

The principles of the Bürgi–Dunitz nucleophilic approach necessitate structural rearrangements in Schiff-base-forming enzymes. Presented as a case study, substrate- and reaction intermediate-bound crystal complexes reveal that substrate conformational changes achieve the required rearrangements in transaldolase Schiff-base formation.
Keywords: Bürgi–Dunitz angle, imines, induced fit, nucleophiles, pentose phosphate pathway, Schiff bases
Abstract
The Bürgi–Dunitz angle (αBD) describes the trajectory of approach of a nucleophile to an electrophile. The adoption of a stereoelectronically favorable αBD can necessitate significant reactive-group repositioning over the course of bond formation. In the context of enzyme catalysis, interactions with the protein constrain substrate rotation, which could necessitate structural transformations during bond formation. To probe this theoretical framework vis-à-vis biocatalysis, Schiff-base formation was analysed in Francisella tularensis transaldolase (TAL). Crystal structures of wild-type and Lys→Met mutant TAL in covalent and noncovalent complexes with fructose 6-phosphate and sedoheptulose 7-phosphate clarify the mechanism of catalysis and reveal that substrate keto moieties undergo significant conformational changes during Schiff-base formation. Structural changes compelled by the trajectory considerations discussed here bear relevance to bond formation in a variety of constrained enzymic/engineered systems and can inform the design of covalent therapeutics.
1. Introduction
In physical organic chemistry theory, the Bürgi–Dunitz angle (αBD) defines the trajectory of a nucleophile towards electrophilic sp 2 reaction partners. In the 1970s, Hans-Beat Bürgi and Jack Dunitz analyzed small-molecule crystal structures that contained an amine and a proximal carbonyl (Bürgi et al., 1973 ▶; Bürgi, Dunitz et al., 1974 ▶; Fleming, 2010 ▶). These analyses revealed that the amine was reliably positioned at an obtuse angle (105 ± 5°) relative to the plane of the carbonyl bond. The closer the amine was to the carbonyl C atom, the greater the sp 3 character of the carbonyl (as assessed based on geometry and bond length), leading to the proposal that the structures captured snapshots of nucleophilic approach (Bürgi et al., 1973 ▶; Bürgi, Dunitz et al., 1974 ▶; Fleming, 2010 ▶). These structural insights, coupled with subsequent molecular-orbital theory-based computational work, established an obtuse αBD as the favored nucleophilic approach trajectory (Fig. 1 ▶; Bürgi, Lehn et al., 1974 ▶; Fleming, 2010 ▶).
Figure 1.
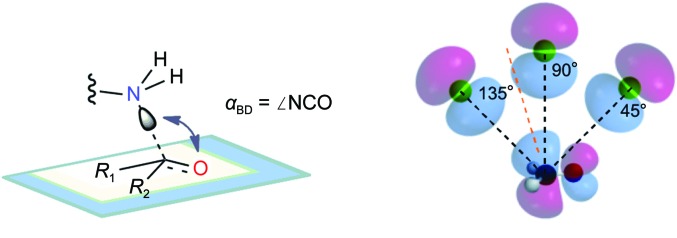
Physical organic chemistry concepts regarding the trajectory of the approach of a nucleophile to an electrophilic center. (a) The Bürgi–Dunitz angle (αBD) describes the trajectory of approach of a nucleophile (the lysine N∊ atom in the TAL reaction) to an electrophilic center (the substrate carbonyl C atom in TAL). Atoms and groups of a carbonyl R 1C(O)R 2 are positioned on a blue-grey plane and an approaching N atom is poised above. The αBD is estimated as ∠N∊⋯C=O (arrow). (b) The αBD originates in HOMO–LUMO overlap. After Bürgi, Lehn et al. (1974 ▶) and Fleming (2010 ▶), a side-on cartoon view of the approach of the two lobes of the p-type highest occupied molecular orbital (HOMO) of nitrogen (green) is shown at three hypothetical positions (αBD = 135, 90 and 45°). Poses are relative to the four lobes of the antibonding π*-type lowest unoccupied molecular orbital (LUMO) of formaldehyde, H2C=O, the simplest carbonyl electrophile (standard atom colors). Per current theory, carbinolamine C—N and C—O single bonds result when the nitrogen trajectory follows an obtuse value of αBD, allowing HOMO–LUMO overlap by wavefunctions (lobes) of like sign (shading) such that this attractive potential overcomes other repulsive potentials (Bürgi, Dunitz et al., 1974 ▶; Fleming, 2010 ▶). An approximate optimum computed for addition to formaldehyde of the simplest nucleophile, hydride anion (Hψ), is indicated by the orange dashed line (αBD ≃ 107°, SCF calculations; Bürgi et al., 1973 ▶).
