

This study presents the crystal structure of a ∼320 Å long protein fiber generated by in-frame extension of its repeated helical coiled-coil core.
Keywords: protein fiber, protein engineering, bacteriophage P22, tail needle gp26, α-helical coiled coil
Abstract
Protein fibers are widespread in nature, but only a limited number of high-resolution structures have been determined experimentally. Unlike globular proteins, fibers are usually recalcitrant to form three-dimensional crystals, preventing single-crystal X-ray diffraction analysis. In the absence of three-dimensional crystals, X-ray fiber diffraction is a powerful tool to determine the internal symmetry of a fiber, but it rarely yields atomic resolution structural information on complex protein fibers. An 85-residue-long minimal coiled-coil repeat unit (MiCRU) was previously identified in the trimeric helical core of tail needle gp26, a fibrous protein emanating from the tail apparatus of the bacteriophage P22 virion. Here, evidence is provided that an MiCRU can be inserted in frame inside the gp26 helical core to generate a rationally extended fiber (gp26-2M) which, like gp26, retains a trimeric quaternary structure in solution. The 2.7 Å resolution crystal structure of this engineered fiber, which measures ∼320 Å in length and is only 20–35 Å wide, was determined. This structure, the longest for a trimeric protein fiber to be determined to such a high resolution, reveals the architecture of 22 consecutive trimerization heptads and provides a framework to decipher the structural determinants for protein fiber assembly, stability and flexibility.
1. Introduction
Fibrous proteins such as collagens, adhesins and elastins contain highly repetitive amino-acid sequences that promote self-assembly to form elongated structures of extraordinary flexibility and resistance (Mitraki & van Raaij, 2005 ▶). This property has been exploited in protein nanotechnology to build nanoscale subunits that can be programmed to assemble into elongated structures (Hyman et al., 2002 ▶). Likewise, protein fibers are commonly found in viruses and bacteriophages, which use them as sensing devices and as structural components of capsids and tails (Conley & Wood, 1975 ▶; Veesler & Cambillau, 2011 ▶; Lander et al., 2006 ▶). A well studied example of a protein fiber is found in the tail machine of phage P22 (Bhardwaj et al., 2013 ▶), a prototypical member of the Podoviridae family of short-tailed phages (Casjens & Molineux, 2012 ▶). The P22 tail machine consists of five polypeptide chains, each of which is present as several copies (Olia et al., 2006 ▶; Bhardwaj et al., 2007 ▶; Tang et al., 2011 ▶); one of these components, the tail needle protein gp26, forms a ∼240 Å long trimeric coiled-coil fiber located at the distal tip of the P22 tail (Olia et al., 2007 ▶, 2009 ▶). The needle is inserted into the portal vertex structure at the end of the DNA-packaging process to stabilize the highly condensed genome inside the capsid (Strauss & King, 1984 ▶; Botstein et al., 1973 ▶; Berget & Poteete, 1980 ▶). During infection, gp26 is released from the virions, suggesting a role in genome ejection (Israel, 1976 ▶, 1978 ▶).
The three-dimensional structure and domain organization of the tail needle gp26 has been elucidated using crystallographic methods (Olia et al., 2007 ▶, 2009 ▶) and biochemical mapping analysis (Bhardwaj et al., 2007 ▶). The N-terminal tip of gp26 (residues 1–60) binds to tail protein gp10 and forms the plug that closes the P22 portal channel (Olia et al., 2011 ▶) after packaging. This is followed by an ∼100-residue-long trimeric α-helical coiled-coil core, which spans three quarters of the length of the gp26 needle and whose average diameter is only 25 Å, thinner than most known α-helical coiled-coil structures (Olia et al., 2007 ▶, 2009 ▶). Downstream of the gp26 helical core, at the virion distal tip, is a short triple β-helix connected to an inverted helical coiled coil (Olia et al., 2007 ▶, 2009 ▶). This domain is replaced by a β-stranded knob in some other members of the Podoviridae family that share the P22 gp26 helical core (Bhardwaj et al., 2009 ▶, 2011 ▶). The structural and conformational stability of gp26 have been studied both in vitro (Bhardwaj et al., 2007 ▶, 2009 ▶) and in vivo (Leavitt et al., 2013 ▶). The trimeric fiber is remarkably stable; it remains folded in the absence of water or in the presence of 10% SDS at room temperature. In solution, the subunits separate and denature irreversibly with an apparent midpoint of guanidine half concentration (C M) of 6.4 M and a melting temperature of ∼85°C (Bhardwaj et al., 2007 ▶, 2009 ▶; Botstein et al., 1973 ▶). Replacing the gp26 C-terminal domain (residues 141–233) with the ‘foldon’ domain of bacteriophage T4 fibritin results in a fiber of high stability which unfolds in a completely reversible manner (Bhardwaj et al., 2008 ▶).
We previously identified an ∼55-residue minimal coiled-coil repeat unit (MiCRU) spanning residues 84–139 of the gp26 helical core (Fig. 1 ▶; Bhardwaj et al., 2009 ▶). In-frame insertion of a MiCRU between heptad 6 and 7 of the gp26 helical core allowed us to extend gp26 modularly, generating rationally engineered fibers of increased length (named gp26-2M, gp26-3M etc.) and enhanced structural stability (Bhardwaj et al., 2009 ▶). In this work, we have determined the crystal structure of gp26-2M, a first-generation fiber that contains two tandemly repeated MiCRUs (Fig. 1 ▶ a). This fiber structure, