz PJ, Vanoli E, Crotti L, Spazzolini C, Ferrandi C, Goosen A, et al. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. J Am Coll Cardiol. 2008;51:920–9. doi: 10.1016/j.jacc.2007.09.069. [DOI] [PubMed] [Google Scholar]
- 74.Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–63. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tomas M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol. 2010;55:2745–52. doi: 10.1016/j.jacc.2009.12.065. [DOI] [PubMed] [Google Scholar]
- 76.Refsgaard L, Holst AG, Sadjadieh G, Haunso S, Nielsen JB, Olesen MS. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur J Hum Genet. 2012 doi: 10.1038/ejhg.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–33. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 78.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Effect of clinical phenotype on yield of long QT syndrome genetic testing. J Am Coll Cardiol. 2006;47:764–8. doi: 10.1016/j.jacc.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 79.Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929–34. doi: 10.1161/01.cir.92.10.2929. [DOI] [PubMed] [Google Scholar]
- 80.Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102:2849–55. doi: 10.1161/01.cir.102.23.2849. [DOI] [PubMed] [Google Scholar]
- 81.Schwartz PJ, Malliani A. Electrical alternation of the T-wave: clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long Q-T syndrome. Am Heart J. 1975;89:45–50. doi: 10.1016/0002-8703(75)90008-3. [DOI] [PubMed] [Google Scholar]
- 82.Taggart NW, Haglund CM, Tester DJ, Ackerman MJ. Diagnostic miscues in congenital long-QT syndrome. Circulation. 2007;115:2613–20. doi: 10.1161/CIRCULATIONAHA.106.661082. [DOI] [PubMed] [Google Scholar]
- 83.Hermosillo AG, Falcon JC, Marquez MF, Arteaga D, Cardenas M. Positive head-up tilt table test in patients with the long QT syndrome. Europace. 1999;1:213–7. doi: 10.1053/eupc.1999.0056. [DOI] [PubMed] [Google Scholar]
- 84.Colman N, Bakker A, Linzer M, Reitsma JB, Wieling W, Wilde AA. Value of history-taking in syncope patients: in whom to suspect long QT syndrome? Europace. 2009;11:937–43. doi: 10.1093/europace/eup101. [DOI] [PubMed] [Google Scholar]
- 85.Rautaharju PM, Surawicz B, Gettes LS, Bailey JJ, Childers R, Deal BJ, et al. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation. 2009;119:e241–50. doi: 10.1161/CIRCULATIONAHA.108.191096. [DOI] [PubMed] [Google Scholar]
- 86.Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88:782–4. doi: 10.1161/01.cir.88.2.782. [DOI] [PubMed] [Google Scholar]
- 87.Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124:2181–4. doi: 10.1161/CIRCULATIONAHA.111.062182. [DOI] [PubMed] [Google Scholar]
- 88.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–74. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 89.Ackerman MJ, Khositseth A, Tester DJ, Hejlik JB, Shen WK, Porter CB. Epinephrine-induced QT interval prolongation: a gene-specific paradoxical response in congenital long QT syndrome. Mayo Clin Proc. 2002;77:413–21. doi: 10.4065/77.5.413. [DOI] [PubMed] [Google Scholar]
- 90.Wong JA, Gula LJ, Klein GJ, Yee R, Skanes AC, Krahn AD. Utility of treadmill testing in identification and genotype prediction in long-QT syndrome. Circ Arrhythm Electrophysiol. 2010;3:120–5. doi: 10.1161/CIRCEP.109.907865. [DOI] [PubMed] [Google Scholar]
- 91.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–60. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 2011;8:1308–39. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 93.Giudicessi JR, Ackerman MJ. Genetic testing in heritable cardiac arrhythmia syndromes: differentiating pathogenic mutations from background genetic noise. Curr Opin Cardiol. 2013;28:63–71. doi: 10.1097/HCO.0b013e32835b0a41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giudicessi JR, Kapplinger JD, Tester DJ, Alders M, Salisbury BA, Wilde AA, et al. Phylogenetic and physicochemical analyses enhance the classification of rare nonsynonymous single nucleotide variants in type 1 and 2 long-QT syndrome. Circ Cardiovasc Genet. 2012;5:519–28. doi: 10.1161/CIRCGENETICS.112.963785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–90. doi: 10.1161/CIRCULATIONAHA.105.592899. [DOI] [PubMed] [Google Scholar]
