

Graphical abstract
Keywords: Aminoacyl-tRNA synthetase, Drug target, Parasite, Protein translation
Highlights
-
•
Aminoacyl-tRNA synthetases are essential and many aaRS inhibitors kill parasites.
-
•
We examine compound inhibitors tested experimentally against parasite aaRSs.
-
•
Successful inhibitors were discovered by both phenotype and target-based approaches.
-
•
Selectivity and resistance are ongoing challenges for development of parasite drugs.
Abstract
Aminoacyl-tRNA synthetases are central enzymes in protein translation, providing the charged tRNAs needed for appropriate construction of peptide chains. These enzymes have long been pursued as drug targets in bacteria and fungi, but the past decade has seen considerable research on aminoacyl-tRNA synthetases in eukaryotic parasites. Existing inhibitors of bacterial tRNA synthetases have been adapted for parasite use, novel inhibitors have been developed against parasite enzymes, and tRNA synthetases have been identified as the targets for compounds in use or development as antiparasitic drugs. Crystal structures have now been solved for many parasite tRNA synthetases, and opportunities for selective inhibition are becoming apparent. For different biological reasons, tRNA synthetases appear to be promising drug targets against parasites as diverse as Plasmodium (causative agent of malaria), Brugia (causative agent of lymphatic filariasis), and Trypanosoma (causative agents of Chagas disease and human African trypanosomiasis). Here we review recent developments in drug discovery and target characterisation for parasite aminoacyl-tRNA synthetases.
1. Introduction – the need for new antiparasitic drugs
The prevalence and persistence of parasitic infections are both remarkable and troubling phenomena. Approximately one billion people harbour at least one worm infection (nematodes and platyhelminths) (Lustigman et al., 2012) and many individuals are simultaneously infected with multiple parasites from distantly related eukaryotic phyla (Fevre et al., 2008; Gething et al., 2011; Nacher, 2012). These parasites cause diseases that impose a serious burden to the health and economic development of affected countries, and are therefore the subject of many varied prevention and control strategies. No human-licensed vaccine exists for any eukaryotic disease, therefore drugs are a major component of intervention against most parasitic diseases (Prichard et al., 2012). Drug based strategies include treatment of verified infections, mass drug administration to presumptive infected communities or at risk individuals (e.g. pregnant mothers), and sporadic prophylaxis for individuals. In many cases existing drug-based programs are at risk from parasites developing resistance, and therefore rendering ineffective our affordable and effective drugs. Some antiparasitic drugs have already had their effective usage severely restricted in regions due to the development of widespread drug resistance (Baird, 2005; Croft and Olliaro, 2011). The development of future control strategies is threatened by the impending and inevitable emergence of resistance to additional drugs (Geerts and Gryseels, 2000). To deal with existing and future shortcomings of antiparasitic drugs, multiple classes of new drugs are urgently needed for many parasitic diseases.
Parasites cause diverse types of disease, requiring drug treatments that address varying causes of pathogenesis. Apicomplexan parasites include Plasmodium spp., Toxoplasma gondii and Cryptosporidium. All parasites in this phylum are obligate intracellular parasites, but their host range and disease type varies immensely. Plasmodium species cause generally acute disease through proliferation within and destruction of erythrocytes. Most existing anti-malarial drugs work by killing this proliferative intra-erythrocytic stage, though action against the parasite forms that initially infect humans (sporozoites) and the forms that are transmitted to mosquitoes (gametocytes) is highly desirable in addition to disease control purposes (Burrows et al., 2013). Toxoplasma gondii parasites infect many diverse animals and many cell types. In humans, Toxoplasma is normally pathogenic only in immunocompromised individuals or in the human foetus. Drugs are needed to arrest the faster growing tachyzoite stages of Toxoplasma, as well as the latent bradyzoite stages that form cysts in the brain and other organs (Rodriguez and Szajnman, 2012). Cryptosporidium infects epithelial cells of the intestine, causing potentially severe and chronic diarrhea. As with Toxoplasma, the most severe Cryptosporidium cases are in immunocompromised individuals, and the need for drugs is more pressing for treatment of such cases (Rossignol, 2010).
Typanosomatid parasites also cause a broad spectrum of diseases. Trypanosoma brucei, spread by the bite of the tsetse fly, causes human African trypanosomiasis, also known as sleeping sickness. These parasites proliferate extra-cellularly in the bloodstream and lymphatic system and later infect the central nervous system (CNS) (Barrett et al., 2007). This disease is fatal within months to years if not treated, and most existing treatments are difficult to administer, toxic or ineffective. New drugs must overcome the additional challenge of crossing the blood brain barrier to treat parasites in the CNS. Trypanosoma cruzi infections are the cause of the chronic and potentially fatal Chagas disease. Existing drugs to treat T. cruzi are ineffective if not administered early during infection and are highly toxic. Leishmania, the second medically important genus of trypanosomatid parasites, includes species that also cause a range of serious human diseases. In humans, Leishmania parasites invade and grow within phagocytic cells. As with other trypanosomatid parasites, existing drugs are generally toxic, difficult to deliver and subject to parasite resistance (Stuart et al., 2008). Although trypanosomatid parasites kill fewer people than malaria, the lack of effective and safe drugs arguably makes discovery of new drugs even more pressing for these parasites.
Three parasites whose anaerobic metabolism distinguishes them from most other eukaryotes are the extracellular parasites Giardia, Trichomonas, and Entamoeba. In these parasites the mainstays for treatment are the nitroimidazole drugs, which are activated by the parasites’ unusual pyruvate:ferredoxin oxidoreductase enzymes (Ali and Nozaki, 2007). In each of these parasites, resistance to nitroimidazol is possible through altered metabolism and alternative drugs are scarce or ineffective (Upcroft and Upcroft, 2001).
The final parasite discussed below in the context of tRNA synthetase targets is the helminth parasite Brugia. Brugia malayi is a nematode spread between humans by mosquitoes and is one of several parasites to cause human filariasis. Lymphatic filariasis is caused by immunological reaction to the adult worms and the thousands of transmissive microfilaria they produce. Drug discovery against nematodes introduces the added difficulty of selective inhibition between the bilaterian animal parasites and their hosts, although Brugia’s dependence on its bacterial Wolbachia symbiont may offer other potential drug targets (Bandi et al., 2001).
2. Protein translation as a drug target
One biological pathway that has been thoroughly validated as a target for anti-infective compounds in a wide range of microbes is the process of protein translation. Most antibiotics that target protein translation interact with microbial ribosomes themselves—binding directly to the rRNA or ribosomal subunit proteins. However, additional molecules within the broader process of protein translation can act as targets for drugs. One such target for existing and future antimicrobial therapeutics is the aminoacyl-tRNA synthetase (aaRS) family. This family of enzymes catalyses the attachment of amino acids to their cognate tRNAs to produce the aminoacyl tRNAs (also aa-tRNA or charged tRNA) that are the substrates for translation (reviewed by Ibba and Soll, 2000). The aaRSs enzymes are not only responsible for producing the raw materials for translation, but also for ensuring the fidelity of translation from nucleic acid to amino acid information. Disruption of aaRSs therefore interrupts or poisons the process of protein translation. Compounds that inhibit aaRSs have been successfully exploited, with at least one antibacterial drug, mupirocin, currently in clinical use for the topical treatment of Staphylococcus aureus, that acts through the inhibition of the isoleucyl-tRNA synthetase (IleRS) of gram-positive bacteria (Nakama et al., 2001). The pursuit of diverse other aminoacyl-tRNA synthetases has yielded specific aaRS inhibitors (Rock et al., 2007), some of which are currently in clinical trials as antimicrobials (de Jonge et al., 2006; Koon et al., 2011).
Besides the excellent precedence for druggability in bacteria, there are several reasons to support protein translation in general, and aaRSs specifically, as a useful antiparasitic target. First is the dependence of many parasites on abundant protein translation in fast growing cells. Because many parasites constitutively undergo active and continuous proliferation they are heavily reliant on efficient protein translation and may be sensitive to disruptions to the translation machinery. Other parasites pass through quiescent life-stages with relatively little cellular proliferation—these stages (such as the bradyzoite stages of Toxoplasma gondii) are likely to have a reduced requirement for protein turnover and may be less sensitive to translation inhibitors. Such stages present a general problem for chemotherapy, though it is noteworthy that inhibition of housekeeping functions, such as the block of gene expression by rifampicin, has been successfully exploited for slow-growing microbes such as latent stage Mycobacterium tuberculosis (Leung et al., 2011). A second aspect of parasite protein translation that renders it a plausible drug target is the immense evolutionary distance between this process in some parasites and human hosts. Furthermore, several parasites have bacterial-like protein translation pathways that are not shared by humans. Apicomplexan parasites in particular, are dependent on their relict plastid (apicoplast), which retains much of the cyanobacterial protein translation apparatus of plastids’ ancestor (Jackson et al., 2011). Trypanosomatid parasites are highly dependent on protein translation in their unusual kinetoplastid mitochondrion, and the protein translation therein differs in several aspects from the translation found in human mitochondria or cytosol (Schneider, 2001; Niemann et al., 2011). These examples highlight the presence of aaRSs in multiple organelles, all of which may be considered when contemplating drug targets.
A number of recent reviews have detailed drug discovery and development against bacterial and fungal aaRSs (Kim et al., 2003; Ochsner et al., 2007; Vondenhoff and Van Aerschot, 2011; Lv and Zhu, 2012). In this review we discuss prospects for drug development against the aaRSs of eukaryotic parasites, including apicomplexan, trypanosomatid, and metamonad protists as well as parasitic worms.
3. Aminoacyl-tRNA synthetases as drug targets
Before considering specific parasite targets, let us briefly consider what types of activities might be inhibited when focusing on aaRSs. Aminoacyl-tRNA synthetases catalyse a two-step reaction whereby an ATP and amino acid molecule (AA) enter the active site, forming an aminoacyl-adenylate intermediate (1), followed by the esterification of the amino acid to the 3′ end of the tRNA, forming the final ‘charged’ aminoacyl-tRNA (2).
| (1) |
| (2) |
This presents several sites on aaRS enzymes that may be considered for drugging purposes; a binding site for ATP, an adjacent amino acid binding site, and a fold for tRNA recognition and binding (Fig. 1). Most aaRS inhibitors bind to the ATP and amino acid binding sites, in many cases as analogues of ATP, amino acids, or aminoacyl-adenylate intermediates (Vondenhoff and Van Aerschot, 2011). Below we review prospects for antiparasitic drug development from such inhibitors for each of the tRNA synthetases.
Fig. 1.