Proteins within the class I aldolase superfamily have an active-site lysine, the N∊ atom of which reacts with a substrate carbonyl to form a catalytically important Schiff base (Choi et al., 2006 ▶). A number of other proteins, including those that utilize the cofactor pyridoxal phosphate, similarly exploit Schiff bases in the commission of their biological function. Within these enzymes, the Bürgi–Dunitz approach trajectory necessitates significant structural rearrangements over the course of Schiff-base formation. Since the lysine N∊ atom initially approaches the substrate sp 2 carbonyl at an obtuse αBD but after Schiff-base formation forms a component that defines the sp 2 plane, it follows that >90° of angular rotation must occur during the intervening chemistry.
When a small-molecule substrate is bound to an enzyme active site, interactions with the protein constrain its rotational freedom and impact the types of structural rearrangements that it can undergo. We hypothesized that combinations of three classes of active-site rearrangement could achieve the angular rotation required for Schiff-base formation (Fig. 2 ▶).
(i) Rigid protein, rigid substrate is characterized by the entirety of the substrate rotating relative to the protein.
(ii) Flexible protein, rigid substrate is characterized by protein conformational change, resulting in the lysine N∊ atom rotating relative to the substrate.
(iii) Rigid protein, flexible substrate is characterized by substrate conformational change, resulting in the plane of the substrate double bond rotating relative to the protein.
Figure 2.
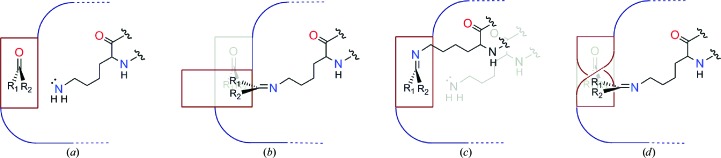
Categories of conformational change that could accompany Schiff-base formation within an active site. The nucleophilic approach of the lysine N∊ atom to a substrate carbonyl is predicted to follow an obtuse αBD (see Fig. 1 ▶). (a) Pre-nucleophilic attack state. The substrate forms an initial interaction with the enzyme that positions its carbonyl at an appropriate αBD relative to the lysine N∊ atom. For the N∊ atom to assume an obtuse αBD but ultimately represent a component of the Schiff base, it follows that >90° of angular rotation must occur in the intervening bond formation. Three categories of positional/conformational change could occur individually or in combination to achieve this rotation. (b) Rigid enzyme, rigid substrate. In this case, the conformations of main/side chains of the enzyme do not change from the pre-nucleophilic attack state positions, but the overall position of the substrate changes via gross rotation toward the lysine N∊ atom, without significant change to internal substrate dihedral angles. (c) Flexible enzyme, rigid substrate. The position and internal dihedrals of the substrate do not change from their pre-nucleophilic attack state values, but significant protein conformational change results in movement of the lysine N∊ atom toward the plane of the substrate carbonyl. (d) Rigid enzyme, flexible substrate. The conformation of the enzyme and the gross position of the substrate do not change, but a significant substrate conformational change (change in internal dihedrals) results in rotation of the plane of the substrate carbonyl towards the lysine N∊ atom. Within the schematic representation, the substructures of the carbonyl substrate, R 1C(O)R 2, are represented by the maroon ribbon, with internal conformational change indicated by a twist in the ribbon. The active site with its reactive lysine is represented within the dark blue body and ledge.
To assess the types of structural rearrangement that occur, we sought to chart the formation of a Schiff base in a representative system.
Transaldolase (TAL), the second enzyme in the non-oxidative part of the pentose phosphate pathway, swaps glyceraldehyde 3-phosphate and erythrose 4-phosphate aldoses to interconvert the six-carbon and seven-carbon keto-phosphosugars fructose 6-phosphate (F6P) and sedoheptulose 7-phosphate (S7P) (Horecker & Smyrniotis, 1953 ▶). A series of biochemical and structural studies have established the TAL reaction mechanism, which contains a covalent Schiff-base intermediate that links substrate to an active-site lysine (Fig. 3 ▶, Supplementary Fig. S11; Lehwess-Litzmann et al., 2011 ▶; Samland & Sprenger, 2009 ▶; Schörken et al., 2001 ▶). Because transaldolase acts on a linear ketone substrate, as is typical of class I aldolases, we reasoned that the enzyme was well suited to serve as a model to probe the types of structural rearrangement that occur during Schiff-base formation.
Figure 3.
