

Abstract
Development of idiosyncratic hepatotoxicity is an intricate process involving both concurrent as well as sequential events determining the direction of the pathways, degree of liver injury and its outcome. Decades of clinical observation have identified a number of drug and host related factors that are associated with an increased risk of antituberculous drug-induced hepatotoxicity, although majority of the studies are retrospective with varied case definitions and sample sizes. Investigations on genetic susceptibility to hepatotoxicity have so far focused on formation and accumulation reactive metabolite as well as factors that contribute to cellular antioxidant defense mechanisms and the environment which can modulate the threshold for hepatocyte death secondary to oxidative stress. Recent advances in pharmacogenetics have promised the development of refined algorithms including drug, host and environmental risk factors that allow better tailoring of medications based on accurate estimates of risk–benefit ratio. Future investigations exploring the pathogenesis of hepatotoxicity should be performed using human tissue and samples whenever possible, so that the novel findings can be translated readily into clinical applications.
Keywords: drug-induced liver injury, hepatotoxicity, tuberculosis, genetic, pathogenesis
Abbreviations: TB, tuberculosis; WHO, World Health Organization; DILI, drug-induced liver injury; ALT, alanine transaminase; AST, aspartate transaminase; ULN, upper limit of normal range; FDA, Food and Drug Administration; INH, isoniazid; NAT2, N-acetyltransferase 2; SH, sulfhydryl; PXR, pregnane X receptor; CYP, cytochrome P450; GST, glutathione S-transferase; ART, anti-retroviral therapy; BSEP, bile salt exporter pump; HBV, hepatitis B virus; HCV, hepatitis C virus; HAART, highly active anti-retroviral therapy; MHC, major histocompatibility complex; ROS, reactive oxygen species; Nrf2, nuclear factor erythroid 2-related factor-2; MnSOD, manganese superoxide dismutase; MPT, mitochondrial permeability transition; ATS, American Thoracic Society; BTS, British Thoracic Society; NICE, National Institute for Clinical Excellence; DOTS, directly observed short-course therapy; NAC, N-acetyl cysteine; OR, odds ratio; BTB, broad complex, tramtrack, bric-a-brac domain; CNC, cap‘n’collar type of basic region; ATP, adenosine triphosphate; HLA, human leukocyte antigen; SNP, single-nucleotide polymorphism
Tuberculosis (TB) remains a major global health problem despite the availability of highly efficacious treatment for decades. World Health Organization (WHO) declared TB a global public health emergency in 1993, at a time when an estimated 7–8 million new cases and 1.3–1.6 million deaths occurred each year. In 2010, there was an estimated 8.8 million new cases reported and 1.4 million deaths including deaths from TB among HIV-positive people. In India, TB is a major public health issue with an estimated prevalence of 256 per 100,000 population and 26 per 100,000 population dying of TB.1 Although about 85% of TB cases are successfully treated, treatment-related adverse events including hepatotoxicity, skin reactions, gastrointestinal and neurological disorders account for significant morbidity leading to reduced effectiveness of therapy. Hepatotoxicity is the commonest of all adverse effect leading to drug discontinuation in 11% of patients treated with combination of isoniazid, rifampicin and pyrazinamide.2
Anti-TB drugs are one of the commonest group underlying idiosyncratic hepatotoxicity worldwide.3–5 The incidence of anti-TB drug induced hepatotoxicity varies widely dependent upon the characteristics of the particular cohort, drug regimens involved, threshold used to define hepatotoxicity, monitoring and reporting practices. Overall, hepatotoxicity attributed to anti-TB drugs has been reported in 5%–28% of people treated with anti-TB drugs.3 However, it is difficult to judge how many of these fit into a more recent international consensus case definition of drug-induced liver injury (DILI).6 Majority of the reports have used an elevated alanine (ALT) or aspartate transaminase (AST) of 3 times upper limit of normal range (ULN) with symptoms (abdominal pain, nausea, vomiting, unexplained fatigue or jaundice) attributable to liver injury or 5 times ULN of ALT or AST without symptoms to define hepatotoxicity.7
Up to 20% of the patients receiving isoniazid either in single or combination therapy develop transient asymptomatic elevation in liver enzymes, which settle with continued use of the drug.8,9 Manifestation of the anti-TB drug induced hepatotoxicity can vary from asymptomatic elevations in the liver enzymes to fulminant liver failure.7,10 DILI generally takes hepatocellular pattern. Burden of anti-TB drug related hepatotoxicity is based not only on its prevalence or incidence, but also on its severity and outcome. The median interval from treatment initiation of drug to development of clinical symptoms is 16 weeks (range 6 weeks–6 months).11–13 Anti-TB drug induced fulminant liver failure appears to have worse outcome when compared with that related to acute viral hepatitis with a case fatality rate between 0.042 and 0.07 per 1000 persons at any given time during therapy.3,4,14–16 Liver biopsy specimens when available reveal lobular hepatitis, sub massive to massive necrosis and hydropic degeneration of hepatocytes in severe cases. In cases associated with rifampicin hepatotoxicity, focal hepatocellular necrosis and apoptosis in zone 3 and cholestasis have been noted on histology.
Despite decades of use and large number of patients exposed to anti-TB drugs worldwide, pathogenesis underlying hepatotoxicity is poorly understood. Investigations aimed at identifying drug related, host genetic and environmental factors associated with susceptibility to hepatotoxicity as well as those exploring the potential mechanisms leading to DILI may allow clinicians to develop strategies to reduce the occurrence of hepatotoxicity and its adverse outcome.
Risk factors associated with hepatotoxicity
Drug Related Factors
It is difficult to estimate the incidence of hepatotoxicity due to individual agents as majority of patients are on combination of medications throughout the course of anti-TB therapy. While isoniazid, rifampicin and pyrazinamide are known to cause hepatotoxicity, ethambutol and streptomycin are considered not to be hepatotoxic. Information related to hepatotoxicity from isoniazid (INH), rifampicin17 and pyrazinamide18,19 are derived from observations made during monotherapy for latent TB or when these drugs were combined with apparently non-hepatotoxic medications. Rifampicin induced DILI has been well documented when it is used to treat pruritus in patients with primary biliary cirrhosis.20 However, this may be an overestimate and excess risk could potentially be related to the underlying liver disease. Other studies where rifampicin has been used alone in prophylaxis in treatment of latent TB has demonstrated relative low risk of DILI due to rifampicin.21,22
INH is the most common drug associated with toxicity. Four large population based observational studies have shown that the incidence of isoniazid hepatotoxicity when used as monotherapy (in treatment of latent infection) to be in the range of 0.1%–0.56%.11,13,23,24 A review based on the data from U.S. Food and Drug Administration (FDA) estimated that 23.2 per 100,000 people die receiving INH based prophylactic therapy.25 In a meta-analysis, isoniazid was more likely to be associated with hepatotoxicity (odds ratio (OR) 1.6) even in the absence of rifampicin, but the combination of these two drugs was associated with higher rate of hepatotoxicity (OR 2.6) when compared to each drug on its own.26 Daily dosing regimens haven't been shown to be associated higher risk of hepatotoxicity than three times a week regimes.27
Intrinsic Toxicity in Animals
Pre-clinical phase of drug development includes toxicity studies in animals. The principle behind these experiments is that the use of high doses of a compound in a group of animals should reveal intrinsic potential toxicity (of a compound) that would occur with a low frequency in patients receiving much smaller therapeutic doses. These experiments generally detect potential hepatotoxicity inherent to the compound and allow elimination of those associated with unacceptable risk. Following these principles, a few animal models of anti-TB DILI have been described.
In male Wister rats, evidence of liver injury appears on treatment with isoniazid at a dose of 100 mg/kg for 21 days and a metabolite of isoniazid, hydrazine plays an important role in liver injury.28 In another example, liver damage was induced by a combination of isoniazid (50 mg/kg) and rifampicin (100 mg/kg) in mice.29 Authors argued that this model supported the hypothesis that mitochondrial redox changes are crucial events in apoptotic liver cell injury in hepatotoxicity due to anti-TB drugs. However, the doses used in this model were about 10 times the human doses on a milligram per kilogram basis. Histological changes seen in the model was that of hepatic steatosis unlike that is seen in human DILI. Hepatocyte necrosis, a prominent finding in human DILI was achieved only by profound glutathione depletion induced by pretreatment with phorone.
Most idiosyncratic DILI by definition are unexpected based on the pharmacological action of a drug.30 As in the case of anti-TB drug induced hepatotoxicity, idiosyncrasy implies an individual's unique response to a particular drug and is expected to be dependent on host factors. Therefore, such combination of circumstances is unlikely to be recreated in animal models. Unsurprisingly, these animal models do not closely reflect the phenotypic features of anti-TB DILI in humans.
Drug Biotransformation, Detoxification and Elimination
Formation of reactive metabolites has been implicated in a range of clinical toxicities including a proportion of those classified as ‘idiosyncratic’ DILI. Reactive metabolites are generally electrophiles. When they escape detoxification, they react with nucleophilic groups such as lysine and cysteine on cellular proteins. Covalently modified cellular proteins can either be repaired or degraded. If these processes fail, drug-metabolite adduct formation itself impairs important cellular function leading to the manifestation target organ injury. Generation of reactive metabolites followed by covalent protein binding can also lead to immune mediated injury.