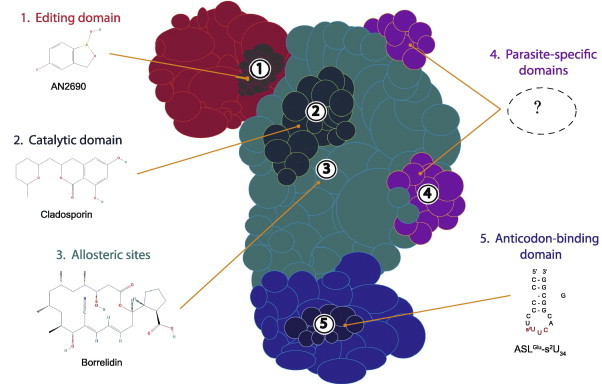
Schematic representation of an aminoacyl-tRNA synthetase. Various aaRS domains are illustrated: the editing domain (red); catalytic domain (cyan); anticodon-binding domain (indigo); and parasite-specific domains (purple). Possible sites of interaction between aaRS and compound (with existing examples) are indicated by numbers: editing site (1); active site (2); allosteric sites (3); parasite-specific domains (4); and anticodon-binding site (5). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Discrimination between different amino acids with similar chemical structures is a biochemical challenge and some aminoacyl-tRNA synthetases are prone to errors in charging. Errors can be reduced by recognition and elimination of misactivated noncognate amino acids, or through editing of misacylated tRNAs. Proof-reading is achieved both through editing domains on the aaRS enzymes themselves, as well as by stand-alone editing enzymes (Ahel et al., 2003; Sokabe et al., 2005). These domains and enzymes have the potential to act as targets for drugs, and several aaRS inhibitors are thought to act via inhibition of the editing process (Rock et al., 2007; Tan et al., 2013).
In addition to their canonical roles in tRNA aminoacylation, these ancient enzymes have also evolved extra functions, in some cases through the acquisition of novel protein domains (Lee et al., 2004; Smirnova et al., 2012; Guo and Schimmel, 2013). In eukaryotes in particular, tRNA synthetases also play roles in non-translation processes including the regulation of transcription, RNA splicing, apoptosis, angiogenesis, immune responses and signalling events. These moonlighting functions may be crucial in some organisms, and some inhibitors that focus specifically on these non protein-translation roles of aaRSs have been explored. Non-canonical roles have been suggested for several parasite tRNA synthetases, and we also review the potential for these enzymes as drug targets below.
4. Existing aaRS inhibitors in parasites
4.1. Alanyl-tRNA synthetase
Alanyl-tRNA synthetase (AlaRS) has been a focus of extensive research due to the presence of an unusual secondary catalytic site with editing activity (Guo et al., 2009; Sokabe et al., 2009). Glycine and serine are common misacylations of tRNAAla, and AlaRS enzymes can edit these products of misacylation to ensure they do not accumulate to toxic levels. Mice with defects in tRNAAla editing have a neurodegeneration phenotype, reinforcing the importance of editing activity (Lee et al., 2006). An AlaX domain found on additional proteins is also involved in the elimination of mischarged tRNAAla and an AlaX domain has been reported in Plasmodium, fused to another tRNA synthetase; PfTrpRS (Khan et al., 2013b). AlaRS has also been characterised in Plasmodium parasites—only one version of this enzyme is encoded by the Plasmodium genome, despite apparent requirements for cytosolic organellar translation. As with the PfGlyRS and PfThrRS, this enzyme is post-translationally targeted to both the Plasmodium falciparun cytosol and the apicoplast, possibly by production of alternatively initiated proteins (Khan et al., 2011; Jackson et al., 2012). Khan et al. (2011) screened several putative inhibitors of the PfAlaRS based on in silico docking against structural homology models. One of these, 4-{2-nitro-1-propenyl}-1,2-benzenediol (Table 1), inhibited parasite growth at low micromolar inhibition, and produced only limited mammalian cytotoxicity at similar concentrations (Khan et al., 2011). Although it is not yet known if these compounds inhibit the parasite PfAlaRS, the dependence of cytosolic and apicoplast translation on this dual targeted enzyme makes it a conceptually attractive target.
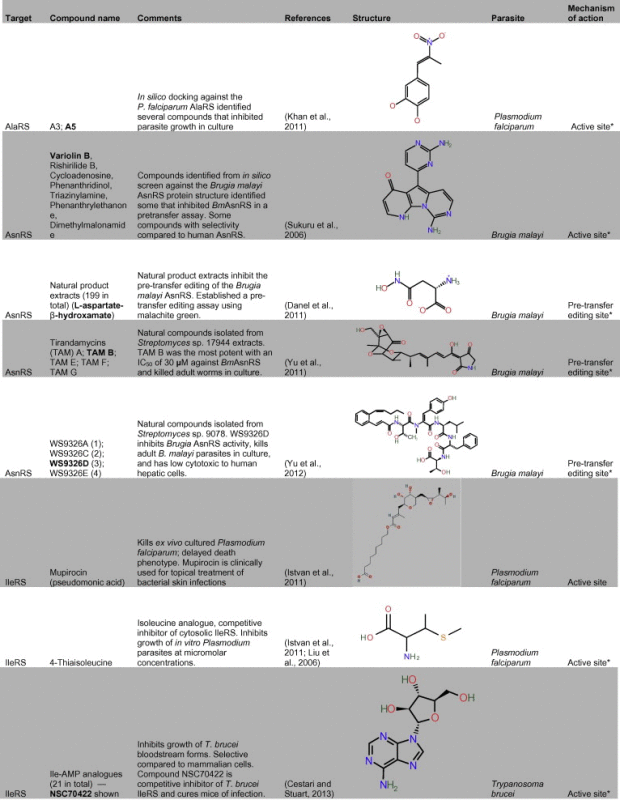
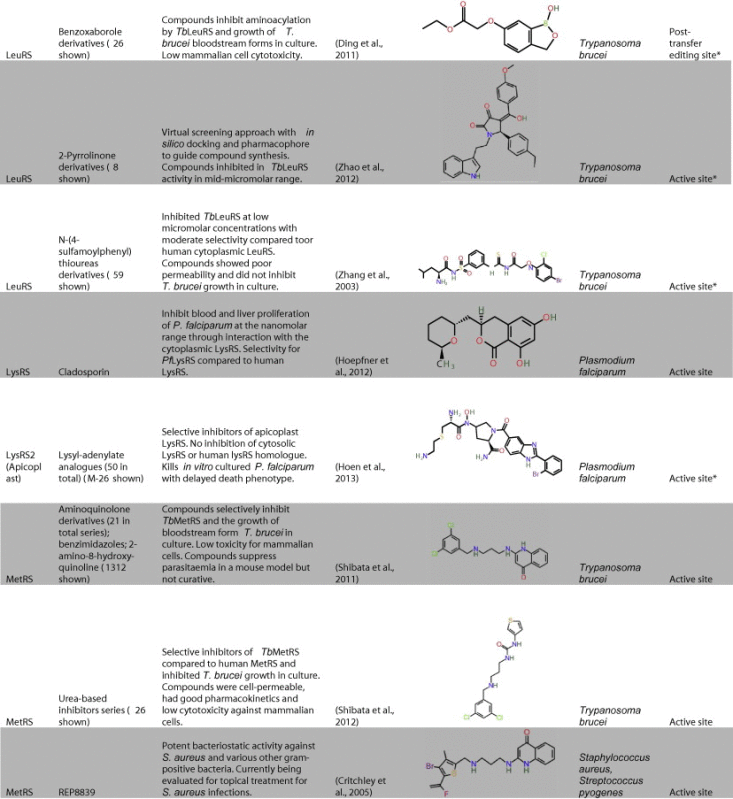
Table 1.
Representative inhibitors of aminoacyl tRNA synthetases in parasites and other infectious agents. The names of the structures shown are indicated in bold text. Structures are derived from PubChem, Chemspider or redrawn from original papers.
 |
 |
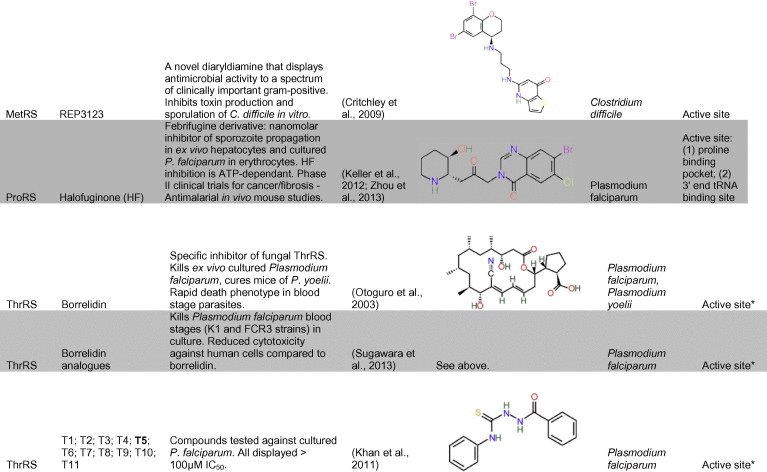 |
a Predicted site of action. (See above-mentioned references for further information.)
4.2. Asparaginyl-tRNA synthetase
The cytoplasmic asparaginyl-tRNA synthetase (AsnRS) has been a long-standing drug target in Brugia malayi, a nematode that is one of the causative agents of lymphatic filariasis. This enzyme is very highly expressed compared to other Brugia aaRSs, and, like other aaRSs mentioned above, appears to have developed non-protein-translation functions in addition to its canonical role. In Brugia, the AsnRS is an immunodominant antigen in human infections (Kron et al., 1995) additionally this enzyme catalyses the production of diadenosine triphosphate (Kron et al., 2003) and exhibits immunomodulatory functions associated with inflammation during host infection (Ramirez et al., 2006; Kron et al., 2012, 2013). Because of these apparently key roles, several drug discovery (and diagnostic) projects have focused on this B. malayi enzyme.
Two distinct strategies have been employed to find inhibitors of the B. malayi enzyme; in silico docking and high throughput screening. In the first approach, compounds from publicly available collections were docked against the B. malayi AsnRS (BmAsnRS). From this docking, 45 compounds were tested for their inhibition of the BmAsnRS, as assayed by a modified malachite green assay, which provides a readout for the first step in aminoacylation reaction. Of the compounds screened, a handful inhibited AsnRS aminoacylation at mid-micromolar IC50s (Table 1) (Sukuru et al., 2006). Subsequent publication of a more detailed structure for the Brugia AsnRS in complex with a substrate analogue that acts as a competitive inhibitor (Crepin et al., 2011) may provide additional information to refine future docking experiments or to rationally improve existing inhibitors.
A second approach makes use of an assay that focuses on AsnRS’s capacity to recognise and edit misacylation prior to transfer. This pre-transfer editing assay initially identified compounds that promoted the editing activity of BmAsnRS, as well as allowing screening of inhibitors that blocked the BmAsnRS (Table 1) (Danel et al., 2011). The assay was then used to experimentally screen for inhibitors of BmAsnRS among tens of thousands of extracts from diverse microbial strains. Further purification and fractionation of these extracts has led to the discovery of two different compound classes—the Tirandamycins (Yu et al., 2011) and the WS9326A derivatives (Yu et al., 2012) (see Table 1)—that each inhibit BmAsnRS aminoacylation and kill adult B. malayi. Both classes appear to show some selectivity for Brugia compared to human AsnRS. Further optimisation and validation of these inhibitors is reportedly underway (Yu et al., 2011; Rateb et al., 2013).
4.3. Isoleucyl-tRNA synthetase
The protist parasite, Trypanosoma brucei is the causative agent of human African trypanosomiasis. There is an urgent need for new, more effective, non-toxic, and cheap antitrypanosomal drugs (Fevre et al., 2008). Recent research validates the Trypanosoma brucei isoleucyl-tRNA synthetase (IleRS) as a potential drug target, with ex vivo and in vivo RNAi knockdowns showing IleRS to be essential for Trypanosoma brucei growth (Cestari and Stuart, 2013). Cestari and Stuart screened 20 compounds from the National Cancer Institute database that were structurally similar to Ile-AMP (the reaction intermediate) for killing of T. brucei bloodstream forms. Several active compounds from this screen, including NSC70442 specifically inhibited activity of recombinant T. brucei isoleucyl-tRNA sythetase (IleRS) and have good selectivity against mammalian cell lines (Table 1). Furthermore, a transgenic T. brucei line that overexpressed IleRS showed reduced sensitivity to NSC70442 and other Ile-AMP analogues, supporting IleRS as the target of these inhibitors. NSC70442 cured in T. brucei-infected mice at low mammalian toxicity (Cestari and Stuart, 2013). There is also evidence that compounds from this chemical class are able to cross the blood brain barrier (Cestari and Stuart, 2013), a very important characteristic for antitrypanosomal drugs that can treat stage 2 trypanosomiasis (Rottenberg et al., 2005)
The most widely-used drug that inhibits aminoacyl-tRNA synthetases targets IleRS. Mupirocin (also known as pseudomonic acid, and marketed as Bactroban®), is used for the topical treatment of Staphylococcus aureus. Crystal structures of mupirocin-bound IleRS indicate that mupirocin inhibits bacterial IleRS by blocking the binding of the Ile-AMP intermediate (Nakama et al., 2001). Mupirocin has now been shown to inhibit blood-stage P. falciparum growth in the nanomolar range (Table 1) (Istvan et al., 2011). Mupirocin resistant Plasmodium parasites have mutations in the apicoplast-located IleRS, indicating that the bacterial type apicoplast IleRS is the target of mupirocin (Istvan et al., 2011). This is supported by specific defects in apicoplast morphology and segregation upon mupirocin, and in the “delayed-death” type mupirocin killing of ex vivo cultured parasites (Jackson et al., 2012), which is characteristic of inhibitors blocking apicoplast maintenance. Mupirocin failed to protect mice from a Plasmodium berghei infection, a likely result of the compound’s well known in vivo instability and its high binding to serum (Casewell and Hill, 1987). Mupirocin itself is therefore very unlikely to serve as a good lead for antiparasitic drug development, but does validate the apicoplast IleRS as a target for specific antimalarial drug research. Istvan and colleagues (Istvan et al., 2011) also showed that the Plasmodium cytosolic IleRS was inhibited by the isoleucine analogue thiaisoleucine (Table 1), which rapidly killed ex vivo cultured parasites. Thiaisoleucine also inhibits mammalian IleRS but its inhibition of Plasmodium growth supports the Plasmodium cytosolic IleRS enzyme as a potential drug target. Another inhibitor of eukaryotic IleRSs, the cyclic beta amino acid, icofungipen, shows good activity against pathogenic fungi, and has been through phase II human trials (Hasenoehrl et al., 2006). We are unaware of any tests on the activity of icofungipen in any parasite.
4.4. Leucyl-tRNA synthetase
LeuRS is a proofreading aaRS that is inhibited by 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2690) by trapping tRNALeu in the editing site (Rock et al., 2007). AN2690 has potent antifungal activity, and was reported to be undergoing clinical trials (Seiradake et al., 2009). Inspired by this success story, inhibitors based on the benzoxaborole core—that contains a boronic acid and forms an adduct with the tRNA—were explored as LeuRS inhibitors for T. brucei. For this purpose, a homology model of the T. brucei CP1 (editing) domain based on the solved Candida albicans LeuRS was used to design a series of benzoxaborole compounds (Ding et al., 2011). Structure–activity relationship was studied, and the best of the compounds (Table 1) inhibited TbLeuRS aminoacylation and T. brucei ex vivo growth at low micromolar IC50s, with low mammalian cell toxicity (Ding et al., 2011). A subsequent TbLeuRS study (Zhao et al., 2012), used a homology model based on the solved Pyrococcus horikoshii LeuRS structure (Fukunaga and Yokoyama, 2005) and instead targeted the enzyme’s synthetic site. This study performed in silico screening with the SPECS chemical library, and tested a range of compounds with a 2-pyrrolinone scaffold against an in vitro TbLeuRS aminoacylation assay. Though a diverse group of analogues showed some structure–activity relationship, inhibition occurred only at rather high concentrations, with the most potent compounds showing IC50s between ∼30 and 100 μM (Table 1) (Zhao et al., 2012).
More recently, a new class of T. brucei LeuRS inhibitors, N-(4-sulfamoylphenyl)thioureas, which targets the synthetic catalytic site, has been discovered through the screening and modification of a small, targeted library of putative aaRS inhibitors (Zhang et al., 2013). This class of inhibitors are designed to inhibit by mimicking the intermediate product, aminoacyl-AMP. To further improve upon the activities of the compounds, TbLeuRS was used to dock inhibitors and guide synthesis of derivatives. The best compound, 59, showed an IC50 of 1.1 μM and is predicted to form four hydrogen bonds and favourable hydrophobic interactions with the synthetic enzyme pocket. These compounds exhibited moderate selectivity (4.5–7.3 fold) compared to human cytoplasmic LeuRS, but none of the compounds optimised for inhibition of the synthetic site inhibited growth of T. brucei in culture at 100 μM (Table 1). Experiments using caco-2 cell permeability assays indicated that these compounds have poor permeability and may explain the poor inhibition seen in the ex vivo bioassays. This new inhibitor class shows early promise as TbLeuRS inhibitors but will require more work to address permeability issues and demonstrate the ability to kill parasites and cure mice infections.
4.5. Lysyl-tRNA synthetase
In a recent comprehensive study, the fungal secondary metabolite cladosporin (Table 1) was shown to inhibit blood and liver proliferation of P. falciparum at the nanomolar range (Hoepfner et al., 2012). Studies in fungi showed that heterozygote mutants of LysRS were more sensitive to cladosporin, and fungi with separate point mutations in LysRS were more resistant to cladosporin. Plasmodium parasites overexpressing the cytosolic PfLysRS are similarly more resistant to cladosporin. In silico docking suggests that cladosporin interacts with the ATP-binding pocket of the LysRS and increasing concentration of ATP in vivo significantly reduced inhibition, consistent with this in silico prediction. Cladosporin only demonstrated weak inhibition of recombinant human LysRS at high micromolar concentrations, which was postulated to be due to steric hindrance within the ATP-binding pocket (Hoepfner et al., 2012). The crystal structure of the cytoplasmic PfLysRS was subsequently solved, and confirms a structural difference in this region is likely to be the basis for this selectivity (Khan et al., 2013a). The structure also describes additional differences that may allow for the design of selective inhibitors that act against the Plasmodium but not human LysRS (Khan et al., 2013a).
Whilst cladosporin inhibits the Plasmodium cytosolic LysRS, Hoen and colleagues (Hoen et al., 2013) have pursued the apicoplast PfLysRS isoform as a potential drug target. They constructed a virtual lysyl-adenylate mimic compound library and screened this through in silico docking against a homology model of P. falciparum apicoplast LysRS. Two of the tested compounds (M-26 and M-37), had potent delayed death inhibition (consistent with apicoplast-specific activity) and inhibited aminoacylation by recombinant apicoplast PfLysRS (Table 1) (Hoen et al., 2013). The availability of specific inhibitors for both the cytoplasmic LysRS and the apicoplast LysRS provides ideal tools for studying the relative importance of organellar and cytosolic tRNA synthetases as well as providing promising drug leads. Cladosporin itself possesses poor oral bioavailability and seems therefore to be a poor drug candidate itself (Hoepfner et al., 2012), but may serve as a chemical starting point for other PfLysRS inhibitors.
4.6. Methionyl-tRNA synthetase
One series of compounds that has been investigated as antitrypanosomal agents was inspired by the success of the bacterial MetRS diaryl diamines inhibitors (Table 1) (Critchley et al., 2009). These bacterial inhibitors are highly selective for bacterial versus mammalian enzymes. Some of these compounds do inhibit recombinant human mitochondrial MetRS, however no cytotoxicity of mammalian cell cultures is apparent (Green et al., 2009). Homology models based on several MetRS structures were used to guide synthesis of related T. brucei MetRS inhibitors (Table 1), and these were tested for binding to TbMetRS (Shibata et al., 2011). Inhibition of aminoacylation activity was assayed at 50 nM and the most interesting of compounds showed >95% inhibition of activity at this concentration. Compounds were also screened using ex vivo cultures of T. brucei (and Trypanosoma cruzi) and the most effective, compound 1, had an EC50 of 4 nM and low toxicity for mammalian cells. Compound 1 was delivered at 25 mg/kg/day for 3 days using subcutaneous osmotic minipump to circumvent issues with bioavailability, and showed initial suppression of parasitaemia but mice later succumbed to disease (Shibata et al., 2011). Partial knockdown of the T. brucei MetRS through RNAi produced a severe growth defect, confirming the importance of this enzyme (Shibata et al., 2011).
Two subsequent studies characterised structures of the Leishmania MetRS (Larson et al., 2011b) and the TbMetRS (Koh et al., 2012) bound to substrates, Met, MetAMP and, in the case of the TbMetRS, inhibitors. This study revealed extensive conformational rearrangement by the TbMetRS structure upon inhibitor binding, suggesting conformation selection as the basis for binding (Koh et al., 2012). Inspection of the structures showed extensive rearrangement of the conformations occurred with introduction of inhibitors—with the compound occupying a pocket that was not present with substrates Met or MetAMP—called the auxillary pocket. The crystal structure of ligand-free TbMetRS1 is very similar to the inhibitor-bound conformation of TbMetRS and supports the idea that conformational selection is the likely model for binding of inhibitors to TbMetRS (Koh et al., 2012). The LmMetRS structure additionally revealed several differences from its human homologs near the active site that might be exploited with inhibitors that could specifically target the parasite enzyme (Larson et al., 2011b).
To improve upon the earlier MetRS inhibitors that exhibited poor PK profiles and poor bioavailability (Jarvest et al., 2002, 2003), the authors made a follow up compound series (Table 1) (Shibata et al., 2012). Guided by structures of inhibitor-bound MetRS, it was rationalised that a urea-based scaffold would increase bioavailability. The enzymes were screened using a thermal shift binding assay then used in in vitro aminoacylation assays to test for inhibition of TbMetRS and HsMitoMetRS. Compounds had similar IC50 and selectivity to the original series and improved pharmacokinetic characteristics, but unfortunately the oral bioavailability remained poor. Nonetheless these authors have demonstrated the importance of this enzyme for parasite growth, as well as the capacity to design specific inhibitors against trypanosomatid MetRS, and this remains a highly promising target.
4.7. Prolyl-tRNA synthetase
One long-used traditional antiparasitic agent, febrifugine, has recently been revealed to inhibit prolyl tRNA synthetase. This quinazolinone alkaloid is a constituent of the Chinese herbal medicine, Chángsān (Dichroa febrifuga). Despite excellent antiparasitic activity, its strong liver toxicity and gastrointestinal side effects have limited the use of febrifugine as a widespread therapeutic. Febrifugine analogues have been synthesised with a reduced capacity to form toxic intermediates and have demonstrated potent inhibition of P. falciparum isolates in ex vivo culture, P. berghei in vivo infection and impressive cure rates in an in vivo Aotus monkey model (Zhu et al., 2010, 2012). One of these analogues, the synthetic halogenated derivative halofuginone (Table 1), potently inhibits cultured erythrocytic and liver stage Plasmodium falciparum (Kobayashi et al., 1999; Derbyshire et al., 2012; Keller et al., 2012). Halofuginone has been a US FDA (Food and Drug Administration) approved drug for apicomplexan parasite (coccidia) infections of chickens and turkeys since the 1980s and is currently involved in both Stage I and Stage II clinical trials for use against proliferative diseases that include carcinoma, advanced solid tumours and AIDs related malignancies (Folz et al., 1988; Elkin et al., 1999; de Jonge et al., 2006; Koon et al., 2011).
Recent papers have demonstrated that halofuginone and other febrifugines act through inhibition of ProRS. In an elegant study, Keller et al. (2012) demonstrated first that halofuginone (which was known to activate the amino acid starvation response) inhibited an in vitro translation assay, and translation was restored only by addition of excess proline. Halofuginone also bound to and inhibited recombinant human ProRS. Addition of excess proline also reduced the sensitivity of P. falciparum parasites to halofuginone (Keller et al., 2012). A subsequent study showed that halofuginone specifically blocks the formation of the Pro-AMP adenylate complex (Zhou et al., 2013). Interestingly, the inhibition is reliant on the presence of ATP to allow high affinity binding of halofuginone to ProRS. ATP directly assists the orientation of halofuginone to enable one end to occupy the proline binding site and consequently, to competitively block activation of this amino acid, whilst the other end simultaneously mimics the 3′ end of the tRNA molecule. This ATP-dependent binding of halofuginone to ProRS has also been modelled with the P. falciparum ProRS, which is predicted to recapitulate these dual site enzyme inhibition interactions (Zhou et al., 2013). The apparent efficacy of febrifugines as antimalarial agents, despite their obvious inhibition of human ProRS and proliferating human cells serves as a reminder that drug selectivity for an acute parasitic infection need not necessarily rely on molecular specificity.
4.8. Threonyl-tRNA synthetase
Borrelidin, first isolated from Streptomyces spp., acts as a non-competitive, selective inhibitor that binds to a unique hydrophobic cluster near the active site of some bacterial and eukaryotic ThrRS enzymes (Nass and Hasenbank, 1970; Ruan et al., 2005; Gao et al., 2012). Borrelidin is an inhibitor of yeast cyclin-dependent kinase (Tsuchiya et al., 2001) and an activator of pro-apoptotic mediators in endothelial cells (Kawamura et al., 2003). Several investigations on the effect of borrelidin on Plasmodium shine light on its presumed target PfThrRS (Table 1). Borrelidin potently inhibits parasite proliferation in culture, with an immediate effect on the first asexual erythrocytic life-cycle after treatment, typical of cytosolic inhibition. This inhibition is unlike the delayed-death seen for apicoplast inhibitors (Ishiyama et al., 2011; Jackson et al., 2012; Azcarate et al., 2013) and borrelidin does not appear to inhibit organellar division (Jackson et al., 2012). Plasmodium possesses only one PfThrRS, a dual-targeted enzyme, which is trafficked to the apicoplast and cytosol (Khan et al., 2011; Jackson et al., 2012). The immediate inhibition seen with Borrelidin is consistent with the requirement of this PfThrRS for cytosolic translation. Although the exact molecular mechanism responsible for the antimalarial effect of borrelidin remains unclear, raised concentrations of l-Threonine in culture reduce parasite sensitivity, thus implicating threonine utilisation and PfThrRS as likely targets of borrelidin (Ishiyama et al., 2011).
In addition to its ex vivo use, borrelidin has been shown to cure mice of rodent malaria infections (Otoguro et al., 2003; Azcarate et al., 2013), with one report of borrelidin-cured mice then acquiring protection from subsequent challenge by Plasmodium yoelii (Azcarate et al., 2013). Although, borrelidin displays some mammalian cytotoxicity, there are efforts to synthesise borrelidin analogues with decreased toxicity (Wilkinson et al., 2006; Sugawara et al., 2013). More recently, Sugawara et al. (2013) generated a borrelidin-like series (Table 1) that reduced the cytotoxicity whilst simultaneously increasing the antimalarial activity, an important step to further progress the development of borrelidin as a future antimalarial drug.
Khan et al. (2011) have also investigated novel inhibitors of the PfThrRS predicted by in silico docking of small molecule compound libraries against homology models of the PfThrRS. The best of these compounds showed only moderate inhibition (IC50 from ∼75 to 150 μM) of ex vivo P. falciparum growth (Table 1) (Khan et al., 2011).
4.9. Tryptophanyl-tRNA synthetase
Considerable divergence in sequence and structure of TrpRSs has previously been described across the three domains of life. This has attracted some interest for TrpRS as a drug target, including in parasites. In trypanosomatid parasites this divergence is compounded by the requirement for an additional TrpRS that charges a non-canonical UGA-recognising tRNATrp required in the parasite’s mitochondria. This tRNA is encoded by the same gene as that used for cytosolic translation, but the version used in the mitochondria is first chemically altered through thiol modification and C to U nucleotide editing at the first position of the anticodon (Alfonzo et al., 1999). T. brucei encodes two TrpRS enzymes – one that recognises only the canonical tRNA in the cytosol, plus a second that recognises the altered tRNA in the mitochondrion (Charriere et al., 2006). This later TbTrpRS is lineage specific, and presents opportunities for selective inhibition. A similar scenario may exist in the apicoplast of apicomplexan parasites, where the UGA in the apicoplast genome also appears to be partially decoded as tryptophan rather than a stop codon (Wilson, 2002).
Structural analysis of TrpRS enzymes from various parasites also appears to offer opportunities for selective drug development. Analysis of the crystal structure of TrpRS from Giardia lamblia revealed a 16-residue α-helix instead of the hydrophobic ß-hairpin that stabilises the bond between tryptophan and the enzyme (Arakaki et al., 2010). The Toxoplasma and Trichomonas sequences also diverge from the human sequence in this area (Arakaki et al., 2010). These are important sub-domains of the human enzyme, so these marked differences may provide a means of selectively inhibiting the parasite homologues. Additional structures for the T. brucei cytosolic TrpRS (Merritt et al., 2011), the Cryptosporidium parvum TrpRS (Merritt et al., 2011), and the P. falciparum TrpRS (Khan et al., 2013b; Koh et al., 2013) also reveal some additional parasite specific sub-domains or structural differences that might be exploited for drug design purposes. A structure has also been solved for a divergent member of three identified Entamoeba histolytica TrpRS homologues, but this enzyme lacks Trp binding and is unlikely to charge tRNATrp (Merritt et al., 2011).
Although no inhibitors have thus far been reported for parasite TrpRSs, several chemical starting points exist for exploration; bacterial TrpRS are inhibited by natural products and tryptophan analogues such as indolmycin (Rao, 1960; Kanamaru et al., 2001) and chuangxinmycin (Brown et al., 2002).
4.10. Tyrosyl-tRNA synthetase
To our knowledge tyrosyl-tRNA synthetases (TyrRS) have yet to be experimentally investigated as targets for drug development in parasites. However, previous studies have identified inhibitors active against bacterial TyrRS including the naturally derived SB-219383 (Berge et al., 2000; Stefanska et al., 2000; Greenwood and Gentry, 2002) and several synthetic compounds (Jarvest et al., 1999; Xiao et al., 2011a,b; Wang et al., 2013). The few parasite-specific studies on TyrRS focused on the structural aspects of the enzyme. Bhatt and colleagues (Bhatt et al., 2011), localised the cytosolic PfTyrRS to the parasite cytoplasm and noted the additional presence of PfTyrRS within infected erythrocytes. These authors also detected extracellular activity of PfTyrRS through mimicry of host cytokines to induce host immune system pro-inflammatory responses (Bhatt et al., 2011). In addition to this intriguing discovery, Bhatt et al. solved the crystal structure of PfTyrRS in complex with tyrosyl-adenylate (Bhatt et al., 2011). Structural comparisons between the Plasmodium and human TyrRS revealed many differences such as the organisation of loop structures and included sequence level differences at 11 residues that contribute to binding of substrate (Bhatt et al., 2011). The crystal structure of the unusual double-length TyrRS orthologue from Leishmania major suggest a pseudo-dimer that is formed by asymmetric domains (Larson et al., 2011a) that also differs from the host TyrRS. Taken together these differences could potentially be exploited for design of structure-based inhibitors of parasite TyrRSs.
5. Concluding remarks
As evident in the list of targets above, much of the research on parasite aminoacyl-tRNA synthetases has taken place over the course of the last decade. Building on earlier studies using classical biochemistry, and a small initial number of chemical starting points, the research community is now using a myraid of technologies to investigate aaRSs and has built up an array of inhibitors that represents diverse chemical space. An encouraging development is the recent discovery of new chemicals that inhibit aaRS in eukaryotic parasites including Brugia, Trypanosoma and Plasmodium parasites (Table 1). Detailed structural research has shed important light on the structural basis for aminoacylation, and provides us with insights into the number of ways in which these enzymes can be chemically inhibited. In silico docking, rational design of compounds to fit into active sites, as well as high throughput screening have all resulted in the identification of compounds that potently inhibit aaRSs. Several combinatorial and medicinal chemistry programs have modified these starting compounds to develop compounds with acceptable pharmaceutical properties, and there are promising animal-model data for aaRS inhibitors for several parasites. Future investigations will need to consider whether these can genuinely be developed as drug like compounds, and if so, whether this can be achieved cheaply – a prime consideration for neglected parasitic diseases.
Advances across a number of systems biology platforms are accelerating the ability to make connections between inhibitors and molecular targets in parasites, and to subsequently validate these targets. Identifying resistance mutations that shed light on modes of action has been a long standing means of interrogating aaRS inhibitors. Alongside the current availability and reduced cost of next-generation sequencing technologies now means that target identification has become feasible and affordable for the larger eukaryotic parasite genomes. A number of resistance mapping studies for aaRS inhibitors in Plasmodium set a standard for future investigations in this genre (Istvan et al., 2011; Hoepfner et al., 2012).
Two concerns, selectivity and resistance, will remain major challenges for the development of antiparasitic aaRS inhibitors. Despite these parasites being only very distantly related to each other, and cause very different diseases, they share the chemotherapeutic challenge of finding drugs that select between one eukaryote and another (humans). Selectivity is a major issue because the human genome encodes for 36 aminoacyl-tRNA synthetase, (16 cytoplasmic, 17 mitochondrial, 3 dual-targeted) (see review by Antonellis and Green (2008)) that have eukaryotic and bacterial origins (mitochondria). Avoiding inhibition of the most conserved homologs is a challenge in aaRS inhibition to avoid potential cytotoxicity. Strategies to circumvent host toxicity include exploitation of organellar, bacterial-type aaRSs where the human homologue is divergent (such as the Leishmania AsnRS with a bacterial origin (Gowri et al., 2012)) or exploitation of parasite-specific modifications (Bour et al., 2009). It should also be kept in mind that for the protist parasites at least, they are separated by a billion years of evolution from their mammalian hosts, so molecular divergence abounds. In all of these cases, structural information is often key to identifying exploitable differences between host and parasite (Bunjun et al., 2000; Larson et al., 2011a). It is noteworthy that it is sometimes possible to design aaRS inhibitors with good selectivity even where few differences exist in active site residues between parasite and host (Shibata et al., 2011).
Resistance to antiparasitic drugs is a major concern. Eventual resistance to aaRS inhibitors is inevitable, and can only be hoped to be delayed, but this unfortunate outcome is often overlooked in the preclinical stages of drug discovery where the focus is on the optimisation of efficacy, cytotoxicity and pharmacokinetic properties. Since mupirocin, the IleRS inhibitor, was first introduced for clinical use, resistance has developed and in some cases results in mupirocin treatment failure against S. aureus (Patel et al., 2009). Parasite drug resistance to aaRS inhibitors has been used in studies characterising aaRS inhibitors in Plasmodium (Istvan et al., 2011) and Trypanosoma (Ranade et al., 2013). These studies are helpful not only in informing mode of action but also useful in the prediction of development and extent of parasite drug resistance. They enable discussion of relative fitness and ultimately facilitate the design of inhibitors to which resistance is not so easily generated.
Conflicts of interest
The authors declare they have no conflicts of interest.
Acknowledgements
JSP is supported by an Australian Postgraduate Award. KEJ is supported by a CASS foundation grant. SAR is supported by an Australian Research Council Future Fellowship (FT0990350).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
James S. Pham, Email: phj@student.unimelb.edu.au.
Karen L. Dawson, Email: k.dawson@student.unimelb.edu.au.
Katherine E. Jackson, Email: kputnam@unimelb.edu.au.
Erin E. Lim, Email: eel@unimelb.edu.au.
Charisse Flerida A. Pasaje, Email: cpasaje@student.unimelb.edu.au.
Kelsey E.C. Turner, Email: turnerke@student.unimelb.edu.au.
Stuart A. Ralph, Email: saralph@unimelb.edu.au.
References
- Ahel I., Korencic D., Ibba M., Söll D. Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15422–15427. doi: 10.1073/pnas.2136934100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali V., Nozaki T. Current therapeutics, their problems, and sulfur-containing-amino-acid metabolism as a novel target against infections by “amitochondriate” protozoan parasites. Clin. Microbiol. Rev. 2007;20:164–187. doi: 10.1128/CMR.00019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonzo J.D., Blanc V., Estevez A.M., Rubio M.A., Simpson L. C to U editing of the anticodon of imported mitochondrial tRNA(Trp) allows decoding of the UGA stop codon in Leishmania tarentolae. EMBO J. 1999;18:7056–7062. doi: 10.1093/emboj/18.24.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis A., Green E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genom. Human Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- Arakaki T.L., Carter M., Napuli A.J., Verlinde C.L.M.J., Fan E., Zucker F., Buckner F.S., Van Voorhis W.C., Hol W.G.J., Merritt E.A. The structure of tryptophanyl-tRNA synthetase from Giardia lamblia reveals divergence from eukaryotic homologs. J. Struct. Biol. 2010;171:238–243. doi: 10.1016/j.jsb.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcarate I.G., Marin-Garcia P., Camacho N., Perez-Benavente S., Puyet A., Diez A., Ribas de Pouplana L., Bautista J.M. Insights into the preclinical treatment of blood-stage malaria by the antibiotic borrelidin. Br. J. Pharmacol. 2013;169:645–658. doi: 10.1111/bph.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird J.K. Effectiveness of antimalarial drugs. N. Engl. J. Med. 2005;352:1565–1577. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- Bandi C., Trees A.J., Brattig N.W. Wolbachia in filarial nematodes: evolutionary aspects and implications for the pathogenesis and treatment of filarial diseases. Vet. Parasitol. 2001;98:215–238. doi: 10.1016/s0304-4017(01)00432-0. [DOI] [PubMed] [Google Scholar]
- Barrett M.P., Boykin D.W., Brun R., Tidwell R.R. Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br. J. Pharmacol. 2007;152:1155–1171. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge J.M., Copley R.C., Eggleston D.S., Hamprecht D.W., Jarvest R.L., Mensah L.M., O’Hanlon P.J., Pope A.J. Inhibitors of bacterial tyrosyl tRNA synthetase: synthesis of four stereoisomeric analogues of the natural product SB-219383. Bioorg. Med. Chem. Lett. 2000;10:1811–1814. doi: 10.1016/s0960-894x(00)00348-6. [DOI] [PubMed] [Google Scholar]
- Bhatt T.K., Khan S., Dwivedi V.P., Banday M.M., Sharma A., Chandele A., Camacho N., de Pouplana L.R., Wu Y., Craig A.G., Mikkonen A.T., Maier A.G., Yogavel M. Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nat. Commun. 2011;2:530. doi: 10.1038/ncomms1522. [DOI] [PubMed] [Google Scholar]
- Bour T., Akaddar A., Lorber B., Blais S., Balg C., Candolfi E., Frugier M. Plasmodial aspartyl-tRNA synthetases and peculiarities in Plasmodium falciparum. J. Biol. Chem. 2009;284:18893–18903. doi: 10.1074/jbc.M109.015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.J., Carter P.S., Fenwick A.S., Fosberry A.P., Hamprecht D.W., Hibbs M.J., Jarvest R.L., Mensah L., Milner P.H., O’Hanlon P.J., Pope A.J., Richardson C.M., West A., Witty D.R. The antimicrobial natural product chuangxinmycin and some synthetic analogues are potent and selective inhibitors of bacterial tryptophanyl tRNA synthetase. Bioorg. Med. Chem. Lett. 2002;12:3171–3174. doi: 10.1016/s0960-894x(02)00604-2. [DOI] [PubMed] [Google Scholar]
- Bunjun S., Stathopoulos C., Graham D., Min B., Kitabatake M., Wang A.L., Wang C.C., Vivares C.P., Weiss L.M., Soll D. A dual-specificity aminoacyl-tRNA synthetase in the deep-rooted eukaryote Giardia lamblia. Proc. Natl. Acad. Sci. U.S.A. 2000;97:12997–13002. doi: 10.1073/pnas.230444397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J.N., Burlot E., Campo B., Cherbuin S., Jeanneret S., Leroy D., Spangenberg T., Waterson D., Wells T.N., Willis P. Antimalarial drug discovery - the path towards eradication. Parasitology. 2013:1–12. doi: 10.1017/S0031182013000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casewell M.W., Hill R.L. Mupirocin (‘pseudomonic acid’)–a promising new topical antimicrobial agent. J. Antimicrob. Chemother. 1987;19:1–5. doi: 10.1093/jac/19.1.1. [DOI] [PubMed] [Google Scholar]
- Cestari I., Stuart K. Inhibition of isoleucyl-tRNA synthetase as a potential treatment for human African trypanosomiasis. J. Biol. Chem. 2013;288:14256–14263. doi: 10.1074/jbc.M112.447441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charriere F., Helgadottir S., Horn E.K., Soll D., Schneider A. Dual targeting of a single tRNA(Trp) requires two different tryptophanyl-tRNA synthetases in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6847–6852. doi: 10.1073/pnas.0602362103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepin T., Peterson F., Haertlein M., Jensen D., Wang C., Cusack S., Kron M. A hybrid structural model of the complete Brugia malayi cytoplasmic asparaginyl-tRNA synthetase. J. Mol. Biol. 2011;405:1056–1069. doi: 10.1016/j.jmb.2010.11.049. [DOI] [PubMed] [Google Scholar]
- Critchley I.A., Young C.L., Stone K.C., Ochsner U.A., Guiles J., Tarasow T., Janjic N. Antibacterial activity of REP8839, a new antibiotic for topical use. Antimicrob. Agents Chemother. 2005;49:4247–4252. doi: 10.1128/AAC.49.10.4247-4252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley I.A., Green L.S., Young C.L., Bullard J.M., Evans R.J., Price M., Jarvis T.C., Guiles J.W., Janjic N., Ochsner U.A. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J. Antimicrob. Chemother. 2009;63:954–963. doi: 10.1093/jac/dkp041. [DOI] [PubMed] [Google Scholar]
- Croft S.L., Olliaro P. Leishmaniasis chemotherapy—challenges and opportunities. Clin. Microbiol. Infect. 2011;17:1478–1483. doi: 10.1111/j.1469-0691.2011.03630.x. [DOI] [PubMed] [Google Scholar]
- Danel F., Caspers P., Nuoffer C., Hartlein M., Kron M.A., Page M.G. Asparaginyl-tRNA synthetase pre-transfer editing assay. Curr. Drug Disc. Technol. 2011;8:66–75. doi: 10.2174/157016311794519947. [DOI] [PubMed] [Google Scholar]
- de Jonge M.J., Dumez H., Verweij J., Yarkoni S., Snyder D., Lacombe D., Marreaud S., Yamaguchi T., Punt C.J., van Oosterom A. Phase I and pharmacokinetic study of halofuginone, an oral quinazolinone derivative in patients with advanced solid tumours. Eur. J. Cancer. 2006;42:1768–1774. doi: 10.1016/j.ejca.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Derbyshire E.R., Mazitschek R., Clardy J. Characterization of Plasmodium liver stage inhibition by halofuginone. ChemMedChem. 2012;7:844–849. doi: 10.1002/cmdc.201200045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D., Meng Q., Gao G., Zhao Y., Wang Q., Nare B., Jacobs R., Rock F., Alley M.R., Plattner J.J., Chen G., Li D., Zhou H. Design, synthesis, and structure–activity relationship of Trypanosoma brucei leucyl-tRNA synthetase inhibitors as antitrypanosomal agents. J. Med. Chem. 2011;54:1276–1287. doi: 10.1021/jm101225g. [DOI] [PubMed] [Google Scholar]
- Elkin M., Ariel I., Miao H.Q., Nagler A., Pines M., de-Groot N., Hochberg A., Vlodavsky I. Inhibition of bladder carcinoma angiogenesis, stromal support, and tumor growth by halofuginone. Cancer Res. 1999;59:4111–4118. [PubMed] [Google Scholar]
- Fevre E.M., Wissmann B.V., Welburn S.C., Lutumba P. The burden of human African trypanosomiasis. PLoS Negl. Trop. Dis. 2008;2:e333. doi: 10.1371/journal.pntd.0000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folz S.D., Lee B.L., Nowakowski L.H., Conder G.A. Anticoccidial evaluation of halofuginone, lasalocid, maduramicin, monensin and salinomycin. Vet. Parasitol. 1988;28:1–9. doi: 10.1016/0304-4017(88)90013-1. [DOI] [PubMed] [Google Scholar]
- Fukunaga R., Yokoyama S. Aminoacylation complex structures of leucyl-tRNA synthetase and tRNALeu reveal two modes of discriminator-base recognition. Nat. Struct. Mol. Biol. 2005;12:915–922. doi: 10.1038/nsmb985. [DOI] [PubMed] [Google Scholar]
- Gao Y.M., Wang X.J., Zhang J., Li M., Liu C.X., An J., Jiang L., Xiang W.S. Borrelidin, a potent antifungal agent: insight into the antifungal mechanism against Phytophthora sojae. J. Agric. Food Chem. 2012;60:9874–9881. doi: 10.1021/jf302857x. [DOI] [PubMed] [Google Scholar]
- Geerts S., Gryseels B. Drug resistance in human helminths: current situation and lessons from livestock. Clin. Microbiol. Rev. 2000;13:207–222. doi: 10.1128/cmr.13.2.207-222.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething P.W., Patil A.P., Smith D.L., Guerra C.A., Elyazar I.R., Johnston G.L., Tatem A.J., Hay S.I. A new world malaria map: plasmodium falciparum endemicity in 2010. Malar. J. 2011;10:378. doi: 10.1186/1475-2875-10-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowri V.S., Ghosh I., Sharma A., Madhubala R. Unusual domain architecture of aminoacyl tRNA synthetases and their paralogs from Leishmania major. BMC Genom. 2012;13:621. doi: 10.1186/1471-2164-13-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green L.S., Bullard J.M., Ribble W., Dean F., Ayers D.F., Ochsner U.A., Janjic N., Jarvis T.C. Inhibition of methionyl-tRNA synthetase by REP8839 and effects of resistance mutations on enzyme activity. Antimicrob. Agents Chemother. 2009;53:86–94. doi: 10.1128/AAC.00275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood R.C., Gentry D.R. Confirmation of the antibacterial mode of action of SB-219383, a novel tyrosyl tRNA synthetase inhibitor from a Micromonospora sp. J. Antibiot. (Tokyo) 2002;55:423–426. doi: 10.7164/antibiotics.55.423. [DOI] [PubMed] [Google Scholar]
- Guo M., Chong Y.E., Beebe K., Shapiro R., Yang X.L., Schimmel P. The C-Ala domain brings together editing and aminoacylation functions on one tRNA. Science. 2009;325:744–747. doi: 10.1126/science.1174343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M., Schimmel P. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013;9:145–153. doi: 10.1038/nchembio.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenoehrl A., Galic T., Ergovic G., Marsic N., Skerlev M., Mittendorf J., Geschke U., Schmidt A., Schoenfeld W. In vitro activity and in vivo efficacy of icofungipen (PLD-118), a novel oral antifungal agent, against the pathogenic yeast Candida albicans. Antimicrob. Agents Chemother. 2006;50:3011–3018. doi: 10.1128/AAC.00254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoen R., Novoa E.M., Lopez A., Camacho N., Cubells L., Vieira P., Santos M., Marin-Garcia P., Bautista J.M., Cortes A., Ribas de Pouplana L., Royo M. Selective inhibition of an apicoplastic aminoacyl-tRNA synthetase from Plasmodium falciparum. ChemBioChem. 2013;14:499–509. doi: 10.1002/cbic.201200620. [DOI] [PubMed] [Google Scholar]
- Hoepfner D., McNamara C.W., Lim C.S., Studer C., Riedl R., Aust T., McCormack S.L., Plouffe D.M., Meister S., Schuierer S., Plikat U., Hartmann N., Staedtler F., Cotesta S., Schmitt E.K., Petersen F., Supek F., Glynne R.J., Tallarico J.A., Porter J.A., Fishman M.C., Bodenreider C., Diagana T.T., Movva N.R., Winzeler E.A. Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–663. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M., Soll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- Ishiyama A., Iwatsuki M., Namatame M., Nishihara-Tsukashima A., Sunazuka T., Takahashi Y., Omura S., Otoguro K. Borrelidin, a potent antimalarial: stage-specific inhibition profile of synchronized cultures of Plasmodium falciparum. J. Antibiot. (Tokyo) 2011;64:381–384. doi: 10.1038/ja.2011.6. [DOI] [PubMed] [Google Scholar]
- Istvan E.S., Dharia N.V., Bopp S.E., Gluzman I., Winzeler E.A., Goldberg D.E. Validation of isoleucine utilization targets in Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 2011;108:1627–1632. doi: 10.1073/pnas.1011560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson K.E., Habib S., Frugier M., Hoen R., Khan S., Pham J.S., Ribas de Pouplana L., Royo M., Santos M.A., Sharma A., Ralph S.A. Protein translation in Plasmodium parasites. Trends Parasitol. 2011;27:467–476. doi: 10.1016/j.pt.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Jackson K.E., Pham J.S., Kwek M., De Silva N.S., Allen S.M., Goodman C.D., McFadden G.I., Ribas de Pouplana L., Ralph S.A. Dual targeting of aminoacyl-tRNA synthetases to the apicoplast and cytosol in Plasmodium falciparum. Int. J. Parasitol. 2012;42:177–186. doi: 10.1016/j.ijpara.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Jarvest R.L., Berge J.M., Houge-Frydrych C.S., Janson C., Mensah L.M., O’Hanlon P.J., Pope A., Saldanha A., Qiu X. Interaction of tyrosyl aryl dipeptides with S. aureus tyrosyl tRNA synthetase: inhibition and crystal structure of a complex. Bioorg. Med. Chem. Lett. 1999;9:2859–2862. doi: 10.1016/s0960-894x(99)00488-6. [DOI] [PubMed] [Google Scholar]
- Jarvest R.L., Berge J.M., Berry V., Boyd H.F., Brown M.J., Elder J.S., Forrest A.K., Fosberry A.P., Gentry D.R., Hibbs M.J., Jaworski D.D., O’Hanlon P.J., Pope A.J., Rittenhouse S., Sheppard R.J., Slater-Radosti C., Worby A. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against gram-positive pathogens. J. Med. Chem. 2002;45:1959–1962. doi: 10.1021/jm025502x. [DOI] [PubMed] [Google Scholar]
- Jarvest R.L., Berge J.M., Brown M.J., Brown P., Elder J.S., Forrest A.K., Houge-Frydrych C.S., O’Hanlon P.J., McNair D.J., Rittenhouse S., Sheppard R.J. Optimisation of aryl substitution leading to potent methionyl tRNA synthetase inhibitors with excellent gram-positive antibacterial activity. Bioorg. Med. Chem. Lett. 2003;13:665–668. doi: 10.1016/s0960-894x(02)01027-2. [DOI] [PubMed] [Google Scholar]
- Kanamaru T., Nakano Y., Toyoda Y., Miyagawa K.I., Tada M., Kaisho T., Nakao M. In vitro and in vivo antibacterial activities of TAK-083, an agent for treatment of Helicobacter pylori infection. Antimicrob. Agents Chemother. 2001;45:2455–2459. doi: 10.1128/AAC.45.9.2455-2459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T., Liu D., Towle M.J., Kageyama R., Tsukahara N., Wakabayashi T., Littlefield B.A. Anti-angiogenesis effects of borrelidin are mediated through distinct pathways: threonyl-tRNA synthetase and caspases are independently involved in suppression of proliferation and induction of apoptosis in endothelial cells. J. Antibiot. (Tokyo) 2003;56:709–715. doi: 10.7164/antibiotics.56.709. [DOI] [PubMed] [Google Scholar]
- Keller T.L., Zocco D., Sundrud M.S., Hendrick M., Edenius M., Yum J., Kim Y.J., Lee H.K., Cortese J.F., Wirth D.F., Dignam J.D., Rao A., Yeo C.Y., Mazitschek R., Whitman M. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012;8:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S., Sharma A., Jamwal A., Sharma V., Pole A.K., Thakur K.K. Uneven spread of cis- and trans-editing aminoacyl-tRNA synthetase domains within translational compartments of P. falciparum. Sci. Rep. 2011;1:188. doi: 10.1038/srep00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S., Garg A., Camacho N., Van Rooyen J., Kumar Pole A., Belrhali H., Ribas de Pouplana L., Sharma V., Sharma A. Structural analysis of malaria-parasite lysyl-tRNA synthetase provides a platform for drug development. Acta Crystallogr. D Biol. Crystallogr. 2013;69:785–795. doi: 10.1107/S0907444913001923. [DOI] [PubMed] [Google Scholar]
- Khan S., Garg A., Sharma A., Camacho N., Picchioni D., Saint-Leger A., Ribas de Pouplana L., Yogavel M. An appended domain results in an unusual architecture for malaria parasite tryptophanyl-tRNA synthetase. PLoS One. 2013;8:e66224. doi: 10.1371/journal.pone.0066224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Lee S.W., Choi E.-C., Choi S.Y. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl. Microbiol. Biotechnol. 2003;61:278–288. doi: 10.1007/s00253-003-1243-5. [DOI] [PubMed] [Google Scholar]
- Kobayashi S., Ueno M., Suzuki R., Ishitani H., Kim H.S., Wataya Y. Catalytic asymmetric synthesis of antimalarial alkaloids febrifugine and isofebrifugine and their biological activity. J. Org. Chem. 1999;64:6833–6841. doi: 10.1021/jo990877k. [DOI] [PubMed] [Google Scholar]
- Koh C.Y., Kim J.E., Shibata S., Ranade R.M., Yu M., Liu J., Gillespie J.R., Buckner F.S., Verlinde C.L., Fan E., Hol W.G. Structure; 2012. Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh C.Y., Kim J.E., Napoli A.J., Verlinde C.L., Fan E., Buckner F.S., Van Voorhis W.C., Hol W.G. Crystal structures of Plasmodium falciparum cytosolic tryptophanyl-tRNA synthetase and its potential as a target for structure-guided drug design. Mol. Biochem. Parasitol. 2013;189:26–32. doi: 10.1016/j.molbiopara.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koon H.B., Fingleton B., Lee J.Y., Geyer J.T., Cesarman E., Parise R.A., Egorin M.J., Dezube B.J., Aboulafia D., Krown S.E. Phase II AIDS Malignancy Consortium trial of topical halofuginone in AIDS-related Kaposi sarcoma. J. Acquir. Immune Defic. Syndr. 2011;56:64–68. doi: 10.1097/QAI.0b013e3181fc0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M., Marquard K., Hartlein M., Price S., Leberman R. An immunodominant antigen of Brugia malayi is an asparaginyl-tRNA synthetase. FEBS Lett. 1995;374:122–124. doi: 10.1016/0014-5793(95)01092-s. [DOI] [PubMed] [Google Scholar]
- Kron M., Petridis M., Milev Y., Leykam J., Hartlein M. Expression, localization and alternative function of cytoplasmic asparaginyl-tRNA synthetase in Brugia malayi. Mol. Biochem. Parasitol. 2003;129:33–39. doi: 10.1016/s0166-6851(03)00080-x. [DOI] [PubMed] [Google Scholar]
- Kron M.A., Wang C., Vodanovic-Jankovic S., Howard O.M., Kuhn L.A. Interleukin-8-like activity in a filarial asparaginyl-tRNA synthetase. Mol. Biochem. Parasitol. 2012;185:66–69. doi: 10.1016/j.molbiopara.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Kron M.A., Metwali A., Vodanovic-Jankovic S., Elliott D. Nematode asparaginyl-tRNA synthetase resolves intestinal inflammation in mice with T-cell transfer colitis. Clin. Vaccine Immunol. 2013;20:276–281. doi: 10.1128/CVI.00594-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson E.T., Kim J.E., Castaneda L.J., Napuli A.J., Zhang Z., Fan E., Zucker F.H., Verlinde C.L., Buckner F.S., Van Voorhis W.C., Hol W.G., Merritt E.A. The double-length tyrosyl-tRNA synthetase from the eukaryote Leishmania major forms an intrinsically asymmetric pseudo-dimer. J. Mol. Biol. 2011;409:159–176. doi: 10.1016/j.jmb.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson E.T., Kim J.E., Zucker F.H., Kelley A., Mueller N., Napuli A.J., Verlinde C.L., Fan E., Buckner F.S., Van Voorhis W.C., Merritt E.A., Hol W.G. Structure of Leishmania major methionyl-tRNA synthetase in complex with intermediate products methionyladenylate and pyrophosphate. Biochimie. 2011;93:570–582. doi: 10.1016/j.biochi.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.W., Beebe K., Nangle L.A., Jang J., Longo-Guess C.M., Cook S.A., Davisson M.T., Sundberg J.P., Schimmel P., Ackerman S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- Lee S.W., Cho B.H., Park S.G., Kim S. Aminoacyl-tRNA synthetase complexes: beyond translation. J. Cell Sci. 2004;117:3725–3734. doi: 10.1242/jcs.01342. [DOI] [PubMed] [Google Scholar]
- Leung C.C., Rieder H.L., Lange C., Yew W.W. Treatment of latent infection with Mycobacterium tuberculosis: update 2010. Eur. Respir. J. 2011;37:690–711. doi: 10.1183/09031936.00079310. [DOI] [PubMed] [Google Scholar]
- Liu J., Istvan E.S., Gluzman I.Y., Gross J., Goldberg D.E. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc. Natl. Acad. Sci. U.S.A. 2006;103:8840–8845. doi: 10.1073/pnas.0601876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustigman S., Prichard R.K., Gazzinelli A., Grant W.N., Boatin B.A., McCarthy J.S., Basanez M.-G. A research agenda for helminth diseases of humans: the problem of helminthiases. PLoS Negl. Trop. Dis. 2012:e1582. doi: 10.1371/journal.pntd.0001582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv P.C., Zhu H.L. Aminoacyl-tRNA synthetase inhibitors as potent antibacterials. Curr. Med. Chem. 2012;19:3550–3563. doi: 10.2174/092986712801323199. [DOI] [PubMed] [Google Scholar]
- Merritt E.A., Arakaki T.L., Gillespie R., Napuli A.J., Kim J.E., Buckner F.S., Van Voorhis W.C., Verlinde C.L., Fan E., Zucker F., Hol W.G. Crystal structures of three protozoan homologs of tryptophanyl-tRNA synthetase. Mol. Biochem. Parasitol. 2011;177:20–28. doi: 10.1016/j.molbiopara.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacher M. Helminth-infected patients with malaria: a low profile transmission hub? Malar. J. 2012;11:376. doi: 10.1186/1475-2875-11-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakama T., Nureki O., Yokoyama S. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J. Biol. Chem. 2001;276:47387–47393. doi: 10.1074/jbc.M109089200. [DOI] [PubMed] [Google Scholar]
- Nass G., Hasenbank R. Effect of Borrelidin on the threonyl-tRNA-synthetase activity and the regulation of threonine-biosynthetic enzymes in Saccharomyces cerivisiae. Mol. Gen. Genet. 1970;108:28–32. doi: 10.1007/BF00343181. [DOI] [PubMed] [Google Scholar]
- Niemann M., Schneider A., Cristodero M. Mitochondrial translation in trypanosomatids: a novel target for chemotherapy? Trends Parasitol. 2011;27:429–433. doi: 10.1016/j.pt.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Ochsner U.A., Sun X., Jarvis T., Critchley I., Janjic N. Aminoacyl-tRNA synthetases: essential and still promising targets for new anti-infective agents. Exp. Opin. Invest. Drugs. 2007;16:573–593. doi: 10.1517/13543784.16.5.573. [DOI] [PubMed] [Google Scholar]
- Otoguro K., Ui H., Ishiyama A., Kobayashi M., Togashi H., Takahashi Y., Masuma R., Tanaka H., Tomoda H., Yamada H., Omura S. In vitro and in vivo antimalarial activities of a non-glycosidic 18-membered macrolide antibiotic, borrelidin, against drug-resistant strains of Plasmodia. J. Antibiot. (Tokyo) 2003;56:727–729. doi: 10.7164/antibiotics.56.727. [DOI] [PubMed] [Google Scholar]
- Patel J.B., Gorwitz R.J., Jernigan J.A. Mupirocin resistance. Clin. Infect. Dis. 2009;49:935–941. doi: 10.1086/605495. [DOI] [PubMed] [Google Scholar]
- Prichard R.K., Basanez M.-G., Boatin B.A., McCarthy J.S., Garcia H.H., Yang G.-J., Sripa B., Lustigman S. A research agenda for helminth diseases of humans: intervention for control and elimination. PLoS Negl. Trop. Dis. 2012;6:e1549. doi: 10.1371/journal.pntd.0001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez B.L., Howard O.M., Dong H.F., Edamatsu T., Gao P., Hartlein M., Kron M. Brugia malayi asparaginyl-transfer RNA synthetase induces chemotaxis of human leukocytes and activates G-protein-coupled receptors CXCR1 and CXCR2. J. Infect. Dis. 2006;193:1164–1171. doi: 10.1086/501369. [DOI] [PubMed] [Google Scholar]
- Ranade R.M., Gillespie J.R., Shibata S., Verlinde C.L.M.J., Fan E., Hol W.G.J., Buckner F.S. Induced resistance to methionyl-tRNA synthetase inhibitors in Trypanosoma brucei. Is due to overexpression of the target? Antimicrob. Agents Chemother. 2013;57:3021–3028. doi: 10.1128/AAC.02578-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao K.V. PA 155A: a new antibiotic. Antibiot. Chemother. 1960;10:312–315. [PubMed] [Google Scholar]
- Rateb M.E., Yu Z., Yan Y., Yang D., Huang T., Vodanovic-Jankovic S., Kron M.A., Shen B. Medium optimization of Streptomyces sp. 17944 for tirandamycin B production and isolation and structural elucidation of tirandamycins H, I and J. J. Antibiot. (Tokyo) 2013 doi: 10.1038/ja.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock F.L., Mao W., Yaremchuk A., Tukalo M., Crepin T., Zhou H., Zhang Y.K., Hernandez V., Akama T., Baker S.J., Plattner J.J., Shapiro L., Martinis S.A., Benkovic S.J., Cusack S., Alley M.R. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- Rodriguez J.B., Szajnman S.H. New antibacterials for the treatment of toxoplasmosis; a patent review. Expert. Opin. Ther. Pat. 2012;22:311–333. doi: 10.1517/13543776.2012.668886. [DOI] [PubMed] [Google Scholar]
- Rossignol J.F. Cryptosporidium and Giardia: treatment options and prospects for new drugs. Exp. Parasitol. 2010;124:45–53. doi: 10.1016/j.exppara.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Rottenberg M.E., Masocha W., Ferella M., Petitto-Assis F., Goto H., Kristensson K., McCaffrey R., Wigzell H. Treatment of African trypanosomiasis with cordycepin and adenosine deaminase inhibitors in a mouse model. J. Infect. Dis. 2005;192:1658–1665. doi: 10.1086/496896. [DOI] [PubMed] [Google Scholar]
- Ruan B., Bovee M.L., Sacher M., Stathopoulos C., Poralla K., Francklyn C.S., Söll D. A unique hydrophobic cluster near the active site contributes to differences in borrelidin inhibition among threonyl-tRNA synthetases. J. Biol. Chem. 2005;280:571–577. doi: 10.1074/jbc.M411039200. [DOI] [PubMed] [Google Scholar]
- Schneider A. Unique aspects of mitochondrial biogenesis in trypanosomatids. Int. J. Parasitol. 2001;31:1403–1415. doi: 10.1016/s0020-7519(01)00296-x. [DOI] [PubMed] [Google Scholar]
- Seiradake E., Mao W., Hernandez V., Baker S.J., Plattner J.J., Alley M.R., Cusack S. Crystal structures of the human and fungal cytosolic Leucyl-tRNA synthetase editing domains: a structural basis for the rational design of antifungal benzoxaboroles. J. Mol. Biol. 2009;390:196–207. doi: 10.1016/j.jmb.2009.04.073. [DOI] [PubMed] [Google Scholar]
- Shibata S., Gillespie J.R., Kelley A.M., Napuli A.J., Zhang Z., Kovzun K.V., Pefley R.M., Lam J., Zucker F., Van Voorhis W.C., Merritt E.A., Hol W.G.J., Verlinde C.L.M.J., Fan E., Buckner F.S. Selective inhibitors of methionyl-tRNA synthetase have potent activity on Trypanosoma brucei infection in mice. Antimicrob. Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S., Gillespie J.R., Ranade R.M., Koh C.Y., Kim J.E., Laydbak J.U., Zucker F.H., Hol W.G., Verlinde C.L., Buckner F.S., Fan E. Urea-based inhibitors of Trypanosoma brucei methionyl-tRNA synthetase: selectivity and in vivo characterization. J. Med. Chem. 2012;55:6342–6351. doi: 10.1021/jm300303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E.V., Lakunina V.A., Tarassov I., Krasheninnikov I.A., Kamenski P.A. Noncanonical functions of aminoacyl-tRNA synthetases. Biochemistry (Mosc) 2012;77:15–25. doi: 10.1134/S0006297912010026. [DOI] [PubMed] [Google Scholar]
- Sokabe M., Okada A., Yao M., Nakashima T., Tanaka I. Molecular basis of alanine discrimination in editing site. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11669–11674. doi: 10.1073/pnas.0502119102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokabe M., Ose T., Nakamura A., Tokunaga K., Nureki O., Yao M., Tanaka I. The structure of alanyl-tRNA synthetase with editing domain. Proc. Natl. Acad. Sci. U.S.A. 2009;106:11028–11033. doi: 10.1073/pnas.0904645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanska A.L., Coates N.J., Mensah L.M., Pope A.J., Ready S.J., Warr S.R. SB-219383, a novel tyrosyl tRNA synthetase inhibitor from a Micromonospora sp. I. Fermentation, isolation and properties. J. Antibiot. (Tokyo) 2000;53:345–350. doi: 10.7164/antibiotics.53.345. [DOI] [PubMed] [Google Scholar]
- Stuart K., Brun R., Croft S., Fairlamb A., Gurtler R.E., McKerrow J., Reed S., Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara A., Tanaka T., Hirose T., Ishiyama A., Iwatsuki M., Takahashi Y., Otoguro K., Ōmura S., Sunazuka T. Borrelidin analogues with antimalarial activity: design, synthesis and biological evaluation against Plasmodium falciparum parasites. Bioorg. Med. Chem. Lett. 2013;23:2302–2305. doi: 10.1016/j.bmcl.2013.02.075. [DOI] [PubMed] [Google Scholar]
- Sukuru S.C., Crepin T., Milev Y., Marsh L.C., Hill J.B., Anderson R.J., Morris J.C., Rohatgi A., O’Mahony G., Grotli M., Danel F., Page M.G., Hartlein M., Cusack S., Kron M.A., Kuhn L.A. Discovering new classes of Brugia malayi asparaginyl-tRNA synthetase inhibitors and relating specificity to conformational change. J. Comput. Aided Mol. Des. 2006;20:159–178. doi: 10.1007/s10822-006-9043-5. [DOI] [PubMed] [Google Scholar]
- Tan M., Wang M., Zhou X.L., Yan W., Eriani G., Wang E.D. The Yin and Yang of tRNA: proper binding of acceptor end determines the catalytic balance of editing and aminoacylation. Nucleic Acids Res. 2013;41:5513–5523. doi: 10.1093/nar/gkt252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya E., Yukawa M., Miyakawa T., Kimura K.I., Takahashi H. Borrelidin inhibits a cyclin-dependent kinase (CDK), Cdc28/Cln2, of Saccharomyces cerevisiae. J. Antibiot. (Tokyo) 2001;54:84–90. doi: 10.7164/antibiotics.54.84. [DOI] [PubMed] [Google Scholar]
- Upcroft P., Upcroft J.A. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin. Microbiol. Rev. 2001;14:150–164. doi: 10.1128/CMR.14.1.150-164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondenhoff G.H., Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur. J. Med. Chem. 2011;46:5227–5236. doi: 10.1016/j.ejmech.2011.08.049. [DOI] [PubMed] [Google Scholar]
- Wang X.D., Deng R.C., Dong J.J., Peng Z.Y., Gao X.M., Li S.T., Lin W.Q., Lu C.L., Xiao Z.P., Zhu H.L. 3-Aryl-4-acyloxyethoxyfuran-2(5H)-ones as inhibitors of tyrosyl-tRNA synthetase: synthesis, molecular docking and antibacterial evaluation. Bioorg. Med. Chem. 2013;21:4914–4922. doi: 10.1016/j.bmc.2013.06.066. [DOI] [PubMed] [Google Scholar]
- Wilkinson B., Gregory M.A., Moss S.J., Carletti I., Sheridan R.M., Kaja A., Ward M., Olano C., Mendez C., Salas J.A., Leadlay P.F., vanGinckel R., Zhang M.Q. Separation of anti-angiogenic and cytotoxic activities of borrelidin by modification at the C17 side chain. Bioorg. Med. Chem. Lett. 2006;16:5814–5817. doi: 10.1016/j.bmcl.2006.08.073. [DOI] [PubMed] [Google Scholar]
- Wilson R.J. Progress with parasite plastids. J. Mol. Biol. 2002;319:257–274. doi: 10.1016/S0022-2836(02)00303-0. [DOI] [PubMed] [Google Scholar]
- Xiao Z.P., He X.B., Peng Z.Y., Xiong T.J., Peng J., Chen L.H., Zhu H.L. Synthesis, structure, molecular docking, and structure–activity relationship analysis of enamines: 3-aryl-4-alkylaminofuran-2(5H)-ones as potential antibacterials. Bioorg. Med. Chem. 2011;19:1571–1579. doi: 10.1016/j.bmc.2011.01.051. [DOI] [PubMed] [Google Scholar]
- Xiao Z.P., Ma T.W., Liao M.L., Feng Y.T., Peng X.C., Li J.L., Li Z.P., Wu Y., Luo Q., Deng Y., Liang X., Zhu H.L. Tyrosyl-tRNA synthetase inhibitors as antibacterial agents: synthesis, molecular docking and structure-activity relationship analysis of 3-aryl-4-arylaminofuran-2(5H)-ones. Eur. J. Med. Chem. 2011;46:4904–4914. doi: 10.1016/j.ejmech.2011.07.047. [DOI] [PubMed] [Google Scholar]
- Yu Z., Vodanovic-Jankovic S., Ledeboer N., Huang S.X., Rajski S.R., Kron M., Shen B. Tirandamycins from Streptomyces sp. 17944 inhibiting the parasite Brugia malayi asparagine tRNA synthetase. Org. Lett. 2011;13:2034–2037. doi: 10.1021/ol200420u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., Vodanovic-Jankovic S., Kron M., Shen B. New WS9326A congeners from Streptomyces sp. 9078 inhibiting Brugia malayi asparaginyl-tRNA synthetase. Org. Lett. 2012;14:4946–4949. doi: 10.1021/ol302298k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.M., Christian T., Newberry K.J., Perona J.J., Hou Y.M. Zinc-mediated amino acid discrimination in cysteinyl-tRNA synthetase. Journal of molecular biology. 2003;327:911–917. doi: 10.1016/s0022-2836(03)00241-9. [DOI] [PubMed] [Google Scholar]
- Zhang F., Du J., Wang Q., Hu Q., Zhang J., Ding D., Zhao Y., Yang F., Wang E., Zhou H. Discovery of N-(4-sulfamoylphenyl)thioureas as Trypanosoma brucei leucyl-tRNA synthetase inhibitors. Org. Biomol. Chem. 2013;11:5310–5324. doi: 10.1039/c3ob40236c. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Wang Q., Meng Q., Ding D., Yang H., Gao G., Li D., Zhu W., Zhou H. Identification of Trypanosoma brucei leucyl-tRNA synthetase inhibitors by pharmacophore- and docking-based virtual screening and synthesis. Bioorg. Med. Chem. 2012;20:1240–1250. doi: 10.1016/j.bmc.2011.12.035. [DOI] [PubMed] [Google Scholar]
- Zhou H., Sun L., Yang X.L., Schimmel P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature. 2013;494:121–124. doi: 10.1038/nature11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Wang J., Chandrashekar G., Smith E., Liu X., Zhang Y. Synthesis and evaluation of 4-quinazolinone compounds as potential antimalarial agents. Eur. J. Med. Chem. 2010;45:3864–3869. doi: 10.1016/j.ejmech.2010.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Chandrashekar G., Meng L., Robinson K., Chatterji D. Febrifugine analogue compounds: synthesis and antimalarial evaluation. Bioorg. Med. Chem. 2012;20:927–932. doi: 10.1016/j.bmc.2011.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]