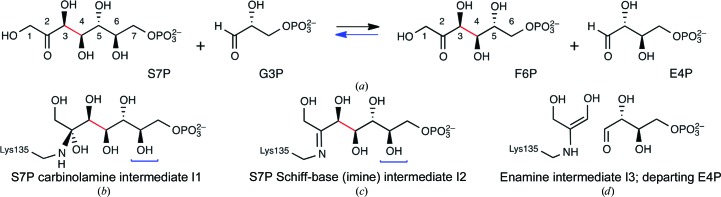
Substrates, products and intermediates in the TAL reaction. (a) Overall transformation of sedoheptulose 7-phosphate (S7P) and glyceraldehyde 3-phosphate (G3P) to fructose 6-phosphate (F6P) and erythrose 4-phosphate (E4P). The carbon–carbon bond involved in the TAL-catalyzed tandem retroaldol/aldol sequence is highlighted in red. The remaining panels show the covalent enzymic intermediates involved: (b) carbinolamine I1, initially formed by nucleophilic attack of the TAL active-site lysine on the S7P ketone carbonyl, (c) the subsequent imine I2, formed by dehydration of the carbinolamine, and (d) hydroxy-enamine (dihydroxyacetone) intermediate I3, the result of the retroaldol reaction of the preceding intermediate, shown with the departing E4P product. After departure of E4P and binding of G3P, the subsequent aldol reaction between I3 and G3P yields F6P; upon F6P release, the free enzyme is regenerated. F6P-type intermediates generated in this direction correspond to structures I1, I2 and I3, but lack the hydroxymethine [–CH(OH)–] unit underscored in the intermediates by a blue bracket. See Supplementary Fig. S1 for a more detailed representation of reaction mechanism.
Two types of TALs have previously been characterized. The more common ‘classical TAL’ (CT) is a 35–40 kDa protein that forms a homodimer, whereas ‘TAL-like’ (TLL) is a 20–25 kDa protein that forms a homodecamer (Supplementary Figs. S2a and S2b; Jia et al., 1996 ▶; Thorell et al., 2000 ▶, 2002 ▶; Reizer et al., 1995 ▶). Despite significant differences in size and oligomeric assembly, CTs and TLLs share highly similar active sites (Supplementary Fig. S2c). F6P and S7P Schiff-base-bound complexes have previously been reported for TLLs but not for the more common CTs (Lehwess-Litzmann et al., 2011 ▶). Structural studies of Francisella tularensis CT thus promised to serve the dual purpose of addressing structural transformations associated with Schiff-base formation and shedding light on distinctions in transaldolase-type function.
2. Materials and methods
2.1. Gene cloning, protein expression and protein purification
The talB gene was amplified from F. tularenesis subspecies tularensis SCHU S4 genomic DNA by PCR, subcloned into the pMCSG7 expression vector and transformed into Escherichia coli BL21 (DE3) cells utilizing previously described standardized methods (Light, Minasov, Shuvalova, Duban et al., 2011 ▶; Light, Minasov, Shuvalova, Peterson et al., 2011 ▶). 1 l TB medium was inoculated with overnight culture and then incubated at 37°C for 4 h. The temperature was then reduced to 25°C and protein overexpression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.5 mM. Following overnight growth, the cells were harvested by centrifugation, resuspended in a buffer consisting of 10 mM Tris–HCl pH 8.3, 500 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol and then lysed by sonication. Following centrifugation, the soluble fraction of the resulting cell lysate was purified by Ni–NTA affinity chromatography. Approximately 60 mg protein was obtained by a step elution with 500 mM imidazole in a buffer consisting of 10 mM Tris–HCl pH 8.3, 500 mM sodium chloride, 5 mM β-mercaptoethanol. A single band consistent with the expected molecular weight of F. tularensis CT was observed by SDS–PAGE chromatography.
2.2. Site-directed mutagenesis
The QuikChange XL Site-Directed Mutagenesis Kit (Agilent) was used to generate the K135M mutation. Successful incorporation of the mutation was confirmed by sequence analysis. The expression and purification steps described for the wild-type protein were followed to isolate the K135M variant.
2.3. Protein crystallization and data collection
Immediately following purification, the protein was concentrated using Amicon centrifugation devices (Millipore) and crystallized by the sitting-drop vapour-diffusion method using a 1:1 ratio of protein (at 11.2 mg ml−1 for the wild type and 9.4 mg ml−1 for the K135M variant) to reservoir. Wild-type and K135M variants were screened in the presence of 2 mM F6P or 2 mM S7P. Crystals were harvested from conditions consisting of 0.2 M magnesium acetate, 20% PEG 3350 (unliganded structure), 0.1 M bis-tris pH 5.5, 25% PEG 3350 (F6P Schiff-base complex), 0.1 M sodium acetate, 25% PEG 4000 (S7P Schiff-base complex), 0.2 M potassium/sodium tartrate, 20% PEG 3350 (K135M F6P complex) or 0.1 M HEPES pH 7.5, 25% PEG 2000 monomethyl ether (K135M S7P complex). Crystals were flash-cooled in liquid nitrogen from mother liquor consisting of the crystallization condition supplemented with 10 mM of the co-crystallization ligand. Diffraction data were collected at −173°C on the Life Sciences Collaborative Access Team beamlines at the Advanced Photon Source, Argonne, Illinois, USA.
2.4. Structure determination and refinement
Data were processed using HKL-3000 for indexing, integration and scaling (Minor et al., 2006 ▶). The structures were determined by molecular replacement in Phaser using the phosphate-bound structure (PDB code 3igx; Center for Structural Genomics of Infectious Diseases, unpublished work) as the starting model (McCoy et al., 2005 ▶). The structures were refined with REFMAC (Murshudov et al., 2011 ▶). Models were displayed in Coot and manually adjusted based on electron-density maps (Emsley & Cowtan, 2004 ▶). The final refined coordinates were deposited in the PDB with accession codes 4e0c (acetate-bound), 3tk7 (F6P Schiff-base complex), 3tno (S7P Schiff-base complex), 3te9 (K135M F6P complex), and 3tkf (K135M S7P complex). All residues within each of the five structures fall within allowed regions of the Ramachandran plot, with the exception of Ser230 (the side chain of which hydrogen bonds to the sugar phosphate group in the complex structures) in a subset of subunits (Table 1 ▶). Structure figures were prepared with PyMOL (v.1.3; Schrödinger).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Acetate | F6P | S7P | K135M–F6P | K135M–S7P | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Unit-cell parameters | |||||
| a (Å) | 54.76 | 56.12 | 56.32 | 54.84 | 54.83 |
| b (Å) | 87.02 | 74.25 | 74.09 | 86.73 | 87.19 |
| c (Å) | 140.69 | 165.00 | 165.42 | 140.49 | 141.14 |
| α = β = γ (°) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| Resolution range (Å) | 29.59–1.80 (1.85–1.80) | 28.99–2.00 (2.05–2.00) | 28.99–1.65 (1.69–1.65) | 29.58–1.80 (1.85–1.80) | 29.67–1.50 (1.54–1.50) |
| Completeness (%) | 98.3 (96.4) | 99.6 (97.1) | 99.9 (99.7) | 99.8 (98.6) | 100.0 (100.0) |
| Multiplicity | 6.6 (6.5) | 4.9 (4.9) | 7.3 (7.3) | 7.2 (7.2) | 7.2 (5.0) |
| 〈I/σ(I)〉 | 20.2 (4.0) | 18.6 (3.6) | 29.0 (4.3) | 24.5 (3.7) | 23.1 (3.7) |
| R merge † (%) | 7.7 (49.1) | 8.4 (47.4) | 6.8 (53.8) | 7.5 (58.2) | 6.9 (45.2) |
| Wilson B factor (Å2) | 20.2 | 21.8 | 19.8 | 24.3 | 17.3 |
| Refinement statistics | |||||
| Resolution range (Å) | 29.59–1.80 (1.85–1.80) | 28.99–2.00 (2.05–2.00) | 28.99–1.65 (1.69–1.65) | 29.58–1.80 (1.85–1.80) | 29.67–1.50 (1.54–1.50) |
| No. of reflections | 62125 (4137) | 47362 (3101) | 84042 (5737) | 62778 (4453) | 108935 (7982) |
| R work/R free ‡ (%) | 15.1/18.9 | 18.8/23.2 | 17.4/20.3 | 16.0/19.2 | 14.5/15.9 |
| No. of atoms | |||||
| Protein | 4872 | 4978 | 4978 | 4867 | 4867 |
| Waters | 753 | 654 | 979 | 610 | 766 |
| F6P/S7P | — | 32 | 36 | 16 | 36 |
| Average B factors (Å2) | |||||
| Protein | 23.3 | 27.2 | 21.6 | 27.1 | 18.5 |
| Waters | 32.9 | 32.2 | 31.8 | 37.5 | 30.4 |
| F6P/S7P | — | 16.9 | 18.2 | 24.9 | 16.5 |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.020 | 0.007 | 0.005 | 0.011 | 0.012 |
| Bond angles (°) | 1.95 | 1.19 | 1.07 | 1.33 | 1.52 |
| Ramachandran analysis | |||||
| Favored regions (%) | 95.1 | 99.3 | 99.0 | 99.0 | 99.0 |
| Allowed regions (%) | 100.0 | 99.7 | 99.6 | 99.9 | 99.7 |
| Disallowed regions (%) | 0.0 | 0.3 | 0.4 | 0.1 | 0.3 |
R
merge = 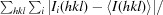
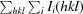 .
.
R
work and R
free are defined as R = 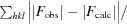
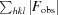 , where hkl are the reflections used in refinement for R
work and 5% of reflections that were not used in refinement for R
free. F
obs and F
calc are structure factors deduced from measured intensities or calculated from the model, respectively.
, where hkl are the reflections used in refinement for R
work and 5% of reflections that were not used in refinement for R
free. F
obs and F
calc are structure factors deduced from measured intensities or calculated from the model, respectively.
