

Abstract
An interest in the schweinfurthins, natural stilbenes with significant anti-proliferative activity, has prompted efforts to prepare a set of indole analogues. To approach the desired compounds through a Horner-Wadsworth-Emmonds condensation, new indole derivatives bearing a phosphonomethyl substituent in the B-ring were required. The parent indole system with the necessary substitution pattern was obtained through a Stobbe condensation and cyclization. A prenyl substituent was incorporated at the C-3 position of a 4,6-disubstituted indole through a highly regioselective electrophilic aromatic substitution reaction, while metalation and alkylation provided the C-2 prenylated indole. After introduction of the phosphonate group through classical reactions, the new indole phosphonates were found to undergo the desired condensation with nonracemic aldehydes representing the schweinfurthin left half. This approach gives facile access to new heteroaromatic analogues of the natural schweinfurthins, and should be applicable to many other natural stilbenes as well.
Keywords: Schweinfurthin, indole, stilbene analogues, phosphonate
Introduction
The schweinfurthins (Figure 1), a small group of rare natural products,1,2 display a novel pattern of differential activity in the National Cancer Institute’s (NCI) 60 cell line screen. Their activity pattern suggests that these compounds act on a novel target or through a new mechanism,1 and thus these compounds can be viewed as potential leads for further drug development. To alleviate the scarcity of these natural products, to access novel analogues, and to explore the limits of the pharmacophore, we have undertaken the synthesis of both natural schweinfurthins and a range of analogues.3–9 After an analysis of new compounds of potential interest, we considered the possibility of incorporating an indole in the stilbene system. The indole substructure is so common in both natural products and pharmaceutical agents that it often is considered a privileged scaffold.10,11 Incorporation of an indole motif might afford analogues with comparable or improved activity while at the same time increasing bioavailability.12,13 Furthermore, the D-ring resorcinol of the natural schweinfurthins may limit the schweinfurthins’ stability, and proper placement of an indole system might improve the chemical stability as well. Based on this rationale, synthesis of indole analogues of the schweinfurthins became a goal of our program.
Figure 1.

Some natural schweinfurthins (1 – 4) and some synthetic analogues (5, 6).
There are multiple ways that an indole moiety could be superimposed upon the D-ring of the natural schweinfurthins. The pattern pursued in this study would view the indole nitrogen as a replacement for one of the resorcinol oxygens, and incorporate the remainder of the indole ring as a substituent on the position para to the stilbene olefin (Figure 2). These structures would exploit the known flexibility of the para position to modification with preservation of biological activity.4,7,8 Furthermore, preparation of intermediates leading to structures 7 and 8 might be readily modified to allow addition of isoprenoid substituents to the 5-membered ring, via electrophilic aromatic substitution (which is favored at C-3 of indole itself14 and would lead to compound 9) or via anion chemistry (which can be directed to C-2 in N-substituted indoles and would provide compound 10).15,16 Because both compounds 9 and 10 represent modest deviations from the natural products in terms of the position of the prenyl group both series were of interest, and a strategy that could diverge to both isomers at a later stage would be particularly attractive.
Figure 2.

First generation indole targets.
Our foray into schweinfurthin studies began with synthesis of schweinfurthin C,17 and that early effort established the strategy of a late stage Horner-Wadsworth-Emmons (HWE) condensation for construction of the trans-stilbene olefin. To take advantage of intermediates already in hand from previous research, especially the now readily available R,R,R-aldehydes 11 and 12 that carry all of the schweinfurthin stereogenic centers (Scheme 1), would require an indole phosphonate such as compound 13. Given the vast number of known indoles it was somewhat surprising to find that apparently only C-218 and C-319 phosphonomethyl compounds have been prepared. Based on the assumption that phosphonate 13 could be prepared from the corresponding alcohol 14, which in turn should be available from the ester 15, routes to these two potential intermediates were considered. The presence of the “benzylic” alcohol of compound 14 might not be tolerated by many of the classical methods20 for de novo indole synthesis because of their reliance on acidic conditions, and the recent Kraus indole synthesis appears to be better suited for preparation of 2-substituted or 2,3-disubstituted compounds.21,22 However, preparation of the substituted indole 15 has been reported through an approach based on a Stobbe condensation of a succinate diester (17) and 2-pyrrole carboxaldehyde (18) followed by cyclization of the intermediate acid 16.23 While the initial report did not provide a complete characterization of the product, a more recent study from the Vedejs labs placed this approach on a solid foundation and proved that it does afford the desired substitution pattern.24 Therefore we began an effort to obtain the targeted schweinfurthin analogues with preparation of several indoles based on this strategy.
Scheme 1.

Retrosynthetic analysis.
Results and Discussion
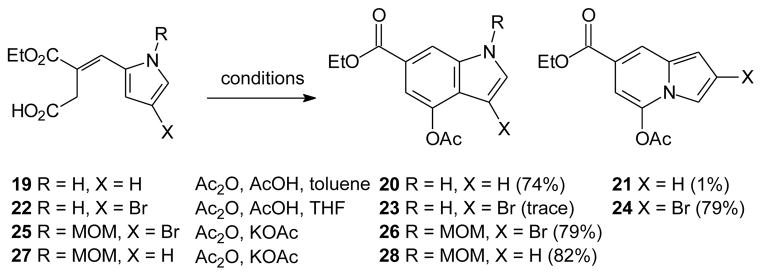
The Stobbe condensation of diethyl succinate with 2-pyrrole carboxaldehyde (18) smoothly gave the half ester 19 as expected.24 Without extensive purification, this material was treated with a mixture of acetic anhydride and acetic acid (6:1) in refluxing toluene to induce cyclization (Scheme 2). These conditions resulted in formation of the acetate-protected indole 20 (74%) accompanied by small amounts of the indolizine 21 (~1%), also as expected,24 while a parallel reaction in THF at reflux gave a less favorable product ratio (42% and 19%, respectively). Attempts to extend this approach to the brominated pyrrole 22, which might be useful for elaboration of the final products through halogen-metal exchange or cross coupling reactions,25 were more complex. While the desired half ester 22 was readily prepared by a Stobbe condensation, treatment of compound 22 under standard cyclization conditions gave only trace amounts of the desired indole 23 and afforded the indolizine 24 as the major product instead. Compound 24 is highly fluorescent and might be useful for synthesis of new types of fluorescent schweinfurthin analogues.26 However, for the immediate goal, N-protection of the pyrrole aldehyde would circumvent this issue as observed with N-methyl pyrrole.27 Because previous syntheses of schweinfurthin analogues employed MOM-protected phenols, the half ester 25 was prepared by Stobbe condensation of the MOM-protected aldehyde. In this case, cyclization under the standard conditions afforded only the desired indole product 26. In a similar sense, after the pyrrole 18 was protected as its MOM derivative 27, cyclization of the Stobbe product now gave only the desired indole 28. Because a late stage deprotection of the indole MOM group ultimately proved more difficult than expected (vide infra), pyrrole aldehyde 18 also was protected as its tosyl derivative. However, in this case attempted Stobbe condensation proved problematic, so introduction of this group at this stage of the sequence was not pursued further.
Scheme 2.
Cyclization to indoles and indolizines
After hydrolysis of the acetate group of indole 20, treatment of the resulting phenol 29 with NaH and MOMCl in THF gave the desired MOM-protected indole 30 along with a significant amount of a C-alkylated product, tentatively assigned as the C-5 isomer 31 (Scheme 3). Addition of DMF to the solvent system improved the ratio of desired to undesired product from ~1.3:1 to ~9:1. Reduction of ester 30 proceeded in quantitative yield, but attempts at conversion to the phosphonate were somewhat frustrating. The reaction proceeded via the corresponding bromide, although the Arbuzov reaction of that bromide with (EtO)3P in refluxing toluene gave the desired phosphonate 33 in modest yield.
Scheme 3.

Synthesis and HWE condensation of indole phosphonate 33.
The HWE coupling of the hexahydroxanthene aldehyde 1228 with phosphonate 33 smoothly gave the protected analogue 34. Unfortunately, attempted hydrolysis of the three MOM groups by treatment with TsOH/MeOH gave compound 35, where both of the phenolic MOM groups had been cleaved but the indole nitrogen was still protected. Attempts to remove this remaining MOM group under more vigorous conditions29–31 proved unsuccessful, and gave only decomposition.
To circumvent this difficult hydrolysis, a new strategy based upon early formation of a differentially protected indole was explored. Selective MOM protection of the phenol 29 gave indole 36 (Scheme 4) and different N-protecting groups then could be introduced easily. For example, treatment of compound 36 with base and Boc2O gave the carbamate 37, and selective reduction of the ethyl ester gave the primary alcohol 38 in good yield. Under standard conditions for formation of the phosphonates (i.e. initial formation of the mesylate followed by treatment with LiBr and then neat (EtO)3P at reflux), formation of the C-P bond was accompanied by cleavage of the Boc group32 to afford phosphonate 39 as the major product. The Boc group was easily re-installed through treatment of phosphonate 39 with Boc2O to give phosphonate 40, or phosphonate 40 could be obtained more directly from the alcohol 38 in a reasonable yield (61%) if the Arbuzov reaction were conducted at a lower temperature (~95 °C) instead of reflux (~165 °C). Alternatively, a tosylate protecting group could be installed through treatment of indole 36 with TsCl and base, and the intermediate carboxylic acid ester was reduced selectively to the alcohol 41 in good yield. The tosyl group proved stable to standard conditions for formation of the phosphonate, and compound 42 was obtained smoothly.
Scheme 4.

Synthesis of new indole phosphonates.
Of the new indole phosphonates 39, 40, and 42, the HWE condensation of compound 39 with an aldehyde representing the schweinfurthin left half would be most advantageous because it would avoid an N-deprotection step of the product at a later stage. In the limited number of condensations between an indole phosphonate and an aldehyde, an N-protected indole always was employed.33–36 Nevertheless, because aldehyde 12 has been used in similar HWE reactions,3,6,13 condensations were attempted between this aldehyde and phosphonate 39. At best just trace amounts of a possible stilbene product were observed in this case, even though p-methoxybenzaldehyde reacted smoothly with phosphonate 39.37 Attempted condensation of aldehyde 12 with phosphonate 40 also was problematic. In this case, little or no condensation was observed and TLC analysis suggested that Boc cleavage had taken place instead. Fortunately, the HWE condensation of phosphonate 42 with aldehyde 12 at reflux gave a mixture of stilbene products in very good total yield (Scheme 5). Somewhat to our surprise, analysis of the 1H and 13C NMR spectra showed that the major product 43 carried a tosylate as an A-ring ester, while the minor product 44 did not have an A-ring tosylate, but already had undergone cleavage of the N-tosyl group. The hindered tosylate ester 43 proved resistant to standard hydrolysis,38–45 but reduction with LiAlH446,47 converted the major HWE product (43) to the minor product (44) in low yield. Final hydrolysis of the MOM groups gave the stilbene 7, the first schweinfurthin G analogue that incorporates an indole system.
Scheme 5.

Synthesis of an indole analogue of schweinfurthin G.
To prepare the analogous schweinfurthin F analogue, phosphonate 42 was allowed to react with aldehyde 113 and base (Scheme 6). When the reaction was conducted at reflux in THF, the only stilbene product (56%) again reflected transfer of the tosyl group from the indole nitrogen to the A-ring alcohol. Treatment of this hindered tosylate ester with LiAlH4 did afford the free alcohol 46 in modest yield. Compound 46 undergoes hydrolysis of the phenolic MOM group under standard conditions to afford the schweinfurthin F analogue 8.
Scheme 6.

Synthesis of an indole analogue of schweinfurthin F.
Because the natural schweinfurthins contain an isoprene chain as a D-ring substituent, installation of an isoprenoid chain on the indole would afford analogues more closely parallel to the natural products. Our original plan had been to incorporate this chain in a regiospecific manner through halogen-metal exchange on a protected indole derived from bromide 26, but this sequence would become unappealing if the MOM hydrolysis were problematic or the SN2′ product was formed during alkylation with prenyl bromide.48–52 An attractive alternative might be based on an extension of the methodology of Ganesan,53 which relies upon Zn(OTf)2 activation of an allylic halide to bring about only C-3 alkylation through electrophilic aromatic substitution. Among the attractive features of the original study, alkylation of indole itself with prenyl halides generally gave only the C-3 alkylated product, proceeded in ~60% yield, and did not give the products of SN2′ reaction (i.e. “reversed” prenyl substituents) that are frequently observed with other methods.48,49 However, it was unclear whether this approach could be applied to access the substituted indole required here, where both C-6 and C-4 groups that might impact reactivity were required. In particular, a C-6 ethoxycarbonyl group would add an electron withdrawing substituent system, while reduction of this group to the corresponding alcohol might invite polymerization reactions given the known reactivity of benzyl alcohol under these conditions.53 Furthermore, a MOM substituent at the C-4 position might compete with an isoprenoid halide for complexation with the Zn(OTf)2 or introduce a degree of steric hindrance to the C-3 position. Nevertheless, the brevity of this approach led us to study the process with indole 36. To our delight, the reaction of indole 36 with prenyl bromide in the presence of Zn(OTf)2 gave the desired product 47 in 65% yield (Scheme 7). This yield is comparable to those obtained on indole itself,53 despite the presence of the B-ring substituents.
Scheme 7.

Synthesis of the prenylated indole schweinfurthin 9.
Once ester 47 was in hand, the remaining steps in the sequence proceeded in a fashion parallel to those employed for preparation of the earlier analogues. Protection of the indole nitrogen as the tosylate proceeded smoothly. Then, after selective reduction of the carboxylic acid ester with DIBAL, the resulting alcohol 48 was readily converted to phosphonate 49. An HWE condensation with aldehyde 11 afforded a mixture of N-tosyl intermediate 50 and the free indole 51. After partial purification, treatment with NaH in a mixture of THF and i-PrOH afforded only compound 51. Final hydrolysis of the MOM group proceeded in low yield, but did afford the desired target compound, the schweinfurthin F analogue 9.
To access compound 10 from an intermediate already in hand, indole 41 was protected as its silyl ether 52, and then treated with n-BuLi and prenyl bromide. Despite the presence in the B-ring of two substituents that might participate in directed ortho metallation,54 this sequence gave a single product identified as the C-2 alkylated indole 53. After deprotection to the alcohol 54, and formation of the phosphonate 55 through standard reactions, condensation of phosphonate 55 with aldehyde 11 provided a mixture of the new stilbenes 56 and 57. After partial purification, treatment with 2-propanol and base completed conversion to compound 57, and final deprotection gave the desired schweinfurthin analogue 10.
In preliminary bioassays, compounds 7–10 were tested for their activity against the SF-295 cell line, which is one of those more sensitive to the natural schweinfurthins.1 These new schweinfurthin analogues did show activity in these assays, with EC50’s ranging from ~200 nM to 2.5 μM (Table 1).37 Because the more active compounds show potency comparable to some of the natural schweinfurthins, preparation of additional indole analogues as well as more extensive testing in the 60 cell line assay of the National Cancer Institute would be warranted.
Table 1.
Preliminary bioassays in the SF-295 cell line
| Compound | EC50 (μM) |
|---|---|
| 7 | 0.2 |
| 8 | 2.5 |
| 9 | 0.2 |
| 10 | 2.2 |
In conclusion, we have developed a strategy for synthesis of indole analogues of the natural schweinfurthins. This effort included preparation of several new indoles by cyclization after a Stobbe condensation, and ultimately led to preparation of the first indoles bearing a phosphonomethyl substituent in the indole B-ring. These B-ring phosphonates have been used in HWE reactions with the complex aldehydes 11 and 12, and undergo these condensations smoothly as long as the indole nitrogen is securely protected. With a tosyl group on the indole nitrogen, an unexpected transfer of the tosyl group to an unprotected alcohol was observed. While this transfer undoubtedly could be avoided through use of an alcohol protecting group, instead, because this transfer also deprotected the indole nitrogen, the tosylate ester was isolated and cleaved to the free alcohol, which allowed preparation of indole analogues of the schweinfurthin G and F cores. These studies also have shown that the Zn(OTf)2 mediated alkylation of a 4,6-disubstituted indole is a facile way to introduce a prenyl substituent to C-3 of the indole system, which in turn allowed preparation of a schweinfurthin F analogue complete with a side chain. In this more hindered prenyl indole, an HWE condensation at room temperature did afford the desired stilbene without transfer of the tosyl group, and reductive cleavage of the N-tosyl group was more efficient. Finally, a C-2 prenylated indole was obtained through metalation and alkylation of a tosyl indole intermediate, which allows divergent use of intermediate 35 to obtain either the C-2 or C-3 alkylated compounds. Together these studies have afforded four new indole analogues (7–10) of the natural schweinfurthins, and they define procedures that could be used to prepare analogues of many other natural stilbenes including resveratrol,55 the chiricanines,56 the arachidins and arahypins,57 and the pawhuskins.58 Further research on the biological activity of the new schweinfurthin analogues is underway, and will be reported in due course.
Experimental Section
General Experimental Procedures
THF was freshly distilled from sodium/benzophenone, while CH2Cl2 and Et3N were freshly distilled from CaH2. All reactions in non-aqueous solvents were conducted in oven dried glassware under a positive pressure of argon with magnetic stirring. All commercial reagents were used without further purification unless otherwise stated. NMR spectra were recorded at 300 MHz for 1H, and 75 MHz for 13C or higher with CDCl3 as solvent and (CH3)4Si (1H, 0.00 ppm) or CDCl3 (13C, 77.0 ppm) as internal standards unless otherwise noted. High resolution mass spectra were run with magnet detection unless another method is noted. Elemental analyses were performed by a commercial facility.
2-(1H-Pyrrol-2-ylmethylene)-succinic acid 1-ethyl ester (19)
General Procedure for Stobbe Condensations
According to the procedure of Vedejs24 but in THF (60 mL) instead of benzene, NaH (4.2 g, 105 mmol, 60% dispersion oil) was added slowly to aldehyde 18 (5.01 g, 52.6 mmol) and diethylsuccinate (13.3 mL, 80.2 mmol) at 0 °C. The reaction mixture was allowed to stir overnight and warm to rt. The reaction mixture was cooled to 0 °C, quenched by addition of water and Et2O was added and then extracted with 5% KOH. The combined aqueous layers were acidified with HCl (6 M) and extracted with Et2O. The combined organic extracts were washed with brine, dried (MgSO4), filtered, and the solvent was removed in vacuo to afford acid 19 (11.2 g, 96%) as a red-brown solid: 1H NMR ((CD3)2CO, 400 MHz) δ 10.83 (br s, 1H), 10.63 (br s, 1H), 7.75 (s, 1H), 7.07 – 7.06 (m, 1H), 6.61 – 6.59 (m, 1H), 6.30 – 6.27 (m, 1H), 4.20 (q, J = 7.1 Hz, 2H), 3.65 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR ((CD3)2CO, 400 MHz) δ 172.4, 168.5, 131.8, 128.8, 123.1, 119.2, 114.4, 111.9, 61.3, 34.4, 14.9; HRMS (TOF MS EI) m/z calcd for C11H13NO4 (M+) 223.0845, found 223.0851.
4-Acetoxy-1H-indole-6-carboxylic acid ethyl ester (20) and 5-Acetoxy-indolizine-7-carboxylic acid ethyl ester (21)
To acid 19 (17.1 g, 76.7 mmol) in toluene (800 mL) was added Ac2O (48 mL, 506 mmol) and glacial AcOH (4.62 mL, 80.5 mmol) and the reaction was heated to reflux. The next day the reaction mixture was allowed to cool to rt, quenched by addition of K2CO3 (sat), washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (0 to 50% ethyl acetate in hexanes) afforded indole 20 (14.0 g, 74%) as a light brown solid and indolizine 21 (201 mg, 1%) as a yellow-brown oil. For indole 20: 1H NMR δ 8.98 (br s, 1H), 7.95 (s, 1H), 7.53 (s, 1H), 7.20 – 7.18 (m, 1H), 6.40 (m, 1H), 4.37 (q, J = 7.1 Hz, 2H), 2.40 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H); 13C NMR 169.5, 167.0, 142.8, 136.7, 127.9, 124.8, 124.2, 112.6, 111.8, 99.4, 60.9, 20.9, 14.3. Anal. calcd. for C13H13NO4: C, 63.15; H; 5.30; N, 5.66. Found: C, 62.97; H, 5.31; N, 5.61.
For indolizine 21: 1H NMR δ 8.14 (s, 1H), 7.33 – 7.31 (m, 1H), 6.94 (d, J = 1.4 Hz, 1H), 6.90 (dd, J = 3.9, 2.8 Hz, 1H), 6.79 (dd, J = 4.0, 1.2 Hz, 1H), 4.36 (q, J = 7.2 Hz, 2H), 2.45 (s, 3H), 1.39 (t, J =7.2 Hz, 3H); 13C NMR 166.9, 165.6, 138.8, 133.4, 120.2, 119.2, 115.7, 110.5, 105.6, 99.0, 60.9, 20.6, 14.3; HRMS (TOF MS EI) m/z calcd for C13H13NO4 (M+) 247.0845, found 247.0849.
Alternative route to indole 20 and indolizine 21
To acid 19 (1.00 g, 4.48 mmol) in THF was added Ac2O (5.4 mL, 57.5 mmol) and glacial AcOH (2.2 mL, 5.76 mmol) and the reaction mixture was heated to reflux. The next day the reaction mixture was allowed to cool to rt, poured into Et2O and water, washed with NaHCO3 (sat), dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (15% to 50% Et2O in hexanes) afforded indole 20 (461 mg, 42%) and indolizine 21 (212 mg, 19%).
2-(4-Bromo-1H-pyrrol-2-ylmethylene)-succinic acid 1-ethyl ester (22)
According to the general procedure, a solution of 4-bromo-2-pyrrolecarboxaldehyde (502 mg, 2.89 mmol) and diethyl succinate (0.72 mL, 4.29 mmol) in THF (4 mL) at 0 °C was treated with NaH (266 mg, 6.65 mmol, 60 % dispersion oil). Standard work-up and final purification by flash column chromatography (30% to 40% ethyl acetate in hexanes) afforded acid 22 (316 mg, 36%) as a light brown solid: 1H NMR ((CD3)2CO) δ 10.87 (br s, 1H), 7.67 (s, 1H), 7.14 (dd, J = 2.9, 1.4 Hz, 1H), 6.63 – 6.62 (m, 1H), 4.21 (q, J = 7.1 Hz, 2H), 3.65 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR ((CD3)2CO) δ 172.2, 167.9, 130.7, 129.3, 122.5, 121.5, 115.0, 98.8, 61.3, 34.1, 14.5; HRMS (TOF MS EI) m/z calcd for C11H12Br NO4 (M+) 300.9950, found 300.9954.
5-Acetoxy-2-bromo-indolizine-7-carboxylic acid ethyl ester (24)
To acid 22 (811 mg, 2.68 mmol) in THF was added glacial AcOH (0.19 mL, 3.3 mmol), and Ac2O (3.2 mL 33.8 mmol) and the solution was heated at reflux overnight. The reaction mixture was then allowed to cool to rt, quenched by addition of Na2CO3 (sat), and extracted with ethyl acetate. The combined organic extracts were washed with water and brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (20% Et2O in hexanes) afforded indolizine 24 (687 mg, 79%): 1H NMR δ 8.00 (d, J = 1.4 Hz, 1H), 7.32 (dd, J = 1.5, 0.5 Hz, 1H), 6.96 (d, J = 1.4 Hz, 1H), 6.78 (d, J = 1.5 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 2.44 (s, 3H), 1.28 (t, J = 7.2 Hz, 3H); 13C NMR δ 166.6, 165.1, 138.1, 133.3, 120.6, 118.6, 110.3, 107.2, 105.4, 99.4, 61.2, 20.6, 14.3. Anal. calcd. for C13H12BrNO4: C, 47.88; H; 3.71; N, 4.29. Found: C, 48.10; H, 3.73; N, 4.22.
2-(4-Bromo-1-methoxymethyl-1H-pyrrol-2-ylmethylene)-succinic acid 1-ethyl ester (25)
To 4-bromo-1H-pyrrole-2-carboxaldehyde (1.84 g, 10.6 mmol) in 10:1 THF/DMF (55 mL) at 0 °C was added NaH (525 mg, 7.5 mmol, 60% dispersion oil) and the reaction was allowed to stir for 5 min. To the resulting solution was added MOMCl (0.97 mL, 12.8 mmol) and the reaction was allowed to stir for 2 h and then quenched by addition of NH4Cl (sat), diluted with water, and extracted with Et2O. The combined organic extracts were washed with water and the brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (25% Et2O in hexanes) afforded the protected aldehyde (1.97 g, 86%) as a white solid: 1HMR δ 9.53 (d, J = 1.0 Hz, 1H), 7.13 (dd, J = 1.7, 1.0 Hz, 1H), 6.97 (d, J = 1.9 Hz, 1H), 5.62 (s, 2H), 3.31 (s, 3H); 13C NMR δ 179.0, 131.8, 130.1, 125.8, 98.0, 78.4, 56.3; HRMS (EI) m/z calcd for C7H8BrNO2 (M+) 216.9738, found 216.9740. According to the general procedure, the MOM-protected bromopyrrole aldehyde (1.01 g, 4.63 mmol) in THF (9 mL) at 0 °C was treated with diethyl succinate (1.2 mL, 1.54 mmol), followed by NaH (310 mg, 7.75 mmol). Standard work-up and final purification by flash column chromatography (25% to 40% ethyl acetate in hexanes) afforded acid 25 (425 mg, 27%) as a brown-yellow solid: 1H NMR δ 7.77 (s, 1H), 6.92 (d, J = 1.4 Hz, 1H), 6.61 (d, J = 1.1 Hz, 1H), 5.23 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 3.68 (s, 2H), 3.26 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H); 13C NMR 175.4, 167.6, 128.3, 128.2, 125.2, 122.1, 116.6, 97.9, 78.1, 61.5, 56.0, 34.0, 14.2. Anal. calcd for C13H16BrNO5: C, 45.10; H; 4.66; N, 4.05. Found: C, 45.19; H, 4.69; N, 3.93.
4-Acetoxy-3-bromo-1-methoxymethyl-1H-indole-6-carboxylic acid ethyl ester (26)
To acid 25 (1.084 g, 3.13 mmol) in Ac2O (20 mL) was added KOAc (0.49 g, 5.0 mmol) and the reaction was heated to reflux for 1 h and then allowed to cool to rt. The solution was diluted with ethyl acetate, washed with Na2CO3 (sat), water, and brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (20% ethyl acetate in hexanes) afforded indole 26 (911 mg, 79%) as a brown solid: 1H NMR δ 8.14 (d, J = 1.2 Hz, 1H), 7.56 (d, J = 1.2 Hz, 1H), 7.33 (s, 1H), 5.45 (s, 2H), 4.40 (q, J = 7.1 Hz, 2H), 3.28 (s, 3H), 2.43 (s, 3H), 1.41 (t, J = 7.1 Hz. 3H); 13C NMR δ 179.9, 166.2, 142.9, 137.1, 130.8, 126.1, 123.1, 115.2, 110.8, 88.2, 77.7, 61.2, 56.4, 21.0 14.4. Anal. calcd for C15H16BrNO5: C, 48.67; H, 4.36; N, 3.78. Found: C, 48.84; H, 4.60; N, 3.58.
2-(1-Methoxymethyl-1H-pyrrol-2-ylmethylene)-succinic acid 1-ethyl ester (27)
A solution of N-MOM-2-pyrrolecarboxaldehyde (100 mg, 0.72 mmol) and diethyl succinate (145 mg, 0.84 mmol) in THF at 0 °C was treated with KOt-Bu (120 mg, 1.07 mmol). The solution was allowed to warm to rt overnight and the next day was heated to reflux for one h. The solution was cooled to 0 °C, quenched by addition of water, diluted with Et2O, and extracted with 5% KOH. The combined aqueous extracts were acidified (6M HCl) and extracted with Et2O. The combined organic layers were washed with brine, dried (MgSO4), and filtered, and then the filtrate was concentrated in vacuo. Final purification by flash column chromatograph (30% ethyl acetate in hexanes) afforded acid 27 (60 mg, 31%) as a yellow solid: 1H NMR δ 7.87 (s, 1H), 6.93 (dd, J = 2.7, 1.5 Hz, 1H), 6.67 – 6.66 (m, 1H)), 6.29 – 6.27 (m, 1H), 5.29 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 3.72 (s, 2H), 3.25 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H); 13C NMR δ 176.1, 168.1, 129.3, 127.7, 126.2, 120.0, 115.6, 110.1, 78.0, 61.3, 55.7, 34.2, 14.2. Anal. calcd for C13H17NO5: C, 58.42; H, 6.41. Found: C, 58.49; H, 6.43.
4-Acetoxy-1-methoxymethyl-1H-indole-6-carboxylic acid ethyl ester (28)
To acid 27 (333 mg, 1.25 mmol) in Ac2O (10 mL) was added KOAc (153 mg, 1.56 mol) and the solution was heated at reflux until the reaction was complete as judged by TLC analysis. The solution was allowed to cool to rt and then poured into NaHCO3 (sat) and diluted with Et2O. Once bubbling had ceased, the aqueous layer was extracted with Et2O and the combined organic extracts were washed with NaHCO3 (sat), water, and brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (40% ethyl acetate in hexanes) afforded indole 28 (298 mg, 82%) as a brown-yellow solid: 1H NMR δ 8.14 (dd, J = 1.0, 1.0 Hz, 1H), 7.60 (d, J = 1.1 Hz, 1H), 7.31 (d, J = 3.3 Hz, 1H), 6.46 (dd, J = 3.3, 0.8 Hz, 1H), 5.47 (s, 2H), 4.39 (q, J = 7.1 Hz, 2H), 3.25 (s, 3H), 2.40 (s, 3H), 1.41 (t, J = 7.1 Hz, 3H); 13C NMR δ 169.0, 166.6, 143.0, 137.2, 131.2, 125.9, 124.9, 113.4, 110.3, 99.7, 77.5, 60.9, 56.0, 22.0, 14.3; HRMS (TOF MS EI) m/z calcd for C15H17NO5 (M+) 291.1107, found 291.1104.
4-Hydroxy-1H-indole-6-carboxylic acid ethyl ester (29)
To a solution of acetate 20 (201 mg, 0.81 mmol) in EtOH (20 mL) was added K2CO3 (210 mg, 1.52 mmol) and the resulting mixture was heated to reflux for 90 min. The reaction mixture was cooled to 0 °C, filtered through celite, and then concentrated in vacuo. The resulting residue was dissolved in Et2O and extracted with 2N NaOH. The aqueous extracts were acidified and extracted with Et2O, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50% Et2O in hexanes) afforded phenol 29 (136 mg, 82%) as a light brown solid: 1H NMR (CD3)2CO δ 10.5 (br s, 1H), 8.60 (br s, 1H), 7.76 (dd, J = 1.2, 1.2 Hz, 1H), 7.42 (dd, J = 3.2, 2.5 Hz, 1H), 7.18 (d, J = 1. 3 Hz, 1H), 6.67 (m, 1H), 4.32 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.2 Hz, 3H); 13C NMR δ 167.8, 150.9, 138.2, 127.2, 125.5, 122.8, 107.0, 104.5, 100.1, 60.9, 14.7. Anal. calcd for C11H11NO3 : C, 64.38; H, 5.40; N, 6.83. Found: C, 64.39; H, 5.49; N, 6.66.
4-Methoxymethoxy-1-methoxymethyl-1H-indole-6-carboxylic acid ethyl ester (30) and 4-methoxymethoxy-1,5-bis-methoxymethyl-1H-indole-6-carboxylic acid ethyl ester (31)
To a stirring suspension of NaH (800 mg, 20 mmol, 60% dispersion in oil) in a 6:1 mixture of THF and DMF (35 mL) at 0 °C was added indole 29 (1.61 g, 7.86 mmol) as a THF solution. Next MOMCl (1.5 mL, 20 mmol) was added dropwise and the reaction mixture was allowed to stir for 50 min. The reaction was quenched by addition of water and extracted with Et2O. The combined organic extracts were dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (25 to 50% Et2O in hexanes) afforded indoles 30 (1.82 g, 79%) and 31 (227 mg, 9%). For compound 30: 1H NMR δ 7.94 (dd, J = 0.9, 0.9 Hz, 1H), 7.47 (d, J = 1.1 Hz, 1H), 7.25 (d, J = 3.3 Hz, 1H), 6.69 (dd, J = 3.2, 0.8 Hz, 1H), 5.47 (s, 2H), 5.38 (s, 2H), 4.40 (q, J = 7.2 Hz, 2H), 3.55 (s, 3H), 3.25 (s, 3H), 1.44 (t, J = 7.2 Hz, 3H); 13C NMR δ 167.3, 150.0, 137.1, 129.8, 125.4, 124.0, 106.8, 104.6, 100.2, 94.7, 77.4, 60.8, 56.2, 55.9, 14.4. Anal. calcd for C15H19NO5: C, 61.42; H, 6.53; Found: C, 61.59; H, 6.62. For compound 31: 1H NMR δ 7.82 (d J = 0.6 Hz, 1H), 7.25 (d, J = 3.3 Hz, 1H), 6.68 (dd, J = 3.3, 0.8 Hz, 1H), 5.46 (s, 2H), 5.28 (s, 2 H), 4.93 (s, 2H), 4.40 (q, J = 7.1 Hz, 2H), 3.66 (s, 3H), 3.39 (s, 3H), 3.22 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H); 13C NMR δ 168.4, 150.0, 136.7, 130.1, 126.9, 124.6, 121.1, 109.1, 100.9, 99.5, 77.4, 65.7, 61.0, 58.0, 57.4, 56.0, 14.3. Anal. calcd for C17H23NO6: C, 60.52; H, 6.87; N, 4.15. Found: C, 60.40; H, 7.00; N, 4.00.
(4-Methoxymethoxy-1-methoxymethyl-1H-indol-6-yl)-methanol (32)
To ester 30 (668 mg, 2.28 mmol) in THF at 0 °C was added LiAlH4 (190 mg, 5.0 mmol) and the resulting mixture was allowed to stir for 2 h. The reaction mixture was then quenched by addition of water, acidified, and extracted with Et2O. The combined organic extracts were washed with water, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50% ethyl acetate in hexanes) afforded alcohol 32 (566 mg, 99%) as a white solid: 1H NMR δ 7.17 (s, 1H), 7.09 (d, J = 3.3 Hz, 1H), 6.80 (d, J = 0.9 Hz, 1H), 6.63 (dd, J = 3.2, 0.7 Hz, 1H), 5.39 (s, 2H), 5.32 (s, 2 H), 4.75 (s, 2H), 3.53 (s, 3H), 3.22 (s, 3H), 2.02 (br s, 1H); 13C NMR δ 150.7, 137.9, 136.6, 127.3, 119.9, 103.7, 102.8, 99.8, 94.7, 77.5, 66.1, 56.1, 55.8; HRMS (EI) m/z calcd for C13H17NO4 (M+) 251.1158; found 251.1152.
(4-Methoxymethoxy-1-methoxymethyl-1H-indol-6-ylmethyl)-phosphonic acid diethyl ester (33)
To a solution of alcohol 32 (12 mg, 0.048 mmol) in CH2Cl2 (5 mL) at 0 °C was added Et3N (0.05 mL, 0.38 mmol) and MsCl (0.02 mL, 0.24 mmol) and the reaction was allowed to warm to rt. The following day the reaction was quenched by addition of NH4Cl (sat) and extracted with CH2Cl2. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. The resulting residue was dissolved in acetone (5 mL) at rt, LiBr (33 mg, 0.38 mmol) was added, and the reaction mixture was allowed to stir overnight. The following day the reaction mixture was poured into Et2O, quenched by addition of water, and extracted with Et2O. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. The resulting residue was dissolved in P(OEt)3 (0.5 mL) and toluene (3 mL) and the solution was heated at reflux overnight. The following day the solution was allowed to cool to rt, poured into Et2O, and then quenched by addition of water and extracted with Et2O. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (80% ethyl acetate in hexanes) afforded phosphonate 33 (7 mg, 39% yield) as an oil: 1H NMR δ 7.12 (d, J = 3.2 Hz, 1H), 7.07 (dd, J = 3.2 Hz, 1.0 Hz, 1H), 6.75 (dd, J = 1.7, 1.3 Hz, 1H), 6.61 (dd, J = 3.2 Hz, 0.7 Hz, 1H), 5.40 (s, 2H), 5.32 (s, 2H), 4.06 – 3.96 (m, 4H), 3.53 (s, 3H), 3.25 (d, JHP = 21.3 Hz, 2H), 3.23 (s, 3H), 1.26 (td, J = 7.1 Hz, 0.3 Hz, 6H); 13C NMR δ 150.4 (d, JCP = 2.8 Hz), 138.0 (d, JCP = 3.0 Hz), 127.0 (d, JCP = 1.2 Hz), 126.4 (d, JCP = 9.2 Hz), 119.3 (d, JCP = 2.9 Hz), 106.5 (d, JCP = 5.9 Hz), 105.5 (d, JCP = 7.7 Hz), 99.7 (d, JCP = 1.5 Hz), 94.7, 77.4, 62.0 (d, JCP = 6.6 Hz, 2C), 56.1, 55.8, 34.2 (d, JCP = 138 Hz), 16.3 (d, JCP 6.1 Hz, 2C); 31P NMR δ 27.4; HRMS (EI) m/z calcd for C17H26NO6P (M+) 371.1498; found 371.1497.
5-Methoxymethoxy-7-[2-(4-methoxymethoxy-1-methoxymethyl-1H-indol-6-yl)-vinyl]-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-2-ol (34)
To a suspension of NaH (45 mg, 1.13 mol, 60% dispersion in oil) in THF at 0 °C was added phosphonate 33 (37 mg, 0.10 mmol) as a THF solution followed by aldehyde 1228 (17.6 mg, 0.052 mmol) as a THF solution and the reaction was allowed to warm slowly to rt. The following day the reaction mixture was quenched by addition of water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50 to 70% ethyl acetate in hexanes) afforded stilbene 34 (16 mg, 55%) as an oil: 1H NMR δ 7.24 (s, 1H), 7.16 (d, J = 1.9 Hz, 1H), 7.43 (d, J = 3.2 Hz, 1H), 7.03 – 6.97 (m, 4H), 6.62 (d, J = 3.2 Hz, 1H), 5.44 (s, 2H), 5.39 (s, 2H), 5.25 (d, J = 6.5 Hz, 1H), 5.21 (d, J = 6.6 Hz, 1H), 3.57, (s, 3H) 3.55 (s, 3H), 3.47 – 3.42 (m, 1H), 3.27 (s, 3H), 2.75 – 2.72 (m, 2H), 2.13 – 2.08 (m, 1H), 1.91 – 1.64 (m, 5H), 1.25 (s, 3H), 1.12 (s, 3H), 0.90 (s, 3H); 13C NMR δ 150.8, 146.2, 143.6, 138.2, 133.5, 129.5, 127.7, 127.0,125.5, 123.1, 121.9, 120.1, 113.4, 102.9, 102.5, 100.0, 95.9, 94.8, 78.0, 77.6, 76.9, 56.2, 56.2, 55.9, 46.8, 38.4, 37.7, 28.3, 27.3, 23.2, 19.9, 14.3; HRMS (EI) m/z calcd for C32H41NO7 (M+) 551.2883 found 551.2891.
7-[2-(4-Hydroxy-1-methoxymethyl-1H-indol-6-yl)-vinyl]-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthene-2,5-diol (35)
To MOM-protected compound 34 (16 mg, 0.029 mmol) in MeOH (3 mL) was added TsOH (80 mg, 0.42 mmol) and the solution was allowed to stir at rt. The next day the solution was quenched by addition of NH4Cl (sat), diluted with water, and extracted with ethyl acetate. The combined organics extracts were washed with water, dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50% ethyl acetate in hexanes) afforded the schweinfurthin analogue 35 (9 mg, 67%) as a yellow oil: 1H NMR (CD3OD) δ 7.16 (d, J = 3.3 Hz, 1H), 7.11 (m, 1H), 6.98 (d, J = 16.0 Hz, 1H), 6.91 (d, J = 16.4 Hz, 1H), 6.86 (d, J = 1.9 Hz, 1H), 6.77 (d, J = 1.8 Hz, 1H), 6.73 (d, J = 1.0 Hz, 1H), 6.55 (dd, J = 3.3, 0.7 Hz, 1H), 5.47 (s, 2H), 3.40–3.35 (m, 1H), 3.25 (s, 3H), 2.75–2.71 (m, 2H), 2.09–2.04 (m, 1H), 1.85–1.63 (m, 4H), 1.24 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 151.6, 147.0, 142.1, 140.1, 134.9, 131.4, 128.6, 128.4, 128.0, 124.0, 120.3, 120.2, 111.1, 103.3, 102.2, 100.5, 78.8, 78.3, 78.2, 56.0, ~49 (obscured by solvent), 39.5, 38.9, 29.0, 27.9, 24.0, 20.3, 14.8; HRMS (EI) m/z calcd for C28H33NO5 (M+) 463.2359 found 463.2353.
Preparation of 4-Methoxymethoxy-1H-indole-6-carboxylic acid ethyl ester (36)
To a suspension of phenol 29 (1.18 g, 5.74 mmol) in CH2Cl2 (100 mL) at rt was added DIPEA (4.0 mL, 23.0 mmol) and MOMCl (0.7 mL, 9.2 mmol) and the reaction mixture was allowed to stir overnight. The reaction was quenched by addition of water and extracted with CH2Cl2. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (15 to 25% ethyl acetate in hexanes) afforded indole 36 (1.10 g, 77%) as a light yellow solid: 1H NMR δ 8.95 (br s, 1H), 7.89 (dd, J = 1.0, 1.0 Hz, 1H), 7.43 (d, J = 1.1 Hz, 1H), 7.26 (dd, J = 3.1, 2.5 Hz, 1H), 6.69 (m, 1H), 5.38 (s, 2H), 4.38 (q, J = 7.1 Hz, 2H), 3.54 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H); 13C NMR δ 167.7, 149.9, 136.5, 126.4, 124.7, 123.0, 108.4, 103.8, 100.0, 94.7, 60.8, 56.2, 14.3. Anal. calcd for C13H15NO4: C, 62.64; H, 6.07; N, 5.62. Found: C, 62.83; H, 6.12; N, 5.42.
4-Methoxymethoxy-indole-1,6-dicarboxylic acid 1-tert-butyl ester, 6-ethyl ester (37)
To a solution of indole 36 (1.00 g, 4.01 mmol) in THF (20 mL) at 0 °C was added NaH (200 mg, 5 mmol, 60% dispersion in oil) and Boc2O (960 mg, 4.40 mmol). An additional aliquot of THF was added (8 mL) and after 1 h the reaction mixture was quenched by addition of NH4Cl (sat) and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the solvent was removed in vacuo. Final purification of the resulting material by flash column chromatography (12.5 to 15% Et2O in hexanes) afforded indole 37 (1.23 g, 87%): 1H NMR δ 8.54 (br s, 1H), 7.67 (d, J = 3.7 Hz, 1H), 7.57 (d, J = 1.2 Hz, 1H), 6.74 (dd, J = 3.7, 0.7 Hz, 1H), 5.36 (s, 2H), 4.39 (q, J = 7.1 Hz, 2H), 3.53 (s, 3H), 1.70 (s, 9H), 1.41 (t, J = 7.1 Hz, 3H) 13C NMR 167.0, 149.8, 149.4, 135.7, 127.5, 127.4, 125.4, 111.6, 107.5, 104.2, 94.8, 84.4, 60.9, 56.3, 28.1 (3C), 14.4. Anal. calcd for C18H23NO6: C, 61.88; H, 6.64; N, 4.01. Found: C, 62.00; H, 6.68; N, 4.02.
6-Hydroxymethyl-4-methoxymethoxy-indole-1-carboxylic acid tert-butyl ester (38)
To ester 37 (434 mg, 1.24 mmol) in THF (30 mL) at 0 °C was added DIBAL (4.1 mL, 1M in THF). When judged complete by TLC analysis, the reaction was quenched by addition of NH4Cl (sat), poured into ethyl acetate, acidified, and then extracted with ethyl acetate. The combined organic extracts were washed with NaHCO3 (sat) and brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (25% ethyl acetate in hexanes) afforded alcohol 38 (345 mg, 91%) as a colorless oil: 1H NMR δ 7.84 (s, 1H), 7.48 (d, J = 3.8 Hz, 1H), 6.93 (d, J = 0.9 Hz, 1H), 6.67 (dd, J = 3.8, 0.7 Hz, 1H), 5.30 (s, 2H), 4.75 (s, 2H), 3.51 (s, 3H), 2.16 (br s, 1H), 1.66 (s, 9H) 13C 150.3, 149.7, 138.7, 136.6, 124.8, 121.0, 108.0, 106.3, 104.1, 94.7, 83.7, 66.0, 56.1, 28.1 (3C). Anal. calcd for C16H21NO5: C, 62.53; H, 6.89; N, 4.56. Found: C, 62.30; H, 7.13; N, 4.56.
Preparation of (4-Methoxymethoxy-1H-indol-6-ylmethyl)-phosphonic acid diethyl ester (39) and 6-(Diethoxy-phosphorylmethyl)-4-methoxymethoxy-indole-1-carboxylic acid tert-butyl ester (40)
To LiBr (450 mg, 5.18 mmol) and NEt3 (0.43 mL, 3.09 mmol) in THF at 0 °C was added the benzylic alcohol 38 (312 mg, 1.02 mmol) as a THF solution. The solution was stirred for 5 min and then MsCl (0.16 mL, 2.07 mmol) was added dropwise. The reaction mixture was allowed to stir for 1 h and more LiBr (400 mg, 4.61 mmol) was added. After the reaction was judged complete by TLC analysis it was quenched by addition of NaHCO3 (sat), diluted with water, and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. To the resulting residue was added P(OEt)3 (4 mL) and the solution was heated at reflux overnight. The next day the solution was allowed to cool to rt and then poured into water and extracted with ethyl acetate. The organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50 to 70% ethyl acetate in hexanes) afforded indole phosphonate 40 (18 mg, 4%) as an oil and the parent indole phosphonate 39 (194 mg, 58%) as an oil.
For phosphonate 39: 1H NMR δ 9.61 (s, 1H), 7.05 (d, J = 2.9 Hz, 1H), 6.99 (t, J = 2.3 Hz, 1H), 6.66 (s, 1H), 6.54, (t, J = 2.2 Hz, 1H), 5.29 (s, 2H), 4.44 – 3.96 (m, 4H), 3.50 (s, 3H), 3.21 (d, JPH = 21.1 Hz, 2H), 1.24 (t, J = 7.0 Hz, 6H); 13C NMR δ 150.2 (d, JCP = 2.7 Hz), 137.7 (d, JCP = 2.9 Hz), 124.8 (d, JCP = 9.4 Hz), 123.5, 118.2 (d, JCP = 2.7 Hz), 107.1 (d, JCP = 7.4 Hz), 105.6 (d, JCP = 5.8 Hz), 98.7, 94.7, 62.1 (d, JCP = 6.8 Hz, 2C), 55.9, 33.9 (d, JCP = 138 Hz), 16.2 (d, JCP = 6.1 Hz, 2C); 31P NMR δ 28.2; HRMS (EI) m/z calcd for C15H22NO5P (M+) 327.1236; found 327.1229.
Boc protection of phosphonate 39
To phosphonate 39 (194 mg, 0.593 mmol) in CH2Cl2 (10 mL) was added DMAP (8 mg, 0.065 mmol) and Boc2O (150 mg, 0.69 mmol). The reaction was allowed to stir for 2 h and then checked by TLC analysis. After an additional amount of Boc2O was added (50 mg, 0.23 mmol), the reaction was allowed to proceed for another hour. The reaction mixture was quenched by addition of water and extracted with CH2Cl2. The combined organic extracts were dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (80% ethyl acetate in hexanes) afforded the Boc-protected indole 40 (183 mg, 72%) with 1H and 13C NMR spectra consistent with material prepared via the route below.
Preparation of phosphonate 40 at reduced temperature
To alcohol 38 (147 mg, 0.48 mmol) in THF (10 mL) was added LiBr (250 mg, 2.9 mmol) and NEt3 (0.2 mL, 1.4 mmol), the solution was cooled to 0 °C, and then was allowed to stir. After 10 min, MsCl (0.08 mL, 2.07 mmol) was added dropwise and the reaction mixture was allowed to stir for 2 h. The reaction was then quenched by addition of NH4Cl (sat), diluted with water, and extracted with ethyl acetate. The combined organic extracts were dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. To the residue was added P(OEt)3 and the resulting solution was heated to 95 °C and allowed to stir overnight. The next day the solution was allowed to cool to rt, and then concentrated in vacuo. Final purification by flash column chromatography (1.5% EtOH in Et2O) afforded phosphonate 40 (125 mg, 61%) as an oil: 1H NMR δ 7.78 (br s, 1H), 7.48 (d, J = 3.5 Hz, 1H), 6.88 (m, 1H), 6.66 (d, J = 3.7 Hz, 1H), 5.30 (s, 2H), 4.09–4.00 (m, 4H), 3.51 (s, 3H), 3.26 (d, JPH = 21.6 Hz, 2H), 1.66, (s 9H), 1.27 (t, J = 7.1 Hz, 6H); 13C NMR δ 150.0 (d, JCP = 2.9 Hz), 149.6, 128.7 (d, JCP = 9.5 Hz), 124.6, 120.3, 110.7 (d, JCP = 7.9 Hz), 108.9 (d, JCP = 5.7 Hz), 104.0 (d, JCP =1.6 Hz), 94.7, 83.6, 62.0 (d, JCP = 6.6 Hz, 2C), 56.3, 34.3 (d, JCP = 138 Hz), 28.1 (3C), 16.3 (d, JCP = 6.3 Hz, 2C); 31P NMR δ 27.3; HRMS (EI) m/z calcd for C20H30NO7P (M+) 427.1760; found 427.1757
[4-Methoxymethoxy-1-(toluene-4-sulfonyl)-1H-indol-6-yl]-methanol (41)
To indole 36 (805 mg, 3.23 mmol) in THF (30 mL) at 0 °C was added NaH (170 mg, 4.2 mmol, 60% dispersion in oil) followed after 10 min by TsCl (700 mg, 3.61 mmol). After 30 min, DIBAL (1.45 mL, 8.1 mmol) was added and the reaction was allowed to stir for an additional 30 min. It then was quenched by addition of NH4Cl (sat), poured into ethyl acetate, acidified, and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50% ethyl acetate in hexanes) afforded benzylic alcohol 41 (1.02 g, 87% overall yield): 1H NMR ((CD3)2CO) δ 7.84 (d, J = 8.3 Hz, 2H), 7.78 (s, 1H), 7.59 (d, J = 3.6 Hz, 1H), 7.22, (d, J = 8.5 Hz, 2H), 6.99 (s, 1H), 6.81, (dd, J = 3.7, 0.7 Hz, 1H), 5.27 (s, 2H), 4.78 (s, 2H), 4.53 (br s, 1H), 3.41 (s, 3H), 2.23 (s, 3H); 13C NMR δ 151.2, 146.0, 141.8, 136.9, 135.8, 130.7 (2C), 127.5 (2C), 126.0, 121.5, 107.2, 106.7, 105.9, 95.2, 65.0, 56.2, 21.3; HRMS (EI) m/z calcd for C18H19NO5S (M+) 361.0984; found 361.0992.
[4-Methoxymethoxy-1-(toluene-4-sulfonyl)-1H-indol-6-ylmethyl]-phosphonic acid diethyl ester (42)
To alcohol 41 (118 mg, 0.33 mmol) in THF (10 mL) at 0 °C was added LiBr (226 mg, 2.62 mmol) and NEt3 (0.18 mL, 1.30 mmol). The reaction was allowed to stir for 5 min and then MsCl (0.06 mL, 0.78 mmol) was added dropwise. The reaction was allowed to warm to rt and after 3 h it was quenched by addition of NaHCO3 (sat) and extracted with ethyl acetate. The organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. The resulting residue was dissolved in P(OEt)3 (3 mL) and heated to reflux. The next day the reaction was allowed to cool to rt, poured into water, and extracted with ethyl acetate. The organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (2.5 to 3% EtOH in Et2O) afforded phosphonate 42 (133 mg, 85%) as a white solid: 1H NMR δ 7.78 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 2.8 Hz, 1H), 7.44 (dd, J = 3.7, 0.9 Hz, 1H), 7.22, (d, J = 8.0 Hz, 2H), 6.86 (m, 1H), 6.73 (d, J = 3.7 Hz, 1H), 5.25 (s, 2H), 4.05 – 3.95 (m, 4H), 3.47 (s, 3H), 3.25 (d, JPH = 21.5 Hz, 2H), 2.33 (s, 3H), 1.24 (t, J = 7.1 Hz, 6H); 13C NMR δ 150.2 (d, JCP = 2.9 Hz), 144.8, 136.1 (d, JCP = 3.1 Hz), 135.1, 129.7 (2C), 129.3 (d, JCP = 9.2 Hz), 126.8 (2C), 125.0 (d, JCP = 1.4 Hz), 120.6 (d, JCP = 3.1 Hz), 109.3 (d, JCP = 6.0 Hz), 108.6 (d, JCP = 7.5 Hz), 105.8 (d, JCP = 1.5 Hz), 94.6, 62.0 (d, JCP = 6.7 Hz, 2C), 56.2, 34.2 (d, JCP = 138.1 Hz), 21.5, 16.3 (d, JCP = 6.1 Hz, 2C); 31P NMR δ 27.3; HRMS (EI) m/z calcd for C22H28NO7PS (M+) 481.1324; found 481.1315.
Preparation of Toluene-4-sulfonic acid 5-methoxymethoxy-7-[2-(4-methoxymethoxy-1H-indol-6-yl)-vinyl]-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-2-yl ester (43) and 5-methoxymethoxy-7-[2-(4-methoxymethoxy-1H-indol-6-yl)-vinyl]-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-2-ol (44)
To phosphonate 42 (40 mg, 0.83 mmol) and aldehyde 1228 (18 mg, 0.54 mmol) in THF (3 mL) at rt was added NaH (60 mg, 1.5 mmol, 60% dispersion in oil) and 15-crown-5 (3 drops) and the resulting solution was heated to reflux. After 30 min the reaction mixture was allowed to cool to rt and quenched by addition of NH4Cl (sat), diluted with water, and extracted with Et2O. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (20 to 40% ethyl acetate in hexanes) afforded the tosylate 43 (24 mg, 67%) along with the alcohol 44 (5 mg, 22%). For tosylate 43: 1H NMR δ 8.24 (br s, 1H), 7.82 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H) 7.14 – 7.11 (m, 3H), 6.98 – 6.92 (m, 4H), 6.63 (m, 1H), 5.38 (s, 2H), 5.23 (d, J = 6.6 Hz, 1H), 5.19 (d, J = 6.6 Hz, 1H), 4.33 (dd, J = 10.6, 4.8 Hz, 1H), 3.57 (s, 3H), 3.53 (s, 3H), 2.69–2.66 (m, 2H), 2.45 (s, 3H), 2.10–2.04 (m, 1H), 1.82 – 1.60 (m, 4H), 1.22 (s, 3H), 0.91 (s, 3H), 0.90 (s, 3H); 13C NMR δ 150.8, 146.1, 144.7, 143.3, 137.7, 134.3, 133.1, 129.8 (3C), 127.9, 127.7 (2C), 126.5, 123.5, 122.6, 121.7, 119.1, 113.4, 104.1, 101.9, 100.1, 95.9, 94.8, 88.4, 76.0, 56.2, 56.2, 47.0, 38.2, 37.4, 27.0, 25.8, 23.1, 21.6, 19.8, 15.1; HRMS (TOF MS ES) m/z calcd for C37H44NO8S ((M+H)+) 662.2788; found 662.2797.
For alcohol 44: 1H NMR δ 8.30 (br s, 1H), 7.15 – 7.11 (m, 3H), 7.05 – 6.92 (m, 4H), 6.64 (m, 1H), 5.39 (s, 2H), 5.24 (d, J = 6.4 Hz, 1H), 5.20 (d, J = 6.5 Hz, 1H), 3.57 (s, 3H), 3.35 (s, 3H), 3.43 (dd, J = 11.5, 3.8 Hz, 1H), 2.75 – 2.71 (m, 2H), 2.11 – 2.04 (m, 1H), 1.90–1.54 (m, 5H), 1.25 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 150.8, 146.1, 143.6, 137.7, 133.2, 129.6, 127.8, 126.7, 123.5, 123.2, 121.9, 119.1, 113.5, 104.1, 102.0, 100.1, 96.0, 94.8, 78.0, 76.9. 56.2, 56.2, 46.8, 38.4, 37.7, 28.3, 27.3, 23.2, 19.9, 14.2; HRMS (EI) m/z calcd for C30H37NO6 (M+) 507.2621; found 507.2620.
Reduction of tosylate 43
To the MOM-protected tosylate 43 (19.0 mg, 0.03 mmol) in THF (3 mL) at 0 °C was added LiAlH4 (14 mg, 0.40 mmol) and the reaction mixture was allowed to warm to rt overnight. The following morning the reaction was quenched by addition of NH4Cl (sat), diluted with water, and extracted with Et2O. The combined organic layers were washed with brine, dried (MgSO4) and filtered, and the solvent was removed in vacuo. Final purification by preparative TLC (70% ethyl acetate in hexanes) afforded the desired indole 44 (4.4 mg, 30%) along with recovered starting material (2.7 mg, 14%). The 1H NMR spectra was consistent with that of material prepared above.
7-[ 2-(4-Hydroxy-1H-indol-6-yl)-vinyl]-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthene-2,5-diol (7)
To a methanol solution of protected indole 44 (6 mg, 0.012 mmol) at 0 °C was added TsOH (25 mg, 0.145 mmol). The reaction was allowed to stir overnight, then quenched by addition of water and extracted with ethyl acetate. The combined organic extracts were dried (Mg2SO4), filtered, and concentrated in vacuo. Final purification of the residue by preparative TLC (70% ethyl acetate in hexanes) afforded schweinfurthin analogue 7 (2.9 mg, 58%):1H NMR (CD3OD) δ 7.09 (d, J = 3.3 Hz, 1H), 7.00 (s, 1H), 6.95 (d, J = 16.2 Hz, 1H), 6.87 (d, J = 16.2 Hz, 1H), 6.84 (d, J = 1.6 Hz, 1H), 6.75 (d, J = 1.6 Hz, 1H), 6.66, (d, J = 1.0 Hz, 1H), 6.50 (dd, J = 3.2, 0.9 Hz, 1H), 3.43 (dd, J = 11.5. 3.8 Hz, 1H), 2.74 – 2.71 (m, 2H), 2.09 – 2.04 (m, 1H), 1.83 – 1.63 (m, 4H), 1.24 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 151.2, 147.0, 141.9, 139.8, 133.9, 131.5, 128.9, 127.2, 124.4, 124.0, 120.2, 119.3, 111.0, 103.8, 101.8, 99.7, 78.8, 78.2, 39.5, 38.9, 29.0, 27.9, 24.0, 20.3, 14.9; HRMS (EI) m/z calcd for C26H29NO4 (M+) 419.2097; found 419.2096.
Toluene-4-sulfonic acid 5-methoxy-7-[2-(4-methoxymethoxy-1H-indol-6-yl)-vinyl]-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-2-yl ester (45)
To aldehyde 113,28 (63 mg, 0.21 mmol) and phosphonate 42 (156 mg, 0.32 mmol in THF (5 mL) at rt was added NaH (80 mg, 2.0 mmol, 60% dispersion in oil) and 15-crown-5 (3 drops). The reaction mixture was slowly heated to reflux for 40 min and then allowed to cool to rt. After the reaction was quenched by addition of NaHCO3 (sat), it was diluted with water, and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (30% ethyl acetate in hexanes) afforded the tosylate 45 (73 mg, 56%): 1H NMR δ 8.25 (br s, 1H), 7.82 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.14 (s, 1H), 7.12 (dd, J = 3.2, 2.4 Hz, 1H), 7.03 (d, J = 16.2 Hz, 1H), 6.99 (d, J = 1.1 Hz, 1H), 6.95 (d, J = 16.3 Hz, 1H), 6.90 (d, J = 1.6 Hz, 1H), 6.83 (d, J = 1.6 Hz, 1H), 6.65 – 6.63 (m, 1H), 5.39 (s, 2H), 4.36 – 4.31 (m, 1H), 3.89 (s, 3H), 3.57 (s, 3H), 2.70 – 2.67 (m, 2H), 2.45 (s, 3H), 2.14 – 2.09 (m, 1H), 2.01 – 1.96 (m, 1H), 1.87 – 1.68 (m, 3H), 1.56 (br s, 1H), 1.23 (s, 3H), 0.91 (m, 6H); 13C δ 150.8, 148.9, 144.6, 142.0, 137.7, 134.3, 133.1, 129.8 (2C), 129.6, 127.8, 127.7 (2C), 126.8, 123.6, 122.0, 120.1, 119.2, 107.0, 104.0, 102.0, 100.1, 94.8, 88.5, 76.0, 56.2, 56.0, 47.0, 38.2, 37.3, 27.1, 25.7, 23.1, 21.6, 19.7, 15.1; HRMS (TOF MS ES) m/z calcd for C36H42NO7S ((M+H)+) 632.2682; found 632.2684.
Toluene-4-sulfonic acid 5-methoxy-7-[2-(4-methoxymethoxy-1H-indol-6-yl)-vinyl]-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-2-yl ester (46)
To the tosylate 45 (73 mg, 0.12 mmol) in THF (3 mL) was added LiAlH4 (45 mg, 1.18 mmol) and the reaction mixture was allowed to stir overnight. The reaction then was quenched by addition of NH4Cl (sat) and extracted with Et2O. The combined organic layers were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (30 to 50% ethyl acetate in hexanes) yielded alcohol 46 (24 mg, 43%): 1H NMR δ 8.25 (br s, 1H), 7.15 (s, 1H), 7.12 (dd, J = 3.1, 2.5 Hz, 1H), 7.04 (d, J = 16.2 Hz, 1H), 7.00 (s, 1H), 6.97 (d, J = 16.2 Hz, 1H), 6.91 (d, J = 2.3 Hz, 1H), 6.88 (d, J = 2.3 Hz, 1H), 6.63 (m, 1H), 5.39 (s, 2H), 3.90 (s, 3H), 3.58 (s, 3H), 3.45 – 3.40 (m, 1H), 2.74 – 2.71 (m, 2H), 2.15 – 2.10 (m, 1H), 1.90 – 1.80 (m, 2H), 1.74 – 1.50 (m, 3H), 1.26 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 150.8, 148.9, 142.3, 137.7, 133.2, 129.4, 127.6, 127.0, 123.5, 122.6, 120.2, 119.1, 106.9, 104.0, 102.0, 100.1, 94.8, 78.0, 77.0, 56.2, 56.0, 46.8, 38.4, 37.7, 28.3, 27.3, 23.2, 19.9, 14.3; HRMS (EI) m/z calcd for C29H35NO5 (M+) 477.2515; found 477.2512.
6-[2-(7-Hydroxy-4-methoxy-8,8,10a-trimethyl-(5R,8aR,10aR)5,7,8,8a,9a,10a- hexahydro-6H-xanthen-2-yl)-vinyl]-1H-indol-4-ol (8)
To the MOM-protected indole 46 (16.0 mg, 0.033 mmol) in MeOH (3 mL) was added HCl (0.15 mL, 6M). The reaction was stirred in a warm water bath for 8.5 h, quenched by dropwise addition of NaHCO3 (sat), and then extracted with Et2O. The combined organic extracts were washed with brine, dried (MgSO4), and filtered through basic alumina, and the filtrate was concentrated in vacuo. Final purification by preparative TLC (70% ethyl acetate in hexanes) afforded indole 8 (9 mg, 62%); 1H NMR δ 8.2 (br s, 1H), 7.13 (dd, J = 3.1, 2.5 Hz, 1H), 7.07 (s, 1H), 7.00 (d, J = 16.2 Hz, 1H), 6.94 (d, J = 16.4 Hz, 1H), 6.90 (d, J = 1.8 Hz, 1H), 6.85 (d, J = 1.7 Hz, 1H), 6.77 (d, J = 0.9 Hz, 1H), 6.59 – 6.57 (m, 1H), 5.22 (br s, 1H), 3.90 (s, 3H), 3.43 (dd, J = 11.5, 3.7 Hz, 1H), 2.75 – 2.72 (m, 2H), 2.16 – 2.10 (m, 1H), 1.90 – 1.80 (m, 2H), 1.75 – 1.60 (m, 3H), 1.26 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C δ 149.0, 148.9, 142.4, 138.0, 133.4, 129.4, 127.3, 127.2, 123.5, 122.7, 120.4, 117.4, 106.9, 103.1, 102.1, 99.2, 78.1, 77.0, 56.0, 46.8, 38.4, 37.6, 28.3, 27.4, 23.2, 19.9, 14.3; HRMS (EI) m/z calcd for C27H31NO4 (M+) 433.2253; found 433.2245.
4-Methoxymethoxy-3-(3-methyl-but-2-enyl)-1H-indole-6-carboxylic acid ethyl ester (47)
To indole 36 (1.00 g, 4.01 mmol), TBAI (739 mg, 2.00 mmol), and Zn(OTf)2 (878 mg, 2.41 mmol) in a 9:2 mixture of toluene and CH2Cl2 (22 mL) at rt was added DIPEA (0.77 mL, 4.41 mmol). After the reaction mixture was allowed to stir for 10 min, prenyl bromide (298 mg, 2.00 mmol) was added dropwise. After 3 h the reaction mixture was quenched by addition of NH4Cl (sat) and extracted with ethyl acetate. The combined organic extracts were washed with water, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (10 to 15% ethyl acetate in hexanes) afforded prenylated indole 47 (415 mg, 65%) along with recovered starting material 36 (540 mg): 1H NMR δ 8.47 (br s, 1H), 7.79 (d, J = 1.2 Hz, 1H), 7.34 (d, J = 1.1 Hz, 1H), 6.96 (m, 1H), 5.46 (m, 1H), 5.35 (s, 2H), 4.37 (q, J = 7.1 Hz, 2H), 3.65 (d, J = 6.6 Hz, 2H), 3.53 (s, 3H), 1.74 (d, J = 1.0 Hz, 3H), 1.72 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H); 13C δ 167.6, 151.4, 137.4, 131.5, 124.6, 123.8, 123.7, 121.3, 116.7, 108.2, 102.8, 94.2, 60.7, 56.2, 25.7, 25.4, 17.7, 14.4; HRMS (EI) m/z calcd for C18H23NO4 (M+) 317.1627; found 317.1631.
[4-Methoxymethoxy-3-(3-methyl-but-2-enyl)-1-(toluene-4-sulfonyl)-1H-indol-6-yl]-methanol (48)
To indole 47 (315 mg, 0.99 mmol) in THF at 0 °C was added NaH (50 mg, 1.25 mmol, 60% dispersion oil) and the reaction mixture was allowed to stir for 10 min. After TsCl (230 mg, 1.21 mmol) was added, the solution was stirred for 30 min and then DIBAL (0.71 mL, 4.0 mmol) was added dropwise. After an additional 30 min the reaction was quenched by addition of NH4Cl (sat), acidified with HCl, and extracted with ethyl acetate. The combined organic extracts were washed with Na2CO3 (sat) and brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (34% ethyl acetate in hexanes) afforded benzylic alcohol 48 (348 mg, 82%): 1H NMR δ 7.71 (d, J = 8.4 Hz, 2H), 7.60 (s, 1H), 7.16 (d, J = 8.2 Hz, 2H), 7.13 (m, 1H), 6.85 (d, J = 0.6 Hz, 1H), 5.41 – 5.39 (m, 1H), 5.22 (s, 2H), 4.71 (s, 2H), 3.51 (d, J = 7.1 Hz, 2H) 3.46 (s, 3H), 2.37 (br s, 1H), 2.30 (s, 3H), 1.76 (d, J = 0.8 Hz, 3H), 1.68 (s, 3H); 13C NMR δ 151.8, 144.6, 139.1, 137.0, 135.2, 132.9, 129.7 (2C), 126.6 (2C), 122.7, 121.9, 121.8, 120.2, 105.9, 105.7, 94.1, 65.5, 56.1, 25.7, 25.6, 21.4, 17.7; HRMS (EI) m/z calcd for C23H27NO5S (M+) 429.1610; found 429.1609.
[4-Methoxymethoxy-3-(3-methyl-but-2-enyl)-1-(toluene-4-sulfonyl)-1H-indol-6-ylmethyl]-phosphonic acid diethyl ester (49)
To alcohol 48 (332 mg, 0.77 mmol) in THF (15 mL) at 0 °C was added LiBr (537 mg, 6.18 mmol) and NEt3 (0.43 mL, 3.09 mmol). The solution was stirred for 5 min and then MsCl (0.18 mL, 2.32 mmol) was added dropwise. The reaction was allowed to warm to rt, and after 2 h it was quenched by addition of NaHCO3 (sat.) and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Without further purification, the resulting residue was dissolved in P(OEt)3 (3 mL) and heated to reflux. The next day the solution was allowed to cool to rt and then poured into water and extracted with ethyl acetate. The organic extracts were washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by flash column chromatography (2% EtOH in Et2O) afforded indole phosphonate 49 (374 mg, 88%) as a waxy white solid: 1H NMR δ 7.75 (d, J = 8.4 Hz, 2H), 7.57 (m, 1H), 7.21 (d, J = 8.1 Hz, 2H), 7.10, (d, J = 1.1 Hz, 1H), 6.80 (m, 1H), 5.41 – 5.36 (m, 1H), 5.23 (s, 2H), 4.00 (m, 4H), 3.51 – 3.47 (m, 5H), 3.22 (d, JPH = 21.5 Hz, 2H), 2.33 (s, 3H), 1.77 (s 3H), 1.68 (s, 3H), 1.25 (t, J = 7.0 Hz, 6H); 13C NMR δ 151.6 (d, JCP = 2.9 Hz) 144.8, 137.1 (d, JCP = 3.1 Hz), 135.4, 133.0, 129.7 (2C), 129.2 (d, JCP = 9.3 Hz), 126.8 (2C), 122.7 (d, JCP = 1.6 Hz), 121.8, 121.7 (d, JCP = 1.8 Hz), 119.7 (d, JCP = 3.2 Hz), 108.9 (d, JCP = 5.9 Hz), 108.7 (d, JCP = 7.6 Hz), 94.3, 62.1 (d, JCP = 6.7 Hz, 2C), 56.1, 34.2 (d, JCP = 138.3 Hz), 25.7, 25.6, 21.4, 17.7, 16.3 (d, JCP = 6.0 Hz, 2C); 31P NMR δ 26.9; HRMS (EI) m/z calcd for C27H36NO7PS (M+) 549.1950; found 549.1959.
5-Methoxy-7-{2-[4-methoxymethoxy-3-(3-methyl-but-2-enyl)-1H-indol-6-yl]-vinyl}-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-2-ol (51)
To aldehyde 11 (44 mg, 0.15 mmol) and phosphonate 49 (100 mg, 0.18 mmol) in THF (4 mL) at 0 °C was added NaH (80 mg, 2.0 mmol, 60% dispersion oil) and 15-crown-5 (2 drops), and the reaction mixture was allowed warm to rt. After 2 hrs it was quenched by addition of NH4Cl (sat) and extracted with ethyl acetate. The combined organic layers were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (50% ethyl acetate in hexanes) afforded a mixture of the N-Ts compound 50 and free indole 51 (55 mg) as an oil. This material was dissolved in a mixture of THF and i-PrOH (5 mL, 1:1 mixture) at 0 °C, NaH (150 mg, excess) was added, and the reaction mixture was allowed to warm to rt. The following day the reaction mixture was quenched by addition of water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50% ethyl acetate in hexanes) afforded indole 51 (35 mg, 44% for 2 steps) as an oil: 1H NMR δ 7.95 (br s, 1H), 7.07 (s, 1H), 6.99 – 6.98 (m, 2H), 6.92 – 6.90 (m, 2H), 6.87 (m, 1H), 6.81 (s, 1H), 5.51 – 5.46 (m, 1H), 5.36 (s, 2H), 3.90 (s, 3H), 3.62 (d, J = 7.0 Hz, 2H), 3.57 (s, 3H), 3.43 (dd, J = 11.6, 3.8 Hz, 1H), 2.74 – 2.71 (m, 2H), 2.15 – 2.10 (m, 1H), 1.89 – 1.56 (m, 11H), 1.26 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 152.1, 148.9, 142.3, 138.6, 133.0, 131.2, 129.4, 127.6, 126.8, 124.1, 122.6, 120.9, 120.2, 117.5, 116.7, 106.9, 103.8, 100.9, 94.3, 78.0, 77.0, 56.1, 56.0, 46.8, 38.4, 37.7, 28.3, 27.3, 25.7, 25.6, 23.2, 19.8, 17.7, 14.3; HRMS (EI) m/z calcd for C34H43NO5 (M+) 545.3141; found 545.3135.
6-[2-(7-Hydroxy-4-methoxy-8,8,10a-trimethyl-(5R,8aR,10aR)-5,7,8,8a,9,10a-hexahydro-6H-xanthen-2-yl)-vinyl]-3-(3-methyl-but-2-enyl)-1H-indol-4-ol (9)
To compound 51 (31 mg, 0.057 mmol) in MeOH (2 mL) at rt was added TsOH (75 mg, 0.39 mmol) and the reaction flask was wrapped in foil. After 10 h the reaction was quenched by addition to NaHCO3 (sat) and extracted with ethyl acetate. The combined organic extracts were washed with Na2CO3 (sat), brine, and dried (MgSO4), filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50% ethyl acetate in hexanes) afforded stilbene 9 (8 mg, 28%) as a light yellow oil: 1H NMR δ 7.90 (br s, 1H), 6.99 – 6.96 (m, 3H), 6.89 – 6.85 (m, 3H), 6.74 (s, 1H), 5.91 (br s, 1H), 5.54 (m, 1H), 3.90 (s, 3H), 3.58 (d, J = 6.6 Hz, 2H), 3.44 (dd, J = 11.6, 3.7 Hz, 1H), 2.75 – 2.72 (m, 2H), 2.16 – 2.10 (m, 1H), 1.90 – 1.55 (m, 5H), 1.84 (s, 3H), 1.82 (s, 3H), 1.26 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 150.1, 148.9, 139.2, 135.1, 133.6, 129.8, 129.4, 127.3, 127.1, 125.1, 122.6, 121.0, 120.3, 116.4, 115.2, 106.9, 102.8, 102.8, 78.1, 56.0, 46.8, 38.4, 37.7, 28.3, 27.4, 25.8, 25.7, 23.2, 19.8, 17.7, 14.3; HRMS (EI) m/z calcd for C32H39NO4 (M+) 501.2879; found 501.2874.
6-(tert-Butyl-dimethyl-silanyloxymethyl)-4-methoxymethoxy-1-(toluene-4-sulfonyl)-1H-indole (52)
To alcohol 41 (1.09 g, 3.01 mmol) in CH2Cl2 (50 mL) at 0 °C was added imidazole (502 mg, 7.53 mmol) and TBSCl (500 mg, 3.31 mmol), and then the solution was allowed to warm to rt. The next day the reaction was quenched by addition of NH4Cl (sat) and extracted with CH2Cl2. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (8% ethyl acetate in hexanes) afforded silyl ether 52 (1.39 g, 97%): 1H NMR δ 7.75 (d, J = 8.4 Hz, 2H), 7.63 (m, 1H), 7.45 (d, J = 3.7 Hz, 1H), 7.20, (dd, J = 8.5, 0.6 Hz, 2H), 6.88 (m, 1H), 6.73 (dd, J = 3.7, 0.8 Hz, 1H), 5.24 (s, 2H), 4.81 (s, 2H), 3.47 (s, 3H), 2.33 (s, 3H), 0.97 (s, 9H), 0.12 (s, 6H); 13C δ 150.3, 144.8, 139.8, 136.1, 135.3, 129.8 (2C), 128.8 (2C), 124.9, 120.7, 105.8, 105.9, 104.9, 94.7, 65.2, 56.1, 25.9 (3C), 21.5, 18.3, −5.2 (2C); HRMS (EI) m/z calcd for C24H33NO5SSi (M+) 475.1849; found 475.1856.
6-(tert-Butyl-dimethyl-silanyloxymethyl)-4-methoxymethoxy-2-(3-methyl-but-2-enyl)-1-(toluene-4-sulfonyl)-1H-indole (53)
To the silyl-protected indole 52 (724 mg, 1.52 mmol) in THF was added a few 4 Å molecular sieves and the mixture was cooled to −78 °C. After n-BuLi (0.75 mL, 2.3M in hexanes) was added, the mixture was stirred for 20 min and then prenyl bromide (420 mg, 2.82 mmol) was added. The next day the reaction mixture was quenched by addition of NH4Cl (sat), and extracted with Et2O. The combined organic layers were washed with brine, dried (MgSO4), and filtered, and the filtrated was concentrated in vacuo. Final purification by flash column chromatography (5% ethyl acetate in hexanes) afforded the prenyl indole 53 (560 mg, 68%) as well as recovered starting material 52 (76 mg, 10%): 1H NMR δ 7.91 (d, J = 0.8 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.25, (d, J = 8.5 Hz, 2H), 6.99 (s, 1H), 6.52 (d, J = 0.8 Hz, 1H), 5.47 (m, 1H), 5.31 (s, 2H), 4.90 (s, 2H), 3.74 (d, J = 7.2 Hz, 2H), 3.55 (s, 3H), 2.40 (s, 3H), 1.86 (s, 3H), 1.71 (s, 3H) 1.05 (s, 9H), 0.20 (s, 6H); 13C NMR δ 149.5, 144.5, 139.9, 138.7, 138.6, 136.5, 134.5, 129.7 (2C), 126.3 (2C), 119.8, 119.6, 106.5, 106.3, 105.3, 94.8, 65.5, 56.0, 27.9, 25.9 (3C), 25.7, 21.4, 18.3, 17.7, −5.2 (2C); HRMS (EI) m/z calcd for C29H41NO5SSi (M+) 543.2475; found 543.2476.
[4-Methoxymethoxy-2-(3-methyl-but-2-enyl)-1-(toluene-4-sulfonyl)-1H-indol-6-yl]-methanol (54)
To the silyl ether 53 (682 mg, 1.26 mmol) in THF (20 mL) at rt was added TBAF (1.88 mL, 1.0 M in THF). After 2 h the reaction was quenched by addition of water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (30 to 45% ethyl acetate in hexanes) afforded alcohol 54 (461 mg, 85%): 1H NMR δ 7.84 (s, 1H), 7.74 (d, J = 8.3 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 6.93 (s, 1H), 6.44 (s, 1H), 5.38 (m, 1H), 5.24 (s, 2H), 4.74 (s, 2H), 3.64 (d, J = 7.1 Hz, 2H), 3.46 (s, 3H), 2.60 (br s, 1H), 2.31 (s, 3H), 1.78 (s, 3H), 1.61 (s, 3H); 13C δ 149.5, 144.6, 140.1, 138.5, 138.1, 136.2, 134.7, 129.7 (2C), 126.2 (2C), 119.9, 119.5, 107.2, 106.7, 105.2, 94.5, 65.7, 56.1, 27.8, 25.7, 21.4, 17.6; HRMS (TOF MS EI) calcd m/z for C23H27NO5S (M+) 429.1610; found 429.1622.
[4-Methoxymethoxy-2-(3-methyl-but-2-enyl)-1-(toluene-4-sulfonyl)-1H-indol-6-ylmethyl]-phosphonic acid diethyl ester (55)
To benzylic alcohol 54 (333 mg, 0.78 mmol) in THF was added LiBr (540 mg, 6.20 mmol) and NEt3 (0.44 mL, 3.10 mmol) and the solution was cooled to 0 °C. After 15 min MsCl (0.19 mL, 2.46 mmol) was added dropwise and the reaction was allowed to stir and slowly warm to rt. After 2 h, when the reaction was complete by TLC analysis, it was quenched by addition of water and extracted with Et2O. The organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. To the resulting residue was added P(OEt)3 (3 mL) and the solution was heated at reflux overnight. The next day the solution was allowed to cool to rt and then poured into water and extracted with ethyl acetate. The organic extract was washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50 to 70% ethyl acetate in hexanes) afforded indole phosphonate 55 (384 mg, 90%): 1H NMR δ 7.82 (d, J = 2.8 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.5 Hz, 2H), 6.87 (s, 1H), 6.43 (s, 1H), 5.40 – 5.35 (m, 1H), 5.25 (s, 2H), 4.07 – 3.94 (m, 4H), 3.64 (d, J = 7.2 Hz, 2H), 3.48 (s, 3H), 3.26 (d, JPH = 21.3 Hz, 2H), 2.34 (s, 3H), 1.78 (s, 3H), 1.62 (s, 3H), 1.26 (t, J = 7.1 Hz, 6H); 13C NMR δ 149.3 (d, JCP = 3.1 Hz) 144.6, 140.0 (d, JCP = 1.9 Hz), 138.5 (d, JCP = 3.1 Hz), 136.2, 134.7, 129.9 (2C), 128.1 (d, JCP = 9.3 Hz), 126.3 (2C), 119.5, 119.4 (d, JCP = 3.1 Hz), 109.9 (d, JCP = 7.4 Hz), 109.5 (d, JCP = 6.1 Hz), 105.2, 94.8, 62.2 (d, JCP = 6.9 Hz, 2C), 56.2, 34.2 (d, JCP = 137.7 Hz), 27.8, 25.6, 21.4, 17.7, 16.2 (d, JCP = 5.9 Hz, 2C); 31P NMR δ 27.3; HRMS (EI) m/z calcd for C27H36NO7PS (M+) 549.1950; found 549.1943.
5-Methoxy-7-{2-[4-methoxymethoxy-2-(3-methyl-but-2-enyl)-1H-indol-6-yl]-vinyl}-1,1,4a-trimethyl-(2R,4aR,9aR)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-2-ol (57)
To phosphonate 55 (74 mg, 0.14 mmol) and aldehyde 11 (30 mg, 0.10 mmol) in THF (2 mL) at 0 °C was added NaH (50 mg, 1.25 mmol, 60% dispersion oil) and 15-crown-5 (3 drops). The reaction mixture was allowed to stir for 4 h, then quenched by addition of NH4Cl (sat) and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Purification by flash column chromatography (50% ethyl acetate in hexanes) afforded a mixture of N-tosyl indole 56 and the unprotected indole 57. To the mixed residue in 1:1 THF and i-PrOH (3 mL) at 0 °C was added NaH (120 mg, 3.0 mmol) and the reaction mixture allowed to warm to rt overnight. The next day the reaction mixture was quenched by addition of NH4Cl (sat), diluted with water, and extracted with ethyl acetate. The combined organic extracts were washed with water, brine, and dried (MgSO4), filtered, and then the filtrate was concentrated in vacuo. Final purification by flash column chromatography (50% ethyl acetate in hexanes) afforded indole 57 (20 mg, 37% for the 2 steps) as an oil: 1H NMR δ 7.92 (br s, 1H), 7.08 (m, 1H), 7.02 (d, J = 16.1 Hz, 1H), 6.96 (m, 1H), 6.94 (d, J = 16.1 Hz, 1H), 6.89 (m, 1H), 6.86 (m, 1H), 6.31 (m, 1H), 5.40 (m, 1H), 5.36 (s, 2H), 3.90 (s, 3H), 3.56 (s, 3H), 3.49 – 3.39 (m, 3H), 2.74 – 2.71 (m, 2H), 2.18 – 2.10 (m, 1H), 1.90 – 1.60 (m, 5H), 1.79 (s, 3H), 1.74 (s, 3H), 1.26 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR δ 150.1, 148.9, 142.3, 138.3, 137.5, 134.6, 132.1, 129.5, 127.8, 126.4, 122.6, 120.1, 120.1, 119.9, 107.1, 106.9, 103.5, 102.3, 95.0, 78.1, 77.0, 56.1, 56.0, 46.8, 38.4, 37.7, 28.3, 27.4, 27.1, 25.7, 23.2, 19.9, 17.8, 14.3; HRMS (EI) m/z calcd for C34H43NO5 (M+) 545.3141; found 545.3135.