- 96.Goldenberg I, Moss AJ, Zareba W, McNitt S, Robinson JL, Qi M, et al. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J Cardiovasc Electrophysiol. 2006;17:1161–8. doi: 10.1111/j.1540-8167.2006.00587.x. [DOI] [PubMed] [Google Scholar]
- 97.Sauer AJ, Moss AJ, McNitt S, Peterson DR, Zareba W, Robinson JL, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–37. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 98.Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Risk of life-threatening cardiac events among patients with long QT syndrome and multiple mutations. Heart Rhythm. 2013;10:378–82. doi: 10.1016/j.hrthm.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J. 1975;89:378–90. doi: 10.1016/0002-8703(75)90089-7. [DOI] [PubMed] [Google Scholar]
- 100.Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J. 1985;109:399–411. doi: 10.1016/0002-8703(85)90626-x. [DOI] [PubMed] [Google Scholar]
- 101.Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–23. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- 102.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–4. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 103.Villain E, Denjoy I, Lupoglazoff JM, Guicheney P, Hainque B, Lucet V, et al. Low incidence of cardiac events with beta-blocking therapy in children with long QT syndrome. Eur Heart J. 2004;25:1405–11. doi: 10.1016/j.ehj.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 104.Shimizu W, Tanabe Y, Aiba T, Inagaki M, Kurita T, Suyama K, et al. Differential effects of beta-blockade on dispersion of repolarization in the absence and presence of sympathetic stimulation between the LQT1 and LQT2 forms of congenital long QT syndrome. J Am Coll Cardiol. 2002;39:1984–91. doi: 10.1016/s0735-1097(02)01894-6. [DOI] [PubMed] [Google Scholar]
- 105.Chatrath R, Bell CM, Ackerman MJ. Beta-blocker therapy failures in symptomatic probands with genotyped long-QT syndrome. Pediatr Cardiol. 2004;25:459–65. doi: 10.1007/s00246-003-0567-3. [DOI] [PubMed] [Google Scholar]
- 106.Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol. J Am Coll Cardiol. 2012;60:2092–9. doi: 10.1016/j.jacc.2012.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Odero A, Bozzani A, De Ferrari GM, Schwartz PJ. Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias: the surgical supraclavicular approach to cervicothoracic sympathectomy. Heart Rhythm. 2010;7:1161–5. doi: 10.1016/j.hrthm.2010.03.046. [DOI] [PubMed] [Google Scholar]
- 108.Atallah J, Fynn-Thompson F, Cecchin F, DiBardino DJ, Walsh EP, Berul CI. Video-assisted thoracoscopic cardiac denervation: a potential novel therapeutic option for children with intractable ventricular arrhythmias. Ann Thorac Surg. 2008;86:1620–5. doi: 10.1016/j.athoracsur.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 109.Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752–9. doi: 10.1016/j.hrthm.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 110.Schwartz PJ, Snebold NG, Brown AM. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am J Cardiol. 1976;37:1034–40. doi: 10.1016/0002-9149(76)90420-3. [DOI] [PubMed] [Google Scholar]
- 111.Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826–33. doi: 10.1161/01.CIR.0000125523.14403.1E. [DOI] [PubMed] [Google Scholar]
- 112.Hwang SW, Thomas JG, Whitehead WE, Curry DJ, Dauser RC, Kim ES, et al. Left thorascopic sympathectomy for refractory long QT syndrome in children. J Neurosurg Pediatr. 2011;8:455–9. doi: 10.3171/2011.8.PEDS11164. [DOI] [PubMed] [Google Scholar]