3. Results
3.1. Structure of F. tularensis transaldolase
A structure of F. tularensis CT was determined at a resolution of 1.80 Å in space group P212121 (Table 1 ▶). Two molecules comprising the physiological dimer are present within the crystallographic asymmetric unit (Fig. 4 ▶ a). The core of the enzyme forms a classic TIM-barrel (α/β)8-fold, while six helices inserted at various positions decorate the exterior of the barrel (Fig. 4 ▶ b). The predicted Schiff-base-forming lysine, Lys135, is located near the midpoint of the internal wall of the barrel and towards the bottom of the active-site channel. Inclusion of a biological anion (phosphate, sulfate or acetate) in the crystallization buffer was necessary to obtain high-resolution diffracting crystals. In one of the molecules in the asymmetric unit an acetate anion is observed near the entrance of the active-site channel (Supplementary Fig. S3). A superposition with the E. coli and human TALs reveals that the F. tularensis enzyme is nearly identical to these well studied CTs in tertiary and quaternary structure (Supplementary Fig. S4).
Figure 4.

Structure of F. tularensis transaldolase. (a) Depicted in cartoon representation, the crystallographic asymmetric unit contains the physiological dimer. (b) Colored by secondary structure, the TAL monomer forms a classic TIM barrel. The labeled Schiff-base-forming Lys135 is found near the middle of the internal wall of the barrel.
3.2. F6P and S7P Schiff-base intermediate states
Structures of TAL crystallized in the presence of F6P and S7P were determined at resolutions of 2.0 and 1.65 Å, respectively (Table 1 ▶). An inspection of the resulting maps revealed density for the phosphosugars at the active site. In both cases, the density of the sugar C2 atom is continuous with the Lys135 N∊ atom, demonstrating the presence of a covalent reaction intermediate. As no density that might correspond to the carbinolamine intermediate-state O atom is observed and the planar shape of the lysine–sugar connection is indicative of the trigonal hybridization state, F6P and S7P Schiff-base reaction-intermediate states were modeled and fitted the density nicely (Figs. 5 ▶ a and 5 ▶ b).
Figure 5.

F6P and S7P Schiff-base-bound complexes. (a) Stick model of the F6P Schiff-base-bound active site. (b) Stick model of the S7P Schiff-base-bound active site. The 2F o − F c (blue, contoured at 1.0σ) and F o − F c (red, contoured at 2.0σ) electron-density maps were calculated with ligand omitted from the model. (c) Superposition of the F6P and S7P complexes.
A comparison of the two Schiff-base-bound complexes reveals that the chemically identical portions of the sugars (i.e. C1–C6) superimpose to form very similar interactions with TAL (Fig. 5 ▶ c, Supplementary Fig. S6). The difference in sugar length means that the F6P and S7P phosphates are offset, with the S7P phosphate ∼1.3 Å closer to the entrance of the active-site channel. A comparison of these structures with the corresponding TLL complexes reveals very similar modes of sugar binding in the two TAL classes (Supplementary Fig. S6), but also four particularly notable distinctions. Firstly, in CT but not TLL complexes, Arg232 conformational change adapts the active site to the varying position of the substrate phosphate group. These and associated conformational changes in the CT-specific α9–α10 connecting loop are likely to relate to distinctions in relative substrate affinity that functionally differentiate the two TAL types (see §S1 and Supplementary Fig. S7). Secondly, in the TLL structures the ∼55 most N-terminal residues undergo rigid-body movement in response to substrate binding, closing over the active site (Supplementary Fig. S8a). In contrast, the N-terminus retains a single conformational state in all of the CT structures. This can be explained because one of the CT-specific insertions (Supplementary Fig. S2) extends a shorter TLL helix (α2) and establishes an additional helix (αC) in the CT structure. As this appendage forms a stable interaction with the C-terminal helix, its presence could constrain N-terminal mobility and may explain the differential behavior of the two TAL types (Supplementary Fig. S8b). Thirdly, the 1-hydroxyl group of F6P and S7P adopts multiple (at least two) conformations in TLL complexes, whereas an analysis of electron density and temperature factors suggests a single conformation in CT complexes. Since residues within the vicinity of the hydroxyl are strictly conserved, the source and significance of these conformational distinctions is unclear. Finally, in reporting the TLL F6P and S7P complexes, Lehwess-Litzmann et al. (2011 ▶) posit that the geometry of the protein–substrate Schiff-base linkage deviates from planarity. They suggest that this distortion may be functionally relevant, hypothesizing that it reduces the activation barrier of Schiff-base hydrolysis by destabilizing the Schiff-base-bound intermediate state. When dealing with such subtle distinctions in atom position, caution is advisable to avoid over-interpreting electron density. Despite the comparable resolution of our CT complexes, we find any such distortion in Schiff-base geometry to be unresolvable. While a slight deviation in this bond geometry is present within the deposited structure coordinates, the application of more stringent refinement constraints readily generates an undistorted model that reasonably fits the observed electron density. Moreover, partial substrate occupancy (which would imply the presence of partially occupied active-site water molecules) or alternative substrate conformations (such as those described for the 1-hydroxyl group in the TLL structures) could complicate the model that gives rise to the observed density profile. For these reasons, we conclude that the crystallographic data for the CT F6P and S7P complexes are of insufficient quality to determine unequivocally whether the Schiff base is distorted from planarity.
3.3. F6P and S7P noncovalent complexes of the K135M mutant
It has previously been reported that mutation of the Schiff-base-forming lysine abolishes TAL activity (Banki & Perl, 1996 ▶; Miosga et al., 1993 ▶). To prevent formation of the Schiff base and to facilitate the capture of noncovalent reaction complexes, Lys135 was mutated to methionine (K135M). While disallowing Schiff-base formation, we reasoned that the K135M mutant should faithfully replicate the steric environment of the active site and thus allow capture of the pre-nucleophilic attack state. Structures of the K135M mutant crystallized in the presence of F6P and S7P were determined at resolutions of 1.8 and 1.5 Å, respectively (Table 1 ▶). An analysis of the resulting maps revealed electron density for the sugars at the active site (Figs. 6 ▶ a and 6 ▶ b). In contrast to the wild-type complexes, density for the substrates is discontinuous with the enzyme and the planar 2-carbonyl bonds are clearly visible in the K135M variant.
Figure 6.

The K135M mutant in complex with F6P and S7P. (a) Stick model of the K135M mutant in complex with F6P. (b) Stick model of the K135M mutant in complex with S7P. The 2F o − F c (blue, contoured at 1.0σ) and F o − F c (red, contoured at 2.0σ) electron-density maps were calculated with ligand omitted from the model. (c) Superposition of the K135M F6P and S7P complexes.
A comparison of the K135M F6P-bound and S7P-bound structures reveals that the C1–C3 atoms superimpose, but the path of the sugars diverges slightly at C4, so that by the F6P P atom the chains are separated by ∼0.8 Å. Despite this small difference, protein–sugar interactions are basically preserved across the two structures (Fig. 6 ▶ c, Supplementary Fig. S10, §S1).
In contrast to the Schiff-base-bound structures, in which linkage to Lys135 means that the C2 double bond (Schiff base) is directed towards the residue, in the noncovalent complex the C2 double bond (carbonyl) is rotated away from Met135. Given the extra O atom of the carbonyl and the greater separation between the sugar and residue 135 that is required for the noncovalent state, there simply is insufficient room for the carbonyl to point towards residue 135. Repulsive steric forces must therefore ensure that C2 twists away from its covalently bound conformation. In this position, the carbonyl O atom is within hydrogen-bonding distance of the Thr159 side chain (Fig. 6 ▶ c). Thr159 is a strictly conserved and catalytically essential residue that has been attributed functional roles in coordinating the catalytic water and stabilizing the carbinolamine reaction intermediate (Samland & Sprenger, 2009 ▶; Schörken et al., 2001 ▶). The observed Thr159–carbonyl interaction suggests that this residue may also be important for the initial substrate-binding event, and in particular in orienting the carbonyl bond so as to expose C2 for nucleophilic attack.
Because of the K135M mutation, the pre-nucleophilic approach conformation of Lys135 is not directly revealed by the noncovalent complexes. However, if the lysine retained its unliganded conformation the N∊ atom would sterically clash with the substrate (Supplementary Fig. S10). Thus, the binding of substrate alters the surrounding environment and is likely to induce a conformational change of Lys135. Notably, a subtle movement of Lys135 could resolve the steric clash and place the N∊ atom along a reasonable approach trajectory to the carbonyl bond (Supplementary Fig. S10). Thus, while the structures do not directly reveal the nucleophilic approach trajectory, the observed substrate-binding mode is consistent with a favorable αBD.
4. Discussion
A comparison of noncovalent and Schiff-base structures allows the characterization of structural rearrangements that occur over the course of Schiff-base formation. This analysis reveals that Schiff-base formation is associated with a ∼1.1 Å shift in the C2 atom position and a substantial rotation in the plane of its double bond (Fig. 7 ▶). Despite this rather significant change in C2 bond orientation, the functional groups of the substrate preserve interactions before and after Schiff-base formation. Maintenance of the hydrogen-bonding network throughout bond formation is accomplished by a small rotation around the C3—C4/C4—C5 bonds and larger rotations around the C1—C2/C2—C3 bonds (Fig. 7 ▶).
Figure 7.

Structural changes associated with F6P and S7P Schiff-base formation. Superposition of (a) F6P and (b) S7P noncovalent (beige) and Schiff-base-bound (yellow) complexes. Differences in (c) F6P and (d) S7P noncovalent (top panels) and covalent (bottom panels) conformational states result from a ∼0.5 Å translational shift and the calculated rotations around the bond dihedrals ∠O1—C1—C2—O2 (purple), ∠O2—C2—C3—O3 (red), ∠O3—C3—C4—O4 (marine) and ∠O4—C4—C5—O5 (pink).
In light of these observations, it is clear that Schiff-base formation most closely parallels the substrate conformational change-dominated scenario (Fig. 2 ▶ d). In other words, the sugar substrate initially docks in an orientation that exposes C2 to an obtuse αBD approach of the N∊ atom. As the Schiff base forms, changes in internal substrate dihedrals (specifically within C1—C4 bonds) cause C2 to rotate to its carbinolamine and then its Schiff-base-bound position, while only minimally affecting the position of other atoms in the substrate.
The question then turns to the generalizability of substrate conformational change as a means of accommodating the structural rearrangements necessitated by Bürgi–Dunitz principles. Unfortunately, there are few other adequately characterized Schiff-base-forming reactions to form a basis for reference. Structures of the rabbit muscle fructose-1,6-bisphosphate aldolase, which catalyzes a reaction related to the TAL enzyme (Supplementary Fig. S11a), in complex with the reaction product dihydroxyacetone-phosphate have been reported in both pre-nucleophilic attack and Schiff-base intermediate states (Blom & Sygusch, 1997 ▶; St-Jean & Sygusch, 2007 ▶). An analysis of the noncovalent complex reveals that the orientation of the substrate carbonyl allows a favorable αBD approach of the N∊ atom (Supplementary Fig. S11b). A comparison to the Schiff-base-bound complex reveals that changes in the dihydroxyacetone-phosphate conformation occur as the carbonyl C atom moves to its Schiff-base-bound position and, therefore, similar to TAL, substrate conformational change dominates the structural rearrangement associated with Schiff-base formation (Supplementary Fig. S11b).
In two other cases, bond formation appears to qualitatively differ from the substrate conformational change-dominated scenario apparently present in the transaldolase and fructose-1,6-bisphosphate aldolase reactions. Firstly, cellular retinoic acid-binding protein II has been re-engineered to form a Schiff base with retinal (Vasileiou et al., 2007 ▶). In this artificial system, a comparison of mutants to which retinal binds noncovalently and covalently suggested that an extended portion of the retinal molecule rotates as a rigid body over Schiff-base formation (Vasileiou et al., 2007 ▶). Secondly, dehydroquinate dehydratase has been characterized in noncovalent and Schiff-base-bound states, but the unexpected similarity of these structures combined with an apparently unfavorable αBD nucleophilic approach call into question whether the noncovalent complex accurately reflects the Michaelis state (Light, Minasov, Shuvalova, Duban et al., 2011 ▶). Interestingly, among the characterized Schiff-base-forming enzymes, dehydroquinate dehydratase catalyzes the only reaction in which the reactive carbonyl is contained within a carbocyclic ring. The fact that the carbonyl lies within this ring constrains the allowed rotation of the adjacent bond dihedrals. Rotations of the order of magnitude observed in the TAL and fructose-1,6-bisphosphate aldolase reactions are not possible. Therefore, in effect, the cyclic nature of the dehydroquinate dehydratase substrate disallows extensive substrate conformational changes of the type observed in the other Schiff-base-forming enzymes.
In all probability, the substrate conformational change-dominated mechanism of Schiff-base formation (Fig. 2 ▶ d) utilized by TAL and fructose-1,6-bisphosphate aldolase is common in enzymes acting upon linear ketone substrates. This strategy is likely to represent the most parsimonious mechanism of accomplishing the structural rearrangements necessitated by the Bürgi–Dunitz approach. A mechanism of bond formation dominated by protein conformational change (Fig. 2 ▶ c) would require complex protein motion over the timescale associated with bond formation. Alternatively, a mechanism dominated by rigid-body rotation of the substrate (Fig. 2 ▶ b) would require that the substrate form and break three non-overlapping sets of interactions with the protein while transitioning through noncovalent, carbinolamine and Schiff-base intermediate states. As the mechanism dominated by substrate conformational change is the only one that allows the obtuse αBD approach of the N∊ atom without necessitating complex changes in protein–ligand interactions, it arguably presents the simplest mechanism of accomplishing the required angular rotation with this class of substrate.
By contrast, Schiff-base-forming enzymes with distinctive substrates almost certainly employ different rearrangement strategies. For substrates with a reactive aldehyde, such as pyridoxal phosphate, structural rearrangement may be confined to the aldehyde H atoms and thus minimal gross conformational change may be required. For reactions that involve smaller substrates, such as pyruvate, rigid-body rotation may be necessary and/or less energetically costly. In this way, smaller substrates might be anticipated to behave analogously to non-enzymatic reactions, where, in the absence of motion restricting substrate–active site interactions, the barrier to rigid rotation should be minimal.
In general, it seems that the existence of energetically favorable approach trajectories can influence the mechanics of macromolecular bond formation in ways that have been underappreciated. While Schiff-base-forming enzymes serve as the focus of this paper, the functional implications of the analysis extend beyond this relatively small family of proteins. Nucleophilic approach occurs in as varied biochemical processes as translation and serine protease catalysis. Similar macromolecular and/or substrate conformational rearrangements will be relevant for these and many other reactions. The structural analysis presented here also has practical applications, particularly in the development of covalent inactivators. Accounting for structural rearrangement will be useful for prospectively engineering sufficient flexibility to allow covalent inhibitors to undergo the angular rotation required for inactivating bond formation.
Supplementary Material
PDB reference: transaldolase, acetate-bound, 4e0c
PDB reference: F6P Schiff-base complex, 3tkf
PDB reference: S7P Schiff-base complex, 3tno
PDB reference: K135M F6P complex, 3te9
Supporting Information.. DOI: 10.1107/S1399004713030666/dw5075sup1.pdf
Acknowledgments
We thank Dr Scott Peterson and Dr Keehwan Kwon for providing the talB expression clone and Dr Misty Kuhn and Dr Ludmilla Shuvalova for guidance. The Center for Structural Genomics of Infectious Diseases has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract Nos. HHSN272200700058C and HHSN272201200026C (to WFA). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (grant 085P1000817).
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: DW5075).
References
- Banki, K. & Perl, A. (1996). FEBS Lett. 378, 161–165. [DOI] [PubMed]
- Blom, N. & Sygusch, J. (1997). Nature Struct. Biol. 4, 36–39. [DOI] [PubMed]
- Bürgi, H.-B., Dunitz, J., Lehn, J. M. & Wipff, G. (1974). Tetrahedron, 30, 1563–1572.
- Bürgi, H.-B., Dunitz, J. D. & Shefter, E. (1973). J. Am. Chem. Soc. 95, 5065–5067.
- Bürgi, H.-B., Lehn, J. M. & Wipff, G. (1974). J. Am. Chem. Soc. 96, 1956–1957. [DOI] [PubMed]
- Choi, K. H., Lai, V., Foster, C. E., Morris, A. J., Tolan, D. R. & Allen, K. N. (2006). Biochemistry, 45, 8546–8555. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fleming, I. (2010). Moleculular Orbitals and Organic Chemical Reactions Chichester: John Wiley & Sons.
- Horecker, B. L. & Smyrniotis, P. Z. (1953). J. Am. Chem. Soc. 75, 2021–2022.
- Jia, J., Huang, W., Schörken, U., Sahm, H., Sprenger, G. A., Lindqvist, Y. & Schneider, G. (1996). Structure, 4, 715–724. [DOI] [PubMed]
- Laskowski, R. A. & Swindells, M. B. (2011). J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed]
- Lehwess-Litzmann, A., Neumann, P., Parthier, C., Lüdtke, S., Golbik, R., Ficner, R. & Tittmann, K. (2011). Nature Chem. Biol. 7, 678–684. [DOI] [PubMed]
- Light, S. H., Minasov, G., Shuvalova, L., Duban, M. E., Caffrey, M., Anderson, W. F. & Lavie, A. (2011). J. Biol. Chem. 286, 3531–3539. [DOI] [PMC free article] [PubMed]
- Light, S. H., Minasov, G., Shuvalova, L., Peterson, S. N., Caffrey, M., Anderson, W. F. & Lavie, A. (2011). Biochemistry, 50, 2357–2363. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Miosga, T., Schaaff-Gerstenschlager, I., Franken, E. & Zimmermann, F. K. (1993). Yeast, 9, 1241–1249. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Reizer, J., Reizer, A. & Saier, M. H. Jr (1995). Microbiology, 141, 961–971. [DOI] [PubMed]
- Samland, A. K. & Sprenger, G. A. (2009). Int. J. Biochem. Cell Biol. 41, 1482–1494. [DOI] [PubMed]
- Schörken, U., Thorell, S., Schürmann, M., Jia, J., Sprenger, G. A. & Schneider, G. (2001). Eur. J. Biochem. 268, 2408–2415. [DOI] [PubMed]
- St-Jean, M. & Sygusch, J. (2007). J. Biol. Chem. 282, 31028–31037. [DOI] [PubMed]
- Thorell, S., Gergely, P. Jr, Banki, K., Perl, A. & Schneider, G. (2000). FEBS Lett. 475, 205–208. [DOI] [PubMed]
- Thorell, S., Schürmann, M., Sprenger, G. A. & Schneider, G. (2002). J. Mol. Biol. 319, 161–171. [DOI] [PubMed]
- Vasileiou, C., Vaezeslami, S., Crist, R. M., Rabago-Smith, M., Geiger, J. H. & Borhan, B. (2007). J. Am. Chem. Soc. 129, 6140–6148. [DOI] [PubMed]
- Watt, W. B. & Dean, A. M. (2000). Annu. Rev. Genet. 34, 593–622. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: transaldolase, acetate-bound, 4e0c
PDB reference: F6P Schiff-base complex, 3tkf
PDB reference: S7P Schiff-base complex, 3tno
PDB reference: K135M F6P complex, 3te9
Supporting Information.. DOI: 10.1107/S1399004713030666/dw5075sup1.pdf