6-[2-(7-Hydroxy-4-methoxy-8,8,10a-trimethyl-(5R,8aR,10aR)-5,7,8,8a,9,10a-hexahydro-6H-xanthen-2-yl)-vinyl]-2-(3-methyl-but-2-enyl)-1H-indol-4-ol (10)
To compound 57 (8 mg, 0.015 mmol) in MeOH (0.8 mL) in a foil-wrapped flask was added TsOH (25 mg, 0.13 mmol) and the reaction was allowed to stir at rt. After 10 h the reaction was quenched by addition of NaHCO3 (sat) and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by radial chromatography (50% ethyl acetate in hexanes) afforded compound 10 (5 mg, 68%) as a light yellow oil: 1H NMR (CD3OD) δ 6.99 (d, J = 16.4 Hz, 1H), 6.95 (m, 2H), 6.90 (d, J = 16.2 Hz, 1H), 6.82 (m, 1H), 6.63 (s, 1H), 6.17 (s, 1H), 5.46 – 5.41 (m, 1H), 3.85 (s, 3H), 3.44 (d, J = 7.3 Hz, 2H), 3.37 (dd, J = 10.8, 3.9 Hz, 1H), 2.76 – 2.73 (m, 2H), 2.07 – 2.02 (m, 1H), 1.85 – 1.60 (m, 4H), 1.79 (s, 3H), 1.75 (s, 3H), 1.23 (s, 3H), 1.11 (s, 3H), 0.88 (s, 3H); 13C NMR δ 150.5, 150.1, 143.2, 140.1, 139.4, 134.3, 132.9, 131.4, 129.3, 126.6, 124.0, 122.2, 121.4, 119.4, 108.0, 103.4, 102.0, 96.7, 78.7, 78.1, 56.4, ~49 (obscured by solvent), 39.5, 38.9, 29.0, 28.0, 27.9, 25.9, 24.1, 20.2, 17.8, 14.9; HRMS (TOF MS ES) m/z calcd for C32H39NO4 (M+H)+ 502.2957; found 502.2956.
Supplementary Material
Scheme 8.

Synthesis of the prenylated indole schweinfurthin 10.
Acknowledgments
We thank Dr. Nolan Mente for providing aldehyde 12, and Dr. Craig Kuder and Prof. Raymond J. Hohl (University of Iowa, Department of Internal Medicine) for providing the bioassay data in Table 1. Financial support from the University of Iowa Graduate College (in the form of a Presidential Fellowship to J. G. K.), the Roy J. Carver Charitable Trust, and the National Cancer Institute (R41CA126020 via Terpenoid Therapeutics, Inc.) is gratefully acknowledged.
Footnotes
Supporting Information Available: The 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Beutler JA, Shoemaker RH, Johnson T, Boyd MR. J Nat Prod. 1998;61:1509–1512. doi: 10.1021/np980208m. [DOI] [PubMed] [Google Scholar]
- 2.Yoder BJ, Cao SG, Norris A, Miller JS, Ratovoson F, Razafitsalama J, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2007;70:342–346. doi: 10.1021/np060484y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neighbors JD, Beutler JA, Wiemer DF. J Org Chem. 2005;70:925–931. doi: 10.1021/jo048444r. [DOI] [PubMed] [Google Scholar]
- 4.Neighbors JD, Salnikova MS, Beutler JA, Wiemer DF. Bioorg Med Chem. 2006;14:1771–1784. doi: 10.1016/j.bmc.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 5.Mente NR, Wiemer AJ, Neighbors JD, Beutler JA, Hohl RJ, Wiemer DF. Bioorg Med Chem Lett. 2007;17:911–915. doi: 10.1016/j.bmcl.2006.11.096. [DOI] [PubMed] [Google Scholar]
- 6.Topczewski JJ, Neighbors JD, Wiemer DF. J Org Chem. 2009;74:6965–6972. doi: 10.1021/jo901161m. [DOI] [PubMed] [Google Scholar]
- 7.Kuder CH, Neighbors JD, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2009;17:4718–4723. doi: 10.1016/j.bmc.2009.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulrich NC, Kuder CH, Hohl RJ, Wiemer DF. Bioorg Med Chem Lett. 2010;20:6716–6720. doi: 10.1016/j.bmcl.2010.08.143. [DOI] [PubMed] [Google Scholar]
- 9.Topczewski JJ, Kodet JG, Wiemer DF. J Org Chem. 2011;76:909–919. doi: 10.1021/jo1022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nandy JP, Prakesch M, Khadem S, Reddy PT, Sharma U, Arya P. Chem Rev. 2009;109:1999–2060. doi: 10.1021/cr800188v. [DOI] [PubMed] [Google Scholar]
- 11.Humphrey GR, Kuethe JT. Chem Rev. 2006;106:2875–2911. doi: 10.1021/cr0505270. [DOI] [PubMed] [Google Scholar]
- 12.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Advanced Drug Delivery Reviews. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 13.Vieth M, Siegel MG, Higgs RE, Watson IA, Robertson DH, Savin KA, Durst GL, Hipskind PA. J Med Chem. 2004;47:224–232. doi: 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]
- 14.Kishida N, Kamitanaka T, Fusayasu M, Sunamura T, Matsuda T, Osawa T, Harada T. Tetrahedron. 2010;66:5059–5064. [Google Scholar]
- 15.Katritzky AR, Akutagawa K. Tetrahedron Lett. 1985;26:5935–5938. [Google Scholar]
- 16.Naka H, Akagi Y, Yamada K, Imahori T, Kasahara T, Kondo Y. European Journal of Organic Chemistry. 2007:4635–4637. [Google Scholar]
- 17.Treadwell EM, Cermak SC, Wiemer DF. J Org Chem. 1999;64:8718–8723. [Google Scholar]
- 18.Kannadasan S, Srinivasan PC. Tetrahedron Lett. 2002;43:3149–3150. [Google Scholar]
- 19.Greco MN, Hawkins MJ, Powell ET, Almond HR, de Garavilla L, Hall J, Minor LK, Wang YP, Corcoran TW, Di Cera E, Cantwell AM, Savvides SN, Damiano BP, Maryanoff BE. J Med Chem. 2007;50:1727–1730. doi: 10.1021/jm0700619. [DOI] [PubMed] [Google Scholar]
- 20.Taber DF, Tirunahari PK. Tetrahedron. 2011;67:7195–7210. doi: 10.1016/j.tet.2011.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kraus GA, Guo HT. Organic Letters. 2008;10:3061–3063. doi: 10.1021/ol801034x. [DOI] [PubMed] [Google Scholar]
- 22.Kraus GA, Guo HT. J Org Chem. 2009;74:5337–5341. doi: 10.1021/jo900718g. [DOI] [PubMed] [Google Scholar]
- 23.El-Rayyes NR. J Prakt Chemie. 1973;315:295–299. [Google Scholar]
- 24.Kim M, Vedejs E. J Org Chem. 2004;69:6945–6948. doi: 10.1021/jo040191e. [DOI] [PubMed] [Google Scholar]
- 25.Lee JH, Kim I. J Org Chem. 2013;78:1283–1288. doi: 10.1021/jo302590a. [DOI] [PubMed] [Google Scholar]
- 26.Topczewski JJ, Kuder CH, Neighbors JD, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2010;18:6734–6741. doi: 10.1016/j.bmc.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Rayyes NR, Al-Salman NA. J Prakt Chem. 1976;318:816–822. [Google Scholar]
- 28.Mente NR, Neighbors JD, Wiemer DF. J Org Chem. 2008;73:7963–7970. doi: 10.1021/jo800951q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takatori K, Nishihara M, Nishiyama Y, Kajiwara M. Tetrahedron. 1998;54:15861–15869. [Google Scholar]
- 30.Kelly TR, Zhao YJ, Cavero M, Torneiro M. Organic Letters. 2000;2:3735–3737. doi: 10.1021/ol006649q. [DOI] [PubMed] [Google Scholar]
- 31.Kanekiyo N, Kuwada T, Choshi T, Nobuhiro J, Hibino S. J Org Chem. 2001;66:8793–8798. doi: 10.1021/jo0105585. [DOI] [PubMed] [Google Scholar]
- 32.Rawal VH, Cava MP. Tetrahedron Lett. 1985;26:6141–6142. [Google Scholar]
- 33.Nagarathnam D, Srinivasan PC. Synthesis-Stuttgart. 1982:926–927. [Google Scholar]
- 34.Mohan B, Nagarathnam D, Vedachalam M, Srinivasan PC. Synthesis-Stuttgart. 1985:188–190. [Google Scholar]
- 35.Nagarathnam D. J Heterocycl Chem. 1992;29:1371–1373. [Google Scholar]
- 36.Jesudoss K, Srinivasan PC. Synth Commun. 1994;24:1701–1708. [Google Scholar]
- 37.Kodet JG. Ph D Thesis. University of Iowa; 2010. [Google Scholar]
- 38.Elder AM, Rich DH. Organic Letters. 1999;1:1443–1446. doi: 10.1021/ol990990x. [DOI] [PubMed] [Google Scholar]
- 39.Muratake H, Natsume M. Heterocycles. 1989;29:783–794. [Google Scholar]
- 40.Hutchins RO, Suchismita, Zipkin RE, Taffer IM, Sivakumar R, Monaghan A, Elisseou EM. Tetrahedron Lett. 1989;30:55–56. [Google Scholar]
- 41.Muratake H, Natsume M. Tetrahedron Lett. 1989;30:1815–1818. [Google Scholar]
- 42.Cheng CC. Med Hypotheses. 1986;20:157–172. doi: 10.1016/0306-9877(86)90122-2. [DOI] [PubMed] [Google Scholar]
- 43.Saulnier MG, Gribble GW. J Org Chem. 1982;47:2810–2812. [Google Scholar]
- 44.Kozikowski AP, Chen YY. J Org Chem. 1981;46:5248–5250. [Google Scholar]
- 45.Brubaker AN, Colley M. J Med Chem. 1986;29:1528–1531. doi: 10.1021/jm00158a036. [DOI] [PubMed] [Google Scholar]
- 46.Yang HJ, Li BG, Cai XH, Qi HY, Luo YG, Zhang GL. J Nat Prod. 2006;69:1531–1538. doi: 10.1021/np060159a. [DOI] [PubMed] [Google Scholar]
- 47.Karrer P, Asmis H. Helv Chim Acta. 1952;35:1926–1931. [Google Scholar]
- 48.Westermaier M, Mayr H. Organic Letters. 2006;8:4791–4794. doi: 10.1021/ol0618555. [DOI] [PubMed] [Google Scholar]
- 49.Kimura M, Futamata M, Mukai R, Tamaru Y. J Am Chem Soc. 2005;127:4592–4593. doi: 10.1021/ja0501161. [DOI] [PubMed] [Google Scholar]
- 50.Wenkert E, Angell EC, Ferreira VF, Michelotti EL, Piettre SR, Sheu JH, Swindell CS. J Org Chem. 1986;51:2343–2351. [Google Scholar]
- 51.Amat M, Hadida S, Sathyanarayana S, Bosch J. J Org Chem. 1994;59:10–11. doi: 10.1021/jo952251+. [DOI] [PubMed] [Google Scholar]
- 52.Gruber S, Zaitsev AB, Worle M, Pregosin PS, Veiros LF. Organometallics. 2008;27:3796–3805. [Google Scholar]
- 53.Zhu XW, Ganesan A. J Org Chem. 2002;67:2705–2708. doi: 10.1021/jo010996b. [DOI] [PubMed] [Google Scholar]
- 54.Snieckus V. Chem Rev. 1990;90:879–933. [Google Scholar]
- 55.Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Anticancer Res. 2004;24:2783–2840. [PubMed] [Google Scholar]
- 56.Ioset JR, Marston A, Gupta MP, Hostettmann K. J Nat Prod. 2001;64:710–715. doi: 10.1021/np000597w. [DOI] [PubMed] [Google Scholar]
- 57.Sobolev VS. J Agric Food Chem. 2013;61:1850–1858. doi: 10.1021/jf3054752. [DOI] [PubMed] [Google Scholar]
- 58.Belofsky G, French AN, Wallace DR, Dodson SL. J Nat Prod. 2004;67:26–30. doi: 10.1021/np030258d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.