High levels of reactive metabolite formation in an individual may be due to high levels or increased activities of enzymes involved in the biotransformation of a drug into a reactive metabolite; these are generally phase I cytochrome P450 enzyme involved in oxidation, reduction or hydrolysis. Alternatively, individuals may have low levels or reduced activities of enzymes that detoxify reactive metabolites; usually mediated by phase II enzymes through a process of glucuronidation, sulfation, acetylation or glutathione conjugation. Phase III of drug disposition is mediated by transporter molecules or proteins which facilitate excretion of the water soluble metabolites into bile or systemic circulation. Most first line anti-TB drugs are lipophilic and their biotransformation involves their conversion into water soluble compounds and subsequent elimination. Hepatotoxicity appears to involve reactive metabolite formation and accumulation rather than the direct effect of the parent drug itself.8,31
Isoniazid
INH is metabolized and cleared predominantly in the liver. The key enzymes in the metabolic pathway, N-acetyltransferase 2 (NAT2) and microsomal enzyme cytochrome P4502E1 (CYP2E1) determine the risk of hepatotoxicity. As illustrated in Figure 1, NAT2 is responsible for metabolism of isoniazid to acetyl isoniazid, which in turn is hydrolyzed to acetyl hydrazine. Latter could be oxidized by CYP2E1 to form N-hydroxy-acetyl hydrazine, which further dehydrates to yield acetyl diazine. Acetyl diazine may itself be the toxic metabolite or may break down to reactive acetyl onium ion, acetyl radical and ketene, which could bind covalently with hepatic macromolecules resulting in liver injury. The enzyme NAT2 is also responsible for further acetylation of acetyl hydrazine to non-toxic diacetyl hydrazine. Therefore, slow acetylation results not only in accumulation of the parent compound, but also of mono-acetyl hydrazine. Acetylation of acetyl hydrazine is further suppressed by INH itself. In addition, direct hydrolysis of INH without acetylation produces hydrazine that could cause liver injury.32 INH metabolism through this minor pathway is increased ten-fold in slow acetylators, especially in association with rifampicin.33 The hepatic NAT2 is polymorphic in humans, and the presence of any two of the several variant alleles of the NAT2 gene is associated with slow acetylation phenotype, whereas rapid acetylators have one or more wild-type NAT2*4 alleles.34 Acetylation activity in vitro is progressively reduced in association with NAT2*4 > NAT2*7 > NAT2*6 > NAT2*5 alleles.35 In their first study involving genotyping acetylator status in 224 subjects on anti-TB therapy, Huang, et al found that patients possessing NAT2 genotypes associated with slow acetylation had a four-fold risk of developing INH-induced hepatotoxicity36 and recent meta-analysis of 14 studies involving 474 cases and 1446 controls have drawn similar conclusions with an odds ratio of 4.6 for slow acetylators.37 In addition, slow acetylators were prone to develop more severe hepatotoxicity than rapid acetylators. Those with NAT2*6/6 and NAT2*6/7 genotypes had a significantly higher risk than other genotypes. The same group investigated whether genetic polymorphism of CYP2E1 influenced susceptibility to anti-TB drug induced hepatotoxicity. During the administration of INH, CYP2E1 activity is inhibited. Under this inhibitory effect of INH, subjects who were homozygous for CYP2E1 c1 allele (wild type) had higher enzyme activity compared those with one or more CYP2E1 c2 allele.38 A study including 318 subjects on anti-TB therapy showed that subjects with CYP2E1 c1/c1 were 2.5 times more likely to develop hepatotoxicity when compared with the other genotypes. The risk of hepatotoxicity increased 7-fold when CYP2E1 c1/c1 was combined with slow-acetylator status. A similar study looking at 218 patients receiving ATT by Bose et al in India examined the role of the NAT2 and CYP2E1 (promoter and intron 6 region) polymorphisms in ATT hepatotoxicity. There was a higher prevalence of NAT2*5/*7 and NAT2*6/*7 genotypes (slow-acetylator) genotypes in anti-TB DILI group. CYP2E1 c1/c1 were present in both DILI and non-DILI groups, however CYP2E1 C/D or C/C genotypes 3 times more likely to develop anti-TB DILI. The same study also demonstrated that slow-acetylator status (NAT2 gene polymorphism) and the CYP2E1 C/D or C/C genotype together showed a higher frequency in DILI.39
Figure 1.
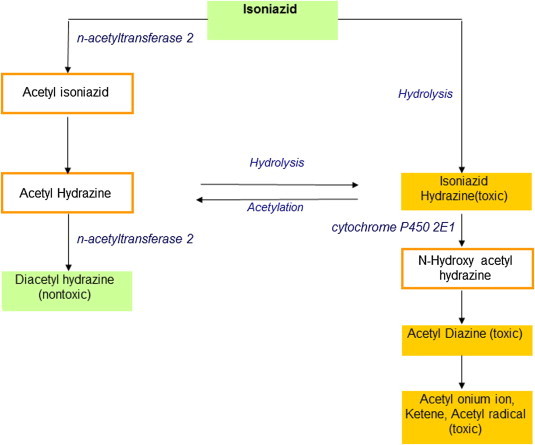
Pathways involved in the metabolism of isoniazid.
Glutathione plays an important protective role as an intracellular free radical scavenger by conjugating with toxic reactive metabolites that are generated from biotransformation of drugs and xenobiotics. Sulfhydryl (SH) conjugation of the metabolites facilitates their elimination from the body, and so reduces the potential for toxicity. Deficiency in GST activity, because of homozygous null mutations at GSTM1 and GSTT1 loci, modulate susceptibility to drug- and xenobiotic-induced hepatotoxicity. In a case control study involving 33 cases and equal number of controls Roy et al examined the frequency of GSTM1 and GSTT1 null mutations and noted GSTM1 null mutations twice as common in the cases with anti-TB DILI.40 In another study involving Caucasian patients, anti-TB DILI was 2.6 times more likely in those with GSTT1 null mutations.41 The data so far suggest that INH hepatotoxicity is caused by the reactive metabolites. A recent review on mechanism of INH hepatotoxicity highlights the role of immune mediated idiosyncrasy as a mechanism that is explained by the adaptive responses of liver to INH and heterogeneity of clinical picture of INH hepatotoxicity.42
Rifampicin
Rifampicin is well absorbed from the stomach and metabolized in the liver by desacetylation to desacetyl rifampicin43,44 and a separate pathway of hydrolysis produces 3-formyl rifampicin.45,46 Desacetyl rifampicin is more polar than the parent compound, and microbiologically active. This metabolite accounts for the majority of the antibacterial activity in the bile. Rifampicin is almost equally excreted in the bile and urine. These metabolites are non-toxic. Rifampicin is associated with hepatocellular pattern of DILI38,43,44 and more often it potentiates the hepatotoxicity of other anti-TB drugs.47,48 The xeno sensing pregnane X receptor (PXR) is a member of the nuclear receptor superfamily of ligand-dependent transcription factors that can be activated by a variety of drugs including rifampicin. Activated PXR binds to response elements in the promoters and up-regulates the transcription of phase I and II drug metabolizing enzymes such as cytochrome P450 (CYP)s and glutathione S-transferases (GSTs), and transporters (involved in phase III). Rifampicin is a potent inducer of several metabolic enzyme pathways in particular cytochrome P450 (CYP3A4) system via the hepatocyte PXR.49,50 This activation of the CYP3A4 leads to increased metabolism of isoniazid yielding toxic metabolites thus explains the potentiating effect of rifampicin in anti-TB drug induced hepatotoxicity. Rifampicin also induces isoniazid hydrolases, leading to increased hydrazine production especially in slow acetylators thus increasing the toxicity when used in combination with isoniazid.33 Processes that are involved in the excretion and elimination of the drug metabolite are grouped as phase III of drug disposition. Transporter ABCB1 is responsible for the transport of many anti-retroviral and anti-TB drugs including rifampicin and ethambutol. ABCB1 3435T variant allele is reported to lower expression level and protein folding thereby altering the structure of substrate binding sites to and decreased transport activity.51 In a study involving patients on treatment with a combination of anti-TB and anti-retroviral therapy (ART), the proportion of those homozygous for ABCB1 3435TT genotype was 3-fold higher in those who developed DILI.52 Rifampicin occasionally interferes with bilirubin uptake and results in transient unconjugated hyperbilirubinemia without hepatocyte damage. However more commonly, it does contribute to conjugated hyperbilirubinemia by interfering with the bilirubin excretion by inhibiting the bile salt exporter pump (BSEP).53–55
Pyrazinamide
Pyrazinamide is a nicotinic acid derivative. It is deamidated to pyrazinoic acid. This is further oxidized by xanthine oxidase to 5-hydroxy pyrazinoic acid.56,57 The metabolites are excreted via the kidneys. The half-life of pyrazinamide is longer than isoniazid and rifampicin; it is prolonged even further in the presence of underlying liver disease and when used with other drugs that inhibit xanthine oxidase such as allopurinol. The toxicity of pyrazinamide is both dose dependent with a higher dose at 40–50 mg/kg being associated with a greater frequency of hepatotoxicity than the doses used in current regimens (25–35 mg/kg). In murine models, pyrazinamide inhibited CYP45058 activity and NAD59 levels were altered in association with free radical species mediated hepatotoxicity.
Fluoroquinolones
Fluoroquinolones have been used as second line agents in the context of multi-drug resistant TB and in the events of hepatotoxicity due to first line agents. Quinolones are either metabolized in the liver (as with ciprofloxacin) or are excreted unchanged by the kidneys (as with levofloxacin). With an exception of trovafloxacin which is now withdrawn, fluoroquinolone induced hepatotoxicity is very rare and identifiable only through large-scale studies or worldwide pharmacovigilance reporting.60 Isolated cases of significant hepatotoxicity have been reported with ciprofloxacin,61 levofloxacin,62 gatifloxacin.63,64 The mechanism of hepatotoxicity is thought to be due to hypersensitivity reaction often associated with peripheral eosinophilia and fever. Introduction of fluoroquinolones didn't cause additional hepatotoxicity when used in patients with hepatitis induced by first line anti-TB drugs.65 In patients with pre-existent liver disease, use of ofloxacin has been found to be safe and effective.66
Host Related Risk Factors
Although several risk factors have been associated with hepatotoxicity, robust conclusions can't be drawn due to marked variations in study design, cohort size and case definitions.
Age
Age has been associated with an increased risk of DILI. In one study including 519 patients on standard anti-TB medications, age over 60 years was associated with a 3.5-fold risk of DILI.2 Another study including 430 patients, pyrazinamide associated adverse events including DILI was 2.6-fold higher in those over the age of 60 years.67 In a cohort of over 3000 patients who were on INH monotherapy, frequency of DILI was higher in those aged 50 years or older.11 The severity of INH hepatotoxicity and consequent mortality has also been reported to be higher after the age of 50.8,16,25 In a case–control study, patients who developed DILI on anti-TB drugs were older (39 years) compared to those who did not (32 years).68 In another study, the incidence of hepatotoxicity was 17% in patients below 35 years of age and 33% in age above 35 years; in a multivariate analysis, age >35 years was the only independent variable for predicting anti-TB DILI.69 Older age is associated with decreased liver blood flow, changes in the drug distribution and metabolism, thus potentially reducing the effective clearance of the drugs.
Contrary to these, a meta-analysis reported a higher incidence of clinical hepatitis (6.9%) in children receiving INH and rifampicin, compared to 2.7% in adults.26 However, the high frequency of DILI in children was primarily derived from the inclusion of 3 small studies each with 22–60 patients, yet reporting a very high frequency of ‘clinical hepatitis’ in 25%–52% of all patients.
Gender
Women are more susceptible to DILI from anti-TB therapy67,70–72 with a reported 4-fold risk.73 Activity of CYP3A is higher in females rendering them more susceptible for hepatotoxicity.74 A trend toward increased incidence of INH hepatotoxicity has been noted in pregnant women in the third trimester and first three months post-partum.75
Nutritional Status
Malnutrition is common in TB and associated with higher incidence of anti-TB drug induced hepatotoxicity.76 A recent retrospective observational study revealed that a weight loss of 2 kg or more developing within 4 weeks during TB treatment is highly significant independent risk factor for DILI.77 An adequate intake of nutrients is important for the integrity of liver metabolism and detoxification of TB drugs, as the cytochrome P450 enzyme system is affected by nutrient intake, fasting and malnourished states.78,79
Alcohol Intake
Alcohol can induce enzymes and has potential to cause liver injury. Several studies have demonstrated that alcohol perpetuates anti-TB drug induced hepatotoxicity.80–82 This risk has been demonstrated even in patients receiving rifampicin for preventative therapy.83,84
Concomitant Infection
The observation that drug hypersensitivity is more common in patients with concomitant viral infection has led to the suggestion that mild inflammatory reaction due to co-existent infection can often act as a ‘danger signal’ which permits the development of initial events in the pathophysiological process into a full blown hepatotoxic reaction.85 Whether chronic infections other than TB increase the risk of DILI during anti-TB therapy has been investigated in several cohorts.
The risk of anti-TB drug induced hepatotoxicity is higher in patients with chronic hepatitis B virus (HBV) patients compared to uninfected subjects (16% vs 4.7% p < 0.001) and the severity was much higher in the HBV patients in this study (4.7% vs 2.5% p < 0.001).86 Studies also have shown that the severity of the hepatotoxicity is directly related to viral load at the time of initiation of anti-TB therapy.86
Similar to HBV infection, approximately 30% of all of hepatitis C virus (HCV) infected patients receiving treatment with anti-TB drugs developed hepatotoxicity compared to 11% in controls with a 5-fold relative risk of developing hepatotoxicity and severity associated directly with the viral load.87 Concomitant HIV infection also increases the risk of anti-TB drug induced hepatotoxicity significantly. This has been observed both before highly active anti-retroviral therapy (HAART) era and during HAART era. The hepatotoxicity in the pre-HAART era ranged from 4 to 15% and in the HAART era ranges from 4 to 27%.88 However, there have been several confounding factors such as intravenous drug use, alcoholism, viral hepatitis, HAART therapy causing hepatotoxicity, drug–drug interactions and liver injury due to immune reconstitution. Overall influence of HIV infection alone on the anti-TB therapy induced hepatotoxicity has been estimated to be 4 times higher than in controls and co-infection with HCV, increases this risk by 14-fold.88
Genetic Susceptibility
Anti-TB DILI remains unpredictable even when variables such as drug regimen and environmental factors are taken into account. Neither the drug related factors nor the concurrent risk factors adequately explain why, in the vast majority of cases, hepatotoxicity occurs during the early phase of anti-TB therapy. In addition, observations such as Asian males have double the rate of isoniazid hepatitis than white males and nearly 14 times that of black males11,12 indicate that genetic susceptibility may contribute substantially to the development of hepatotoxicity.
Single nucleotide polymorphisms in the genes coding for the drug metabolizing enzymes (such as NAT2 and CYP2E1) eventually leading to an excess formation, accumulation of reactive metabolites or their reduced clearance (such as ABCC1) have been associated with increased risk of anti-TB DILI (Table 1). Natural PXR protein variants have been associated with inter-individual variability of CYP3A4 expression89 and may contribute to individuals' susceptibility to anti-TB DILI. Magnitude of impact of reactive metabolites can be modified by cellular response to oxidative stress that is generated. Hence, polymorphisms in the genes which influence antioxidant defense processes (such as BACH1, MAFK, GST1 and MnSOD) appear to determine one's predisposition to DILI.
Table 1.
Studies that have demonstrated association of specific genotypes with anti-TB DILI.
| Drug | Genotype | Hazard ratio | Cases (n) | p Value |
|---|---|---|---|---|
| Isoniazid36 | NAT2*6/6 or NAT2*6/7 | 3.7 | 33 | 0.003 |
| Isoniazid38,39 | CYP2E1 c1/c1 | 2.5 | 49 | 0.009 |
| CYP2E1 C/D or C/C | 3.2 | 41 | 0.005 | |
| Anti-TB drugs and HAART52 | NAT2*4, *12 or *13 | 0.4 | 65 | 0.04 |
| ABCB1 3435T/T | 5.3 | 65 | 0.02 | |
| Isoniazid92 | MnSOD T/C or C/C | 2.5 | 63 | 0.02 |
| Isoniazid40,41,92 | GSTM1 null92 | 2.2 | 63 | 0.03 |
| GSTM1 null40 | 2.1 | 66 | <0.05 | |
| GSTT1 null41 | 2.6 | 95 | 0.03 | |
| Anti-TB drugs90 |
BACH1 C/C at rs2070401 MAFK G/A or A/A at rs4720833 |
9.7a | 100 | 0.0006 |
| Anti-TB drugs81 | HLA-DQB1*0201 | 1.9 | 56 | 0.01 |
| HLA-DQA1*0102 | 0.2 | 56 | <0.001 |
BACH1 gene encodes transcription regulator protein BACH1 (BTB and CNC homology 1).
MAFK gene encodes small Maf basic leucine zipper protein MafK.
Hazard ratio for the any of the two single-nucleotide polymorphism (SNPs).
Although protein binding of reactive metabolites may impair cellular function to cause toxicity, drug-metabolite adducts may interact with the immune system. In order for the drug-metabolite adduct to be recognized by the immune system, they should be presented by antigen presenting cells in conjunction with major histocompatibility complex (MHC) molecules. Consistent with this, studies from India have shown that absence of HLA-DQA1*0102 allele, and presence of HLA-DQB1*0201 allele were independent risk factors for anti-TB drug induced hepatotoxicity.81
Pathophysiology: unifying hypothesis
Development of idiosyncratic DILI is an intricate process involving both concurrent as well as sequential events determining the direction of the pathways, degree of liver injury and its outcome (Figure 2). The key upstream events include drug specific pathways triggered by particular drugs or their metabolites. Increased formation of reactive metabolites generally as a result of phase I metabolism or failure of detoxification usually a function of phase II metabolism is likely to be an initiating event. The expression of these drug metabolizing enzymes and transporters involved in the excretion and elimination of drug metabolites (phase III) are regulated by transcription factors (nuclear hormone receptors) such as pregnane X receptor. Genetic and environmental factors that influence the expression and activities of proteins involved in phase I, II and III of drug disposition or their regulation will determine the rate of formation and accumulation of reactive metabolite.
Figure 2.
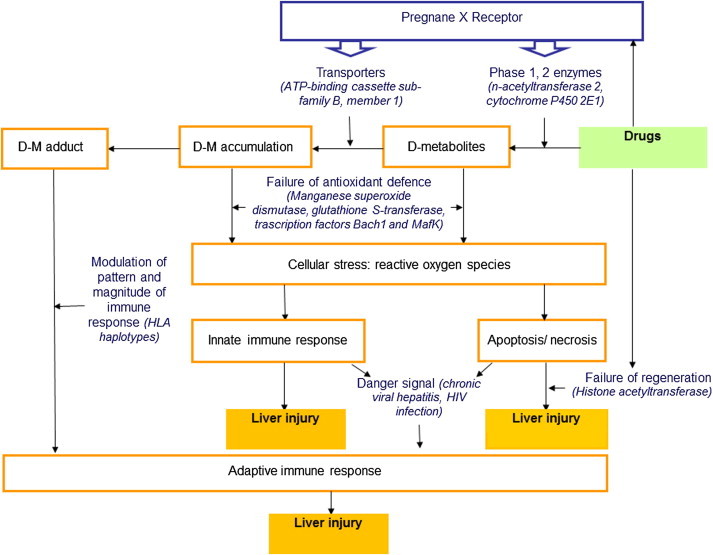
Hypothetical model of DILI due to anti-TB agents with potential drug and host related factors (in blue) involved in the pathogenesis.
These reactive metabolites induce the production of excessive reactive oxygen species (ROS) leading to lipid peroxidation and cell death. Cellular environment can modulate the threshold for hepatocyte death secondary to oxidative stress. Transcription factor Nuclear Factor Erythroid 2-related factor-2 (Nrf2) regulates glutathione synthetic and detoxification enzymes. Heterodimers of Nrf2 and small Maf basic leucine zipper proteins bind to the antioxidant-responsive element in the promoters of cytoprotective gene battery. Nrf2-directed endogenous antioxidant systems in the liver may dampen the injurious effect of reactive metabolites. On the other hand, a heterodimer complex of broad complex, tramtrack, bric-a-brac domain (BTB) and cap‘n’collar type of basic region (CNC) homology 1 (Bach1) and small Maf proteins downregulate the expression of antioxidant enzymes. Therefore, dysregulation of the activator arm (including Nrf2 and small Mafs) and the repressor arm (including Bach1 and small Mafs) of the antioxidant pathway may contribute to the propagation of anti-TB DILI.90
Reactive metabolite may induce cell stress by overwhelming antioxidant defense or alternatively by binding to cellular enzymes, lipids or nucleic acids. Mitochondria have been considered an important target in DILI; inhibition of mitochondrial respiratory chain results in adenosine triphosphate (ATP) depletion and accumulation of reactive oxygen species (ROS). Manganese superoxide dismutase (MnSOD) is a protein encoded by the nucleus and residing in the mitochondrial matrix where it plays a key role in the detoxification of superoxide anion radicals that arise constantly during electron transport.91 In a case–control genetic association study that included 63 patients with anti-TB DILI, those with a variant C allele (genotype T/C or C/C) of MnSOD had 2.5-fold higher risk of hepatotoxicity when compared with those MnSOD TT genotype.92 Mitochondrial inhibition due to lack of defense against superoxide ultimately leads to mitochondrial permeability transition (MPT) with consequent apoptosis or necrosis dependent on presence or absence of ATP.
Although variation of drug metabolism and clearance are certainly the key upstream events determining susceptibility to hepatotoxicity, the occurrence and extent of liver injury could be influenced by events downstream of initial drug metabolism. Innate immune response can promote or inhibit the inflammatory process and thereby determine the progression and severity of DILI. In addition, superimposition of drug metabolizing enzymes and the immune system creates a setting wherein a reactive drug metabolite covalently binds to cellular proteins and this misleads the immune system to mount an attack against the ‘modified self’ resulting in immune mediated destruction of targeted hepatocyte.16 Association of anti-TB DILI with specific human leukocyte antigen (HLA) genotypes81 implies that the susceptible individual has an immune system that would more readily recognize the formed neoantigens. Features of hypersensitivity with multi-system involvement manifested by renal dysfunction, hemolytic anemia, flu like syndrome, arthralgia and rash have been reported with rifampicin.93,94 Rarely pyrazinamide also manifests as granulomatous hepatitis95 with fever, eosinophilia and systemic illness. Alternatively, innate immune system may be involved through cytokines that modulate the degree of hepatic inflammation secondary to toxic injury or the ability of liver to regenerate in response to hepatocyte death. In certain circumstances neither the production of reactive metabolites nor the auto-antibodies generated in response to covalent binding of these metabolites to proteins (haptenization) is sufficient to elicit and immune response. Signal 1 represents the interaction between antigen presenting cell and the T-cell receptor which requires a co-stimulatory trigger, a ‘danger’ signal (signal 2) to prime the adaptive immune system. Signal 3 represents polarizing cytokine environment that drives T-cells to Th1 or Th2 immune response. The ‘danger’ signals are thought to be generated by cytokine release from inflammation associated with chronic viral infections which increase the risk of anti-TB DILI.85 Altered cytokines environment under these circumstances may prime a genetically susceptible adaptive immune system to respond to anti-TB drug-metabolite adduct (hapten) leading to DILI.
The ‘danger’ signals are derived from cells that are stressed by reactive metabolite formation or released from cells dying from necrosis.85 In addition to being a second signal in the adaptive immune response, ‘danger’ signals can also activate signaling pathways for oxidative stress. Therefore, sub-clinical hepatocyte injury manifesting as asymptomatic transaminitis may itself represent the ‘danger’ signal indicating both an individual's vulnerability and act as a determinant of more significant serious DILI.96
More recently, investigations have provided evidence of DILI may potentially be mediated through inhibition of a histone modification.97 Histone acetylation results in an open chromatin structure enabling genes in that region to be read; this acetylation is mediated by enzymes known as histone acetyltransferases.98 Authors hypothesized that histone acetyltransferase can be involved in acetylation of hydralazine derivatives including INH (on long term administration of the drug) leading to exhaustion of the enzyme.97 Considering the fundamental role histone acetylation has in activation of gene transcription, the exhaustion of histone acetyltransferase can lead to failure of hepatocyte regeneration and hence, unrestrained progression of the pathogenic process leading to DILI.
Management
Despite the huge global burden of TB and decades of experience in the use of anti-TB medications, studies investigating DILI lack scientific rigor, consistent methodology and large enough scale to generate the evidence on which recommendations can be based upon. Therefore, approaches to prevent, monitor and manage hepatotoxicity have been based primarily on retrospective observational studies. Recommendations from the American Thoracic Society (ATS),17 the British Thoracic Society (BTS)99 and more recently by National Institute for Clinical Excellence (NICE), UK100 for the assessment, choice of anti-TB drug regimen, patient education, clinical monitoring and interventions in the event of hepatotoxicity have been published. It must be noted that many of these recommendations are reliant on expert opinion only.
Risk Stratification
Pretreatment evaluation whenever feasible should include screening for existing chronic liver disease including history of excess alcohol consumption, intravenous drug abuse and nutritional assessment. Baseline evaluation should also include serology for chronic viral infections (hepatitis B, C and HIV) and appropriate assessment for underlying liver disease.99,100 Assessment of risk–benefit ratio is critical when empirical treatment for TB is being considered as these patients have been shown to have excess risk of adverse outcome.4,5 Individual's risk of DILI should be weighed against that for developing active TB before opting to treat latent TB.
Choice of Drugs and Regimen
There is no evidence to suggest that three times per week regimes are associated with lower risk of hepatotoxicity than daily dosing regimens.27 Guidelines from professional bodies101,102 provide advice on the choice of drugs, combinations and duration of therapy that are considered suitable to different clinical scenarios. Considerations should include the cost, affordability, access as well as efficacy and associated adverse effects.
As isoniazid and rifampicin are highly efficacious, their use in the treatment of latent or active TB infection is desirable whenever possible. However, considering that combination therapy increases the risk of DILI,26 monotherapy with either isoniazid or rifampicin is preferable for the treatment of latent TB when particular individual is at higher risk of hepatotoxicity. In patients with unstable or advanced liver disease, if the serum alanine aminotransferase level is more than 3 times normal at baseline, the following regimens should be considered. The more unstable or severe the liver disease is, the fewer hepatotoxic drugs should be used.
Possible regimens include103:
-
•Two hepatotoxic drugs regimen (rather than the three in the standard regimen):
-
—9 months of isoniazid and rifampicin, plus ethambutol
-
—2 months of isoniazid, rifampicin, streptomycin and ethambutol, followed by 6 months of isoniazid and rifampicin;
-
—6–9 months of rifampicin, pyrazinamide and ethambutol.
-
—
-
•One hepatotoxic drug regimen:
-
—2 months of isoniazid, ethambutol and streptomycin, followed by 10 months of isoniazid and ethambutol.
-
—
-
•No hepatotoxic drug regimen: in patients with advanced cirrhosis or portosystemic encephalopathy
-
—18–24 months treatment with a combination of ethambutol, fluoroquinolone, cycloserine and capreomycin or aminoglycoside has been suggested as an option.103
-
—
Patient Education
Patients should be educated about the importance of adherence to medications, follow up visits for monitoring and symptoms of hepatotoxicity with appropriate reminders wherever possible. In the event of symptoms that are attributable to hepatotoxicity, patients should be forewarned to stop all anti-TB medications and seek medical advice in the event of any symptoms of hepatotoxicity and seek immediate medical advice. One report from a programme of INH based chemoprophylaxis suggested that regular inquiry and reporting of symptoms at monthly visits proved effective in averting serious DILI without the need for routine measurements of liver biochemistry.24 Patients should be advised to refrain from alcohol and to seek medical advice about any prescription or non-prescription medication use as these could potentially increase toxicity leading to DILI.
Monitoring
Regular clinical review of patients is helpful to monitor treatment adherence and directly observed short-course therapy (DOTS) enhances its effectiveness. Therapeutic drug monitoring104–106 has been shown to improve clinical response, but its use in predicting hepatotoxicity remains to be demonstrated. Within the first few weeks of initiation of anti-TB therapy, up to 20% of patients develop transient, asymptomatic elevations in ALT and as these spontaneously resolve these are thought to represent ‘adaptation’107 rather than hepatotoxicity; potential mechanisms underlying the former phenomenon are discussed in the literature.108 The Seattle-King County Public Health Department used a protocol to monitor INH therapy which included advising the patient at each visit to stop the medication and call the clinic if symptoms of hepatotoxicity occurred.24 With careful clinical monitoring without routine laboratory tests, the rate of hepatotoxicity in 11,141 patients was much lower (0.1%–0.15%) than previously expected (1%) and there were no deaths.24 The ATS guidelines do not recommend routine liver biochemistry testing in those without any obvious risk factors; but, liver biochemistry based monitoring should be considered at 2 weekly intervals in the first 2–3 months of therapy in patients with risk factors for developing hepatotoxicity and in those who have abnormal baseline tests.
Interventions
Prompt withdrawal of the offending medication is the most critical intervention in the management of hepatotoxicity. However, considering the lack of specific markers that distinguish transient self-resolving transamintis from potentially serious DILI, the thresholds set for discontinuation of anti-TB drugs remain pragmatic rather than evidence based. The ATS, BTS, NICE and World Health Organization (WHO) and the International Union Against Tuberculosis and Lung Disease guidelines have some minor variations in the interventions for hepatotoxicity. In general, advice is to stop all anti-TB medications when elevation in ALT reaches 3 times ULN with symptoms attributable to hepatotoxicity or ALT level of 5 times ULN is detected. In smear positive TB cases or where discontinuation of therapy is deemed unsafe due to severity of the illness, a non-hepatotoxic anti-TB treatment such as ethambutol, fluoroquinolone or cycloserine could be considered.
Once withdrawn, anti-TB treatment should be withheld ideally until the liver tests normalize, or at least ALT falls below 2 times ULN. Considering that first line anti-TB drugs are highly effective and relatively inexpensive, benefits of re-challenge must outweigh its risks; it is unwise to discard these drugs from the regimen. Therefore, it is acceptable to attempt reintroduction of these medications.109 In 11%–24% of patients, re-exposure to the same drug regimen leads to recurrence of DILI110 and positive re-challenge is not affected by the degree of initial injury.110 Both ATS and BTS advice restarting the anti-TB medications one at a time. The WHO as well as the International Union Against Tuberculosis and Lung Disease advice restarting all the drugs simultaneously; if however, there is a second bout of hepatotoxicity after re-challenge, then the drugs are to be reintroduced consecutively. A study randomized 175 patients with hepatotoxicity into these 3 reintroduction regimens and found no difference in the frequency of recurrent DILI.110 On the contrary a small randomized controlled trial of 45 patients with hepatotoxicity compared restarting of all drugs simultaneously with sequential reintroduction regimen without pyrazinamide showed safety of the latter regimen.111 In the light of such controversial evidence one has to weigh the risks of severe life threatening forms of TB, need for faster sputum conversion, need for reduction in disease transmission in the community and to reduce the risk of developing multi-drug resistance forms against the risks of hepatotoxicity. A sequential regimen with or without pyrazinamide would be suitable in those individuals who have a higher baseline prediction of hepatotoxicity as defined by their phenotype of malnutrition, low albumin, alcoholics and HIV-positive individuals.110
Co-administration of N-acetyl cysteine (NAC)112 was protective against DILI in animals treated with hepatotoxic doses of INH and rifampicin. A report on benefit of oral administration of NAC (600 mg twice daily) in the prevention of anti-TB DILI comes from an open labeled trial including 60 patients of age >60 years; in the study 37.5% developed elevation of both AST (mean 99.4) and ALT (mean 65.8) and mean bilirubin of 1.1 mg/dl within a mean duration of 5 days of starting combination anti-TB therapy when not treated with NAC.113 In contrast, none in the group treated with NAC developed abnormalities of the liver chemistry. It is difficult to put the details of this study in the context of a vast amount of clinical observations described in the literature; significance of these findings and their generalizability are unclear due to the fact, that mean age of the cohorts were 73 years and possibility that the asymptomatic transaminitis could have been tolerance and not clinically significant DILI. Report of reduction of incidence and severity of anti-TB hepatotoxicity by the use herbal formulation114 warrants replication in large clinical trials in independent cohorts.
Future developments
Anti-TB drug induced hepatotoxicity is a serious adverse effect and continues to be a problem worldwide. Efforts at prevention and/or early recognition of anti-TB DILI are severely hampered by limited understanding of its pathogenesis. Future investigations exploring the mechanisms underlying the pathogenesis of anti-TB DILI should be performed using human tissue and samples whenever possible, so that the novel findings can be translated readily into clinical applications. Clear and consistent phenotypic definitions are essential first step115 in those studies seeking to identify risk factors of DILI in large cohorts of patients. Recent investigations focused on genetic susceptibility to ‘idiosyncratic’ DILI have promised the development of refined algorithms including drug, host and environmental risk factors that would allow pre-emption of DILI116 and hence allow better tailoring of medications based on accurate estimates of risk–benefit ratio. Undoubtedly, there is an urgent need for more refined, novel, genetic, proteinaceous and metabolite biomarkers which will detect patients with increased susceptibility to DILI, assist in early diagnosis and monitoring for DILI during therapy. Anti-TB DILI provides an opportunity for research that will have considerable impact on wide areas such as drug discovery/development process, primary and secondary care.
Conflicts of interest
All authors have none to declare.
References
- 1.Global Tuberculosis Control: WHO Report 2011. World Health Organisation; 2011. viii, 246 p. [Google Scholar]
- 2.Schaberg T., Rebhan K., Lode H. Risk factors for side-effects of isoniazid, rifampin and pyrazinamide in patients hospitalized for pulmonary tuberculosis. Eur Respir J. 1996;9:2026–2030. doi: 10.1183/09031936.96.09102026. [DOI] [PubMed] [Google Scholar]
- 3.Ostapowicz G., Fontana R.J., Schiodt F.V. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 4.Kumar R., Bhatia V., Khanal S. Antituberculosis therapy-induced acute liver failure: magnitude, profile, prognosis, and predictors of outcome. Hepatology. 2010;51:1665–1674. doi: 10.1002/hep.23534. [DOI] [PubMed] [Google Scholar]
- 5.Devarbhavi H., Dierkhising R., Kremers W.K. Antituberculosis therapy drug-induced liver injury and acute liver failure. Hepatology. 2010;52:798–799. doi: 10.1002/hep.23805. author reply 799–800. [DOI] [PubMed] [Google Scholar]
- 6.Aithal G.P., Watkins P.B., Andrade R.J. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther. 2011;89:806–815. doi: 10.1038/clpt.2011.58. [DOI] [PubMed] [Google Scholar]
- 7.Tostmann A., Boeree M.J., Aarnoutse R.E. Antituberculosis drug-induced hepatotoxicity: concise up-to-date review. J Gastroenterol Hepatol. 2008;23:192–202. doi: 10.1111/j.1440-1746.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell J.R., Zimmerman H.J., Ishak K.G. Isoniazid liver injury: clinical spectrum, pathology, and probable pathogenesis. Ann Intern Med. 1976;84:181–192. doi: 10.7326/0003-4819-84-2-181. [DOI] [PubMed] [Google Scholar]
- 9.Scharer L., Smith J.P. Serum transaminase elevations and other hepatic abnormalities in patients receiving isoniazid. Ann Intern Med. 1969;71:1113–1120. doi: 10.7326/0003-4819-71-6-1113. [DOI] [PubMed] [Google Scholar]
- 10.Larrey D. Epidemiology and individual susceptibility to adverse drug reactions affecting the liver. Semin Liver Dis. 2002;22:145–155. doi: 10.1055/s-2002-30105. [DOI] [PubMed] [Google Scholar]
- 11.Fountain F.F., Tolley E., Chrisman C.R. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: a 7-year evaluation from a public health tuberculosis clinic. Chest. 2005;128:116–123. doi: 10.1378/chest.128.1.116. [DOI] [PubMed] [Google Scholar]
- 12.Kopanoff D.E., Snider D.E., Jr., Caras G.J. Isoniazid-related hepatitis: a U.S. Public Health Service cooperative surveillance study. Am Rev Respir Dis. 1978;117:991–1001. doi: 10.1164/arrd.1978.117.6.991. [DOI] [PubMed] [Google Scholar]
- 13.International Union Against Tuberculosis Committee on Prophylaxis Efficacy of various durations of isoniazid preventive therapy for tuberculosis: five years of follow-up in the IUAT trial. Bull World Health Organ. 1982;60:555–564. [PMC free article] [PubMed] [Google Scholar]
- 14.Acharya S.K., Dasarathy S., Kumer T.L. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology. 1996;23:1448–1455. doi: 10.1002/hep.510230622. [DOI] [PubMed] [Google Scholar]
- 15.Singh J., Garg P.K., Tandon R.K. Hepatotoxicity due to antituberculosis therapy. Clinical profile and reintroduction of therapy. J Clin Gastroenterol. 1996;22:211–214. doi: 10.1097/00004836-199604000-00012. [DOI] [PubMed] [Google Scholar]
- 16.Millard P.S., Wilcosky T.C., Reade-Christopher S.J. Isoniazid-related fatal hepatitis. West J Med. 1996;164:486–491. [PMC free article] [PubMed] [Google Scholar]
- 17.Saukkonen J.J., Cohn D.L., Jasmer R.M. An official ATS statement: hepatotoxicity of antituberculosis therapy. Am J Respir Crit Care Med. 2006;174:935–952. doi: 10.1164/rccm.200510-1666ST. [DOI] [PubMed] [Google Scholar]
- 18.Younossian A.B., Rochat T., Ketterer J.P. High hepatotoxicity of pyrazinamide and ethambutol for treatment of latent tuberculosis. Eur Respir J. 2005;26:462–464. doi: 10.1183/09031936.05.00006205. [DOI] [PubMed] [Google Scholar]
- 19.Papastavros T., Dolovich L.R., Holbrook A. Adverse events associated with pyrazinamide and levofloxacin in the treatment of latent multidrug-resistant tuberculosis. CMAJ. 2002;167:131–136. [PMC free article] [PubMed] [Google Scholar]
- 20.Prince M.I., Burt A.D., Jones D.E. Hepatitis and liver dysfunction with rifampicin therapy for pruritus in primary biliary cirrhosis. Gut. 2002;50:436–439. doi: 10.1136/gut.50.3.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziakas P.D., Mylonakis E. 4 months of rifampin compared with 9 months of isoniazid for the management of latent tuberculosis infection: a meta-analysis and cost-effectiveness study that focuses on compliance and liver toxicity. Clin Infect Dis. 2009;49:1883–1889. doi: 10.1086/647944. [DOI] [PubMed] [Google Scholar]
- 22.Fountain F.F., Tolley E.A., Jacobs A.R. Rifampin hepatotoxicity associated with treatment of latent tuberculosis infection. Am J Med Sci. 2009;337:317–320. doi: 10.1097/MAJ.0b013e31818c0134. [DOI] [PubMed] [Google Scholar]
- 23.LoBue P.A., Moser K.S. Isoniazid- and rifampin-resistant tuberculosis in San Diego County, California, United States, 1993–2002. Int J Tuberc Lung Dis. 2005;9:501–506. [PubMed] [Google Scholar]
- 24.Nolan C.M., Goldberg S.V., Buskin S.E. Hepatotoxicity associated with isoniazid preventive therapy: a 7-year survey from a public health tuberculosis clinic. J Am Med Assoc. 1999;281:1014–1018. doi: 10.1001/jama.281.11.1014. [DOI] [PubMed] [Google Scholar]
- 25.Snider D.E., Jr., Caras G.J. Isoniazid-associated hepatitis deaths: a review of available information. Am Rev Respir Dis. 1992;145:494–497. doi: 10.1164/ajrccm/145.2_Pt_1.494. [DOI] [PubMed] [Google Scholar]
- 26.Steele M.A., Burk R.F., DesPrez R.M. Toxic hepatitis with isoniazid and rifampin. A meta-analysis. Chest. 1991;99:465–471. doi: 10.1378/chest.99.2.465. [DOI] [PubMed] [Google Scholar]
- 27.Chang K.C., Leung C.C., Yew W.W. Standard anti-tuberculosis treatment and hepatotoxicity: do dosing schedules matter? Eur Respir J. 2007;29:347–351. doi: 10.1183/09031936.00090306. [DOI] [PubMed] [Google Scholar]
- 28.Yue J., Peng R.X., Yang J. CYP2E1 mediated isoniazid-induced hepatotoxicity in rats. Acta Pharmacol Sin. 2004;25:699–704. [PubMed] [Google Scholar]
- 29.Chowdhury A., Santra A., Bhattacharjee K. Mitochondrial oxidative stress and permeability transition in isoniazid and rifampicin induced liver injury in mice. J Hepatol. 2006;45:117–126. doi: 10.1016/j.jhep.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 30.Olson H., Betton G., Robinson D. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol. 2000;32:56–67. doi: 10.1006/rtph.2000.1399. [DOI] [PubMed] [Google Scholar]
- 31.Knowles S.R., Uetrecht J., Shear N.H. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587–1591. doi: 10.1016/S0140-6736(00)03137-8. [DOI] [PubMed] [Google Scholar]
- 32.Scales M.D., Timbrell J.A. Studies on hydrazine hepatotoxicity. 1. Pathological findings. J Toxicol Environ Health. 1982;10:941–953. doi: 10.1080/15287398209530308. [DOI] [PubMed] [Google Scholar]
- 33.Sarma G.R., Immanuel C., Kailasam S. Rifampin-induced release of hydrazine from isoniazid. A possible cause of hepatitis during treatment of tuberculosis with regimens containing isoniazid and rifampin. Am Rev Respir Dis. 1986;133:1072–1075. doi: 10.1164/arrd.1986.133.6.1072. [DOI] [PubMed] [Google Scholar]
- 34.Blum M., Grant D.M., McBride W. Human arylamine N-acetyltransferase genes: isolation, chromosomal localization, and functional expression. DNA Cell Biol. 1990;9:193–203. doi: 10.1089/dna.1990.9.193. [DOI] [PubMed] [Google Scholar]
- 35.Hein D.W., Doll M.A., Rustan T.D. Metabolic activation of N-hydroxyarylamines and N-hydroxyarylamides by 16 recombinant human NAT2 allozymes: effects of 7 specific NAT2 nucleic acid substitutions. Cancer Res. 1995;55:3531–3536. [PubMed] [Google Scholar]
- 36.Huang Y.S., Chern H.D., Su W.J. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology. 2002;35:883–889. doi: 10.1053/jhep.2002.32102. [DOI] [PubMed] [Google Scholar]
- 37.Wang P.Y., Xie S.Y., Hao Q. NAT2 polymorphisms and susceptibility to anti-tuberculosis drug-induced liver injury: a meta-analysis. Int J Tuberc Lung Dis. 2012;16:589–595. doi: 10.5588/ijtld.11.0377. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y.S., Chern H.D., Su W.J. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology. 2003;37:924–930. doi: 10.1053/jhep.2003.50144. [DOI] [PubMed] [Google Scholar]
- 39.Bose P.D., Sarma M.P., Medhi S. Role of polymorphic N-acetyl transferase2 and cytochrome P4502E1 gene in antituberculosis treatment-induced hepatitis. J Gastroenterol Hepatol. 2011;26:312–318. doi: 10.1111/j.1440-1746.2010.06355.x. [DOI] [PubMed] [Google Scholar]
- 40.Roy B., Chowdhury A., Kundu S. Increased risk of antituberculosis drug-induced hepatotoxicity in individuals with glutathione S-transferase M1 ‘null’ mutation. J Gastroenterol Hepatol. 2001;16:1033–1037. doi: 10.1046/j.1440-1746.2001.02585.x. [DOI] [PubMed] [Google Scholar]
- 41.Leiro V., Fernandez-Villar A., Valverde D. Influence of glutathione S-transferase M1 and T1 homozygous null mutations on the risk of antituberculosis drug-induced hepatotoxicity in a Caucasian population. Liver Int. 2008;28:835–839. doi: 10.1111/j.1478-3231.2008.01700.x. [DOI] [PubMed] [Google Scholar]
- 42.Metushi I.G., Cai P., Zhu X. A fresh look at the mechanism of isoniazid-induced hepatotoxicity. Clin Pharmacol Ther. 2011;89:911–914. doi: 10.1038/clpt.2010.355. [DOI] [PubMed] [Google Scholar]
- 43.Nakajima A., Fukami T., Kobayashi Y. Human arylacetamide deacetylase is responsible for deacetylation of rifamycins: rifampicin, rifabutin, and rifapentine. Biochem Pharmacol. 2011;82:1747–1756. doi: 10.1016/j.bcp.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Jamis-Dow C.A., Katki A.G., Collins J.M. Rifampin and rifabutin and their metabolism by human liver esterases. Xenobiotica. 1997;27:1015–1024. doi: 10.1080/004982597239994. [DOI] [PubMed] [Google Scholar]
- 45.Acocella G. Clinical pharmacokinetics of rifampicin. Clin Pharm. 1978;3:108–127. doi: 10.2165/00003088-197803020-00002. [DOI] [PubMed] [Google Scholar]
- 46.Holdiness M.R. Clinical pharmacokinetics of the antituberculosis drugs. Clin Pharm. 1984;9:511–544. doi: 10.2165/00003088-198409060-00003. [DOI] [PubMed] [Google Scholar]
- 47.Menzies D., Dion M.J., Rabinovitch B. Treatment completion and costs of a randomized trial of rifampin for 4 months versus isoniazid for 9 months. Am J Respir Crit Care Med. 2004;170:445–449. doi: 10.1164/rccm.200404-478OC. [DOI] [PubMed] [Google Scholar]
- 48.A double-blind placebo-controlled clinical trial of three antituberculosis chemoprophylaxis regimens in patients with silicosis in Hong Kong. Hong Kong Chest Service/Tuberculosis Research Centre, Madras/British Medical Research Council. Am Rev Respir Dis. 1992;145:36–41. doi: 10.1164/ajrccm/145.1.36. [DOI] [PubMed] [Google Scholar]
- 49.Nannelli A., Chirulli V., Longo V. Expression and induction by rifampicin of CAR- and PXR-regulated CYP2B and CYP3A in liver, kidney and airways of pig. Toxicology. 2008;252:105–112. doi: 10.1016/j.tox.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 50.Burk O., Koch I., Raucy J. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR) J Biol Chem. 2004;279:38379–38385. doi: 10.1074/jbc.M404949200. [DOI] [PubMed] [Google Scholar]
- 51.Kimchi-Sarfaty C., Oh J.M., Kim I.W. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 52.Yimer G., Ueda N., Habtewold A. Pharmacogenetic & pharmacokinetic biomarker for efavirenz based ARV and rifampicin based anti-TB drug induced liver injury in TB-HIV infected patients. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0027810. e27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byrne J.A., Strautnieks S.S., Mieli-Vergani G. The human bile salt export pump: characterization of substrate specificity and identification of inhibitors. Gastroenterology. 2002;123:1649–1658. doi: 10.1053/gast.2002.36591. [DOI] [PubMed] [Google Scholar]
- 54.Capelle P., Dhumeaux D., Mora M. Effect of rifampicin on liver function in man. Gut. 1972;13:366–371. doi: 10.1136/gut.13.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grosset J., Leventis S. Adverse effects of rifampin. Rev Infect Dis. 1983;5(suppl 3):S440–S450. doi: 10.1093/clinids/5.supplement_3.s440. [DOI] [PubMed] [Google Scholar]
- 56.Ellard G.A. Absorption, metabolism and excretion of pyrazinamide in man. Tubercle. 1969;50:144–158. doi: 10.1016/0041-3879(69)90020-8. [DOI] [PubMed] [Google Scholar]
- 57.Lacroix C., Tranvouez J.L., Phan Hoang T. Pharmacokinetics of pyrazinamide and its metabolites in patients with hepatic cirrhotic insufficiency. Arzneimittelforschung. 1990;40:76–79. [PubMed] [Google Scholar]
- 58.Maffei Facino R., Carini M. The inhibitory effect of pyrazinamide on microsomal monooxygenase activities is related to the binding to reduced cytochrome P-450. Pharmacol Res Commun. 1980;12:523–537. doi: 10.1016/s0031-6989(80)80138-x. [DOI] [PubMed] [Google Scholar]
- 59.Shibata K., Fukuwatari T., Sugimoto E. Effects of dietary pyrazinamide, an antituberculosis agent, on the metabolism of tryptophan to niacin and of tryptophan to serotonin in rats. Biosci Biotechnol Biochem. 2001;65:1339–1346. doi: 10.1271/bbb.65.1339. [DOI] [PubMed] [Google Scholar]
- 60.Andrade R.J., Tulkens P.M. Hepatic safety of antibiotics used in primary care. J Antimicrob Chemother. 2011;66:1431–1446. doi: 10.1093/jac/dkr159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Labowitz J.K., Silverman W.B. Cholestatic jaundice induced by ciprofloxacin. Dig Dis Sci. 1997;42:192–194. doi: 10.1023/a:1018870029216. [DOI] [PubMed] [Google Scholar]
- 62.Kahn J.B. Latest industry information on the safety profile of levofloxacin in the US. Chemotherapy. 2001;47(suppl 3):32–37. doi: 10.1159/000057842. discussion 44–48. [DOI] [PubMed] [Google Scholar]
- 63.Wolfson J.S., Hooper D.C. Overview of fluoroquinolone safety. Am J Med. 1991;91:153S–161S. doi: 10.1016/0002-9343(91)90330-z. [DOI] [PubMed] [Google Scholar]
- 64.Coleman C.I., Spencer J.V., Chung J.O. Possible gatifloxacin-induced fulminant hepatic failure. Ann Pharmacother. 2002;36:1162–1167. doi: 10.1345/aph.1A414. [DOI] [PubMed] [Google Scholar]
- 65.Ho C.C., Chen Y.C., Hu F.C. Safety of fluoroquinolone use in patients with hepatotoxicity induced by anti-tuberculosis regimens. Clin Infect Dis. 2009;48:1526–1533. doi: 10.1086/598929. [DOI] [PubMed] [Google Scholar]
- 66.Saigal S., Agarwal S.R., Nandeesh H.P. Safety of an ofloxacin-based antitubercular regimen for the treatment of tuberculosis in patients with underlying chronic liver disease: a preliminary report. J Gastroenterol Hepatol. 2001;16:1028–1032. doi: 10.1046/j.1440-1746.2001.02570.x. [DOI] [PubMed] [Google Scholar]
- 67.Yee D., Valiquette C., Pelletier M. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am J Respir Crit Care Med. 2003;167:1472–1477. doi: 10.1164/rccm.200206-626OC. [DOI] [PubMed] [Google Scholar]
- 68.Pande J.N., Singh S.P., Khilnani G.C. Risk factors for hepatotoxicity from antituberculosis drugs: a case-control study. Thorax. 1996;51:132–136. doi: 10.1136/thx.51.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hwang S.J., Wu J.C., Lee C.N. A prospective clinical study of isoniazid-rifampicin-pyrazinamide-induced liver injury in an area endemic for hepatitis B. J Gastroenterol Hepatol. 1997;12:87–91. doi: 10.1111/j.1440-1746.1997.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 70.Dossing M., Wilcke J.T., Askgaard D.S. Liver injury during antituberculosis treatment: an 11-year study. Tuber Lung Dis. 1996;77:335–340. doi: 10.1016/s0962-8479(96)90098-2. [DOI] [PubMed] [Google Scholar]
- 71.Teleman M.D., Chee C.B., Earnest A. Hepatotoxicity of tuberculosis chemotherapy under general programme conditions in Singapore. Int J Tuberc Lung Dis. 2002;6:699–705. [PubMed] [Google Scholar]
- 72.Shakya R., Rao B.S., Shrestha B. Incidence of hepatotoxicity due to antitubercular medicines and assessment of risk factors. Ann Pharmacother. 2004;38:1074–1079. doi: 10.1345/aph.1D525. [DOI] [PubMed] [Google Scholar]
- 73.Lee A.M., Mennone J.Z., Jones R.C. Risk factors for hepatotoxicity associated with rifampin and pyrazinamide for the treatment of latent tuberculosis infection: experience from three public health tuberculosis clinics. Int J Tuberc Lung Dis. 2002;6:995–1000. [PubMed] [Google Scholar]
- 74.Hunt C.M., Westerkam W.R., Stave G.M. Effect of age and gender on the activity of human hepatic CYP3A. Biochem Pharmacol. 1992;44:275–283. doi: 10.1016/0006-2952(92)90010-g. [DOI] [PubMed] [Google Scholar]
- 75.Franks A.L., Binkin N.J., Snider D.E., Jr. Isoniazid hepatitis among pregnant and postpartum Hispanic patients. Public Health Rep. 1989;104:151–155. [PMC free article] [PubMed] [Google Scholar]
- 76.Singla R., Sharma S.K., Mohan A. Evaluation of risk factors for antituberculosis treatment induced hepatotoxicity. Indian J Med Res. 2010;132:81–86. [PubMed] [Google Scholar]
- 77.Warmelink I., ten Hacken N.H., van der Werf T.S. Weight loss during tuberculosis treatment is an important risk factor for drug-induced hepatotoxicity. Br J Nutr. 2011;105:400–408. doi: 10.1017/S0007114510003636. [DOI] [PubMed] [Google Scholar]
