

Abstract
We report a young man presenting with jaundice and severe debilitating intrahepatic cholestasis 7 months before the diagnosis of Hodgkin's lymphoma. Serum gamma-glutamyl transferase (GGT) activity was not raised. Liver biopsy demonstrated deficiency of canalicular GGT and bile salt export pump expression, which suggested “benign” recurrent intrahepatic cholestasis. Direct sequencing of genomic DNA was therefore undertaken to look for mutations in ATP8B1 and ABCB11. Cholestasis and pruritus are well recognized presenting features of Hodgkin's lymphoma. However, striking in this case is that the intrahepatic cholestasis presented and resolved 7 months before the diagnosis. Furthermore, 4 polymorphisms were identified in ATP8B1 in this patient—c.696T > C (rs319438), c.811A > C (rs319438), c.2855G > A (rs1296811) and c.3454G > A (rs222581)—and two polymorphisms in ABCB11—c.1331T > C (rs2287622) and c.3084A > G (rs497692); 2 of which have been associated with intrahepatic cholestasis of pregnancy. We therefore postulate that these polymorphisms predisposed this patient to the development of intrahepatic cholestasis within the abnormal pro-inflammatory cytokine milieu typical for Hodgkin's lymphoma. This case shows for the first time that some polymorphisms in ABCB11 and ATP8B1 may predispose to the development of intrahepatic cholestasis in Hodgkin's lymphoma. It also demonstrates the importance of close clinical surveillance for the development of Hodgkin's lymphoma in patients presenting with unexplained intrahepatic cholestasis.
Keywords: intrahepatic cholestasis, lymphoma, Hodgkin's, polymorphism
Abbreviations: GGT, gamma-glutamyl transferase; CT, computerized tomography; BSEP, bile salt export pump; BRIC, “benign” recurrent intrahepatic cholestasis; UDCA, ursodeoxycholic acid; PFIC, progressive familial intrahepatic cholestasis; ICP, intrahepatic cholestasis of pregnancy
An obese 17-year-old (BMI 42) man lost 30 kg over six months in a healthy-eating and exercise programme that commenced in January 2008. Following this, he suddenly developed jaundice, pruritus and an urticarial rash. No precipitant was identified and the rash resolved with a 7-day course of chlorphenamine and prednisolone. The persistence of jaundice and debilitating pruritus prompted referral to King's College Hospital Liver Unit in June 2008. Examination was notable for jaundice and excoriations from scratching but there was no evidence of hepatic encephalopathy. Renal biochemistry, full blood count and INR were normal. Liver biochemistry revealed conjugated hyperbilirubinemia (363 μmol/L [normal range < 20]), with elevated serum alkaline phosphatase (596 IU/L [normal range < 130]) and lactate dehydrogenase (298 IU/L [normal range < 240]). Serum transaminases and gamma-glutamyl transferase (GGT) were normal. Infectious, autoimmune and metabolic causes of hepatobiliary disease were excluded. Ultrasonography and computerized tomography (CT) of thorax, abdomen and pelvis were unremarkable, with no organomegaly or lymphadenopathy. Liver biopsy revealed severe canalicular cholestasis with mild inflammation and no evidence of malignancy (Figure 1). Canalicular GGT and bile salt export pump (BSEP) expression was deficient in the biopsy specimen. These findings, in the context of normal serum GGT, suggested “benign” recurrent intrahepatic cholestasis (BRIC). Direct sequencing of genomic DNA was therefore undertaken to look for mutations in ATP8B1 and ABCB11, genes mutated in forms of BRIC, by conventional sequencing of all exons and exon-intron boundaries. Treatment with 20 mg/kg of ursodeoxycholic acid (UDCA) resulted in rapid resolution of the hyperbilirubinemia (Figure 2) and pruritus by September 2008.
Figure 1.

Transjugular liver biopsy specimen. A, Centrilobular cholestasis, with small, tidy hepatocytes and pale bile plugs in canaliculi (“bland cholestasis”). Hematoxylin/eosin. B, Bile salt export pump (BSEP) expression can be demonstrated along canalicular margins. C and D, lack of gamma-glutamyl transferase (GGT) and carcinoembryonic antigen (CEA) expression, respectively, at canaliculi; normal expression pattern (not shown) for both GGT and CEA matches that of BSEP. Leica Bond Max immunostainer protocols using rabbit polyclonal anti-human BSEP (Sigma–Aldrich HPA019035), mouse monoclonal anti-human GGT1 (Abnova H00002678-M01, clone 1F9, and rabbit polyclonal anti-human CEA (DAKO IR526/IS526); EnVision/Dako development; haematoxylin counterstain. Original magnification 200×, all images.
Figure 2.
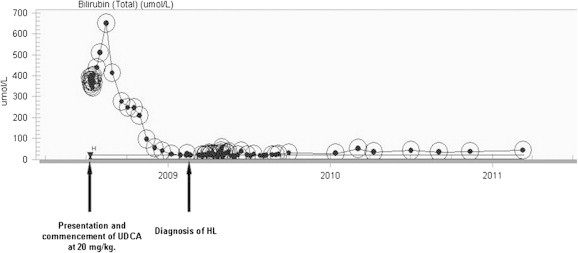
Changes in bilirubin levels over time. Bilirubin peaked at 650 μmol/L in August 2008 before falling dramatically back to normal following commencement of UDCA 20 mg/kg.
Despite resolution of his symptoms, he continued to have an unintentional weight loss of 8 kg by October 2008. This prompted further investigation with a bone marrow biopsy, which revealed reactive changes only. His weight thereafter stabilized and he suffered no recurrence of jaundice or pruritus. In January 2009, he suddenly developed unilateral cervical lymphadenopathy. A lymph node biopsy demonstrated nodular sclerosing Hodgkin's lymphoma. CT showed lymphadenopathy above and below the diaphragm (stage IIIb). He was treated with chemotherapy and involved-field radiotherapy that induced a complete remission that persists (January 2013).
Cholestasis and pruritus are well reported presenting features of Hodgkin's lymphoma. The pruritus is often precipitated by hot baths causing eosinophil release. However, striking in this case is that the intrahepatic cholestasis presented and resolved 7 months before the diagnosis. In Hodgkin's lymphoma -related cholestasis, GGT activity may be high or low1,2; in our patient it was low. Defects in hepatocyte canalicular membrane transport proteins encoded by ATP8B1 and ABCB11 cause low GGT cholestasis (other genes may contribute). Three phenotypes are associated with mutations in these genes: progressive familial intrahepatic cholestasis (PFIC) with severe neonatal jaundice, intractable pruritus and fat malabsorption with progression to cirrhosis; BRIC with intermittent jaundice but no progression to chronic liver disease; and intrahepatic cholestasis of pregnancy (ICP), with mutations that terminate transcription more likely to yield PFIC and substitution mutations more likely to yield BRIC or ICP.
Intrahepatic cholestasis associated with malignancy and linked to mutations in ABCB11 or ATP8B1 has not been published to date. A single nucleotide polymorphism c.1331C > T (V444A) in ABCB11, encoding BSEP, potentially causing jaundice as the presenting symptom of acute lymphoblastic leukemia, has only been reported in abstract form.3
No known pathogenic mutations were found in our patient on sequencing. However, four known polymorphisms were identified in ATP8B1—c.696T > C (rs319438), c.811A > C (rs319438), c.2855G > A (rs1296811) and c.3454G > A (rs222581) —and two known polymorphisms in ABCB11—c.1331T > C (rs2287622) and c.3084A > G (rs497692) (Table 1). Two of these polymorphisms (underlined) have been reported as associated with ICP.4 We therefore postulate that these polymorphisms may have predisposed this patient to the development of intrahepatic cholestasis within the abnormal pro-inflammatory cytokine milieu typical for Hodgkin's lymphoma, which preceded the clinical diagnosis by 7 months. This case extends the existing evidence base and shows for the first time that some of the known single nucleotide polymorphisms in ABCB11 and ATP8B1 may predispose to the development of intrahepatic cholestasis in Hodgkin's lymphoma. It also demonstrates the importance of close clinical surveillance for the development of Hodgkin's lymphoma in patients presenting with unexplained intrahepatic cholestasis.
Table 1.
A summary of genetic findings following full sequencing of ABCB11 and ATP8B1 in this patient. Highlighted rows describe polymorphisms, which have previously been reported to be associated with intrahepatic cholestasis of pregnancy.
| Exon | DNA change | Protein | Zygosity | NCBI dbSNP reference |
|---|---|---|---|---|
| ABCB11 | ||||
| Ex10 | c.909-15A > G | Heterozygous | rs2287618 | |
| Ex13 | c.1331T > C | p.Val444Ala | Heterozygous | rs2287622 |
| Ex14 | c.1638 + 32T > C | Heterozygous | rs2241340 | |
| c.1638 + 80C > T | Heterozygous | rs2241341 | ||
| Ex19 | c.2179-17C > A | Heterozygous | rs853772 | |
| Ex20 | c.2344-157T > G | Heterozygous | # | |
| c.2344-17T > C | Heterozygous | # | ||
| Ex24 | c.3084A > G | p.Ala1028Ala | Heterozygous | rs497692 |
| Ex28 | c.3766-34A > G | Heterozygous | rs579275 | |
| c.*236A > G | Heterozygous | # | ||
| ATP8B1 | ||||
| Ex7 | c.696T > C | p.Asp232Asp | Homozygous | rs319438 |
| Ex9 | c.811A > C | p.Arg271Arg | Homozygous | rs319438 |
| Ex22 | c.2855G > A | p.Arg952Gln | Heterozygous | rs1296811 |
| Ex26 | c.3454G > A | p.Ala1152Thr | Homozygous | rs222581 |
# not on dbSNP but observed frequently by this laboratory.
Conflicts of interest
All authors have none to declare.
References
- 1.Ballonoff A., Kavanagh B., Nash R. Hodgkin lymphoma-related vanishing bile duct syndrome and idiopathic cholestasis: statistical analysis of all published cases and literature review. Acta Oncol. 2008;47(5):962–970. doi: 10.1080/02841860701644078. [DOI] [PubMed] [Google Scholar]
- 2.Barta S.K., Yahalom J., Shia J., Hamlin P.A. Idiopathic cholestasis as a paraneoplastic phenomenon in Hodgkin's lymphoma. Clin Lymphoma Myeloma. 2006 July;7(1):77–82. doi: 10.3816/CLM.2006.n.044. [DOI] [PubMed] [Google Scholar]
- 3.Jahnel J., Deutschmann A., Urban C. The bile salt export transporter (BSEP) polymorphism V444A potentially causes jaundice as presenting symptom of acute lymphoblastic leukemia (ALL) [abstr PO-H-0298] J Pediatr Gastroenterol Nutr. 2011;52:E1–E223. [Google Scholar]
- 4.Dixon P.H., van Mil S.W., Chambers J. Contribution of variant alleles of ABCB11 to susceptibility to intrahepatic cholestasis of pregnancy. Gut. 2009 April;58(4):537–544. doi: 10.1136/gut.2008.159541. [DOI] [PubMed] [Google Scholar]