- 113.Coleman MA, Bos JM, Johnson JN, Owen HJ, Deschamps C, Moir C, et al. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long-QT syndrome. Circ Arrhythm Electrophysiol. 2012;5:782–8. doi: 10.1161/CIRCEP.112.971754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schwartz PJ, Spazzolini C, Priori SG, Crotti L, Vicentini A, Landolina M, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them?: data from the European Long-QT Syndrome Implantable Cardioverter-Defibrillator (LQTS ICD) Registry. Circulation. 2010;122:1272–82. doi: 10.1161/CIRCULATIONAHA.110.950147. [DOI] [PubMed] [Google Scholar]
- 115.Horner JM, Kinoshita M, Webster TL, Haglund CM, Friedman PA, Ackerman MJ. Implantable cardioverter defibrillator therapy for congenital long QT syndrome: a single-center experience. Heart Rhythm. 2010;7:1616–22. doi: 10.1016/j.hrthm.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 116.Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C, et al. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215–21. doi: 10.1161/CIRCULATIONAHA.108.772533. [DOI] [PubMed] [Google Scholar]
- 117.Ackerman MJ, Tester DJ, Porter CJ. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin Proc. 1999;74:1088–94. doi: 10.4065/74.11.1088. [DOI] [PubMed] [Google Scholar]
- 118.Zipes DP, Ackerman MJ, Estes NA, 3rd, Grant AO, Myerburg RJ, Van Hare G. Task Force 7: arrhythmias. J Am Coll Cardiol. 2005;45:1354–63. doi: 10.1016/j.jacc.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 119.Pelliccia A, Fagard R, Bjornstad HH, Anastassakis A, Arbustini E, Assanelli D, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:1422–45. doi: 10.1093/eurheartj/ehi325. [DOI] [PubMed] [Google Scholar]
- 120.Johnson JN, Ackerman MJ. Competitive sports participation in athletes with congenital long QT syndrome. JAMA. 2012;308:764–5. doi: 10.1001/jama.2012.9334. [DOI] [PubMed] [Google Scholar]
- 121.Johnson JN, Ackerman MJ. Return to play? Athletes with congenital long QT syndrome. Br J Sports Med. 2013;47:28–33. doi: 10.1136/bjsports-2012-091751. [DOI] [PubMed] [Google Scholar]
- 122.Wilde AA, Jongbloed RJ, Doevendans PA, Duren DR, Hauer RN, van Langen IM, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1) J Am Coll Cardiol. 1999;33:327–32. doi: 10.1016/s0735-1097(98)00578-6. [DOI] [PubMed] [Google Scholar]
- 123.Rashba EJ, Zareba W, Moss AJ, Hall WJ, Robinson J, Locati EH, et al. Influence of pregnancy on the risk for cardiac events in patients with hereditary long QT syndrome. LQTS Investigators. Circulation. 1998;97:451–6. doi: 10.1161/01.cir.97.5.451. [DOI] [PubMed] [Google Scholar]
- 124.Khositseth A, Tester DJ, Will ML, Bell CM, Ackerman MJ. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004;1:60–4. doi: 10.1016/j.hrthm.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 125.Moss AJ, Windle JR, Hall WJ, Zareba W, Robinson JL, McNitt S, et al. Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (DeltaKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol. 2005;10:59–66. doi: 10.1111/j.1542-474X.2005.00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137–44. doi: 10.1161/CIRCULATIONAHA.107.707877. [DOI] [PubMed] [Google Scholar]
- 127.Wang DW, Crotti L, Shimizu W, Pedrazzini M, Cantu F, De Filippo P, et al. Malignant perinatal variant of long-QT syndrome caused by a profoundly dysfunctional cardiac sodium channel. Circ Arrhythm Electrophysiol. 2008;1:370–8. doi: 10.1161/CIRCEP.108.788349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Priori SG, Napolitano C, Schwartz PJ, Bloise R, Crotti L, Ronchetti E. The elusive link between LQT3 and Brugada syndrome: the role of flecainide challenge. Circulation. 2000;102:945–7. doi: 10.1161/01.cir.102.9.945. [DOI] [PubMed] [Google Scholar]
- 129.Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–93. doi: 10.1111/j.1540-8167.2008.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51–9. doi: 10.1016/j.jacc.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boczek NJ, Best JM, Tester DJ, Giudicessi JR, Kamp TJ, Ackerman MJ. Molecular and Functional Characterization of a Novel Pathogenic Substrate for Autosomal Dominant Long QT Syndrome Discovered by Whole Exome Sequencing, Genomic Triangulation, and Systems Biology. Heart Rhythm. 2012;9:1911–2. [Google Scholar]