- 78.Buchanan N., Eyberg C., Davis M.D. Isoniazid pharmacokinetics in kwashiorkor. S Afr Med J. 1979;56:299–300. [PubMed] [Google Scholar]
- 79.Walter-Sack I., Klotz U. Influence of diet and nutritional status on drug metabolism. Clin Pharm. 1996;31:47–64. doi: 10.2165/00003088-199631010-00004. [DOI] [PubMed] [Google Scholar]
- 80.Fernandez-Villar A., Sopena B., Fernandez-Villar J. The influence of risk factors on the severity of anti-tuberculosis drug-induced hepatotoxicity. Int J Tuberc Lung Dis. 2004;8:1499–1505. [PubMed] [Google Scholar]
- 81.Sharma S.K., Balamurugan A., Saha P.K. Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during antituberculosis treatment. Am J Respir Crit Care Med. 2002;166:916–919. doi: 10.1164/rccm.2108091. [DOI] [PubMed] [Google Scholar]
- 82.Tost J.R., Vidal R., Cayla J. Severe hepatotoxicity due to anti-tuberculosis drugs in Spain. Int J Tuberc Lung Dis. 2005;9:534–540. [PubMed] [Google Scholar]
- 83.White M.C., Tulsky J.P., Lee J.R. Isoniazid vs. rifampin for latent tuberculosis infection in jail inmates: toxicity and adherence. J Correct Health Care. 2012;18:131–142. doi: 10.1177/1078345811435973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Polesky A., Farber H.W., Gottlieb D.J. Rifampin preventive therapy for tuberculosis in Boston's homeless. Am J Respir Crit Care Med. 1996;154:1473–1477. doi: 10.1164/ajrccm.154.5.8912767. [DOI] [PubMed] [Google Scholar]
- 85.Pirmohamed M., Naisbitt D.J., Gordon F. The danger hypothesis–potential role in idiosyncratic drug reactions. Toxicology. 2002;181–182:55–63. doi: 10.1016/s0300-483x(02)00255-x. [DOI] [PubMed] [Google Scholar]
- 86.Wang J.Y., Liu C.H., Hu F.C. Risk factors of hepatitis during anti-tuberculous treatment and implications of hepatitis virus load. J Infect. 2011;62:448–455. doi: 10.1016/j.jinf.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 87.Ungo J.R., Jones D., Ashkin D. Antituberculosis drug-induced hepatotoxicity. The role of hepatitis C virus and the human immunodeficiency virus. Am J Respir Crit Care Med. 1998;157:1871–1876. doi: 10.1164/ajrccm.157.6.9711039. [DOI] [PubMed] [Google Scholar]
- 88.Dworkin M.S., Adams M.R., Cohn D.L. Factors that complicate the treatment of tuberculosis in HIV-infected patients. J Acquir Immune Defic Syndr. 2005;39:464–470. doi: 10.1097/01.qai.0000152400.36723.85. [DOI] [PubMed] [Google Scholar]
- 89.Hustert E., Zibat A., Presecan-Siedel E. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab Dispos. 2001;29:1454–1459. [PubMed] [Google Scholar]
- 90.Nanashima K., Mawatari T., Tahara N. Genetic variants in antioxidant pathway: risk factors for hepatotoxicity in tuberculosis patients. Tuberculosis (Edinb) 2012;92:253–259. doi: 10.1016/j.tube.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 91.Boelsterli U.A., Lim P.L. Mitochondrial abnormalities – a link to idiosyncratic drug hepatotoxicity? Toxicol Appl Pharmacol. 2007;220:92–107. doi: 10.1016/j.taap.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 92.Huang Y.S., Su W.J., Huang Y.H. Genetic polymorphisms of manganese superoxide dismutase, NAD(P)H:quinone oxidoreductase, glutathione S-transferase M1 and T1, and the susceptibility to drug-induced liver injury. J Hepatol. 2007;47:128–134. doi: 10.1016/j.jhep.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 93.Martinez E., Collazos J., Mayo J. Hypersensitivity reactions to rifampin. Pathogenetic mechanisms, clinical manifestations, management strategies, and review of the anaphylactic-like reactions. Medicine (Baltimore) 1999;78:361–369. doi: 10.1097/00005792-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 94.Covic A., Goldsmith D.J., Segall L. Rifampicin-induced acute renal failure: a series of 60 patients. Nephrol Dial Transplant. 1998;13:924–929. doi: 10.1093/ndt/13.4.924. [DOI] [PubMed] [Google Scholar]
- 95.Knobel B., Buyanowsky G., Dan M. Pyrazinamide-induced granulomatous hepatitis. J Clin Gastroenterol. 1997;24:264–266. doi: 10.1097/00004836-199706000-00019. [DOI] [PubMed] [Google Scholar]
- 96.Aithal G.P. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Expert Opin Drug Saf. 2004;3:519–523. doi: 10.1517/14740338.3.6.519. [DOI] [PubMed] [Google Scholar]
- 97.Murata K., Hamada M., Sugimoto K. A novel mechanism for drug-induced liver failure: inhibition of histone acetylation by hydralazine derivatives. J Hepatol. 2007;46:322–329. doi: 10.1016/j.jhep.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 98.Elsharkawy A.M. Todralazine hepatotoxicity: a sting in the histone tail. J Hepatol. 2007;46:189–192. doi: 10.1016/j.jhep.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 99.Chemotherapy and management of tuberculosis in the United Kingdom: recommendations 1998. Joint Tuberculosis Committee of the British Thoracic Society. Thorax. 1998;53:536–548. [PMC free article] [PubMed] [Google Scholar]
- 100.Tuberculosis: Clinical Diagnosis and Management of Tuberculosis, and Measures for Its Prevention and Control; National Institute for Health and Clinical Excellence: Guidance, London. 2011. [PubMed] [Google Scholar]
- 101.Update: adverse event data and revised American Thoracic Society/CDC recommendations against the use of rifampin and pyrazinamide for treatment of latent tuberculosis infection – United States, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:735–739. [PubMed] [Google Scholar]
- 102.Blumberg H.M., Burman W.J., Chaisson R.E. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603–662. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 103.Treatment of Tuberculosis: Guidelines. 4th ed. WHO/HTM/TB/2009.420.
- 104.Peloquin C.A. Therapeutic drug monitoring in the treatment of tuberculosis. Drugs. 2002;62:2169–2183. doi: 10.2165/00003495-200262150-00001. [DOI] [PubMed] [Google Scholar]
- 105.Magis-Escurra C., van den Boogaard J., Ijdema D. Therapeutic drug monitoring in the treatment of tuberculosis patients. Pulm Pharmacol Ther. 2012;25:83–86. doi: 10.1016/j.pupt.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 106.Babalik A., Babalik A., Mannix S. Therapeutic drug monitoring in the treatment of active tuberculosis. Can Respir J. 2011;18:225–229. doi: 10.1155/2011/307150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ormerod L.P., Horsfield N. Frequency and type of reactions to antituberculosis drugs: observations in routine treatment. Tuber Lung Dis. 1996;77:37–42. doi: 10.1016/s0962-8479(96)90073-8. [DOI] [PubMed] [Google Scholar]
- 108.Burham P.C. Molecular basis for adaptive responses during chemically induced hepatotoxicity. Toxicol Sci. 2006;89:349–351. doi: 10.1093/toxsci/kfj059. [DOI] [PubMed] [Google Scholar]
- 109.Hunt C.M. Mitochondrial and immunoallergic injury increase risk of positive drug rechallenge after drug-induced liver injury: a systematic review. Hepatology. 2010;52:2216–2222. doi: 10.1002/hep.24022. [DOI] [PubMed] [Google Scholar]
- 110.Sharma S.K., Singla R., Sarda P. Safety of 3 different reintroduction regimens of antituberculosis drugs after development of antituberculosis treatment-induced hepatotoxicity. Clin Infect Dis. 2010;50:833–839. doi: 10.1086/650576. [DOI] [PubMed] [Google Scholar]
- 111.Tahaoglu K., Atac G., Sevim T. The management of anti-tuberculosis drug-induced hepatotoxicity. Int J Tuberc Lung Dis. 2001;5:65–69. [PubMed] [Google Scholar]
- 112.Attri S., Rana S.V., Vaiphei K. Isoniazid- and rifampicin-induced oxidative hepatic injury – protection by N-acetylcysteine. Hum Exp Toxicol. 2000;19:517–522. doi: 10.1191/096032700674230830. [DOI] [PubMed] [Google Scholar]
- 113.Baniasadi S., Eftekhari P., Tabarsi P. Protective effect of N-acetylcysteine on antituberculosis drug-induced hepatotoxicity. Eur J Gastroenterol Hepatol. 2010;22:1235–1238. doi: 10.1097/MEG.0b013e32833aa11b. [DOI] [PubMed] [Google Scholar]
- 114.Adhvaryu M.R., Reddy N., Vakharia B.C. Prevention of hepatotoxicity due to anti tuberculosis treatment: a novel integrative approach. World J Gastroenterol. 2008;14:4753–4762. doi: 10.3748/wjg.14.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pirmohamed M., Aithal G.P., Behr E. The phenotype standardization project: improving pharmacogenetic studies of serious adverse drug reactions. Clin Pharmacol Ther. 2011;89:784–785. doi: 10.1038/clpt.2011.30. [DOI] [PubMed] [Google Scholar]
- 116.Aithal G.P., Daly A.K. Preempting and preventing drug-induced liver injury. Nat Genet. 2010;42:650–651. doi: 10.1038/ng0810-650. [DOI] [PubMed] [Google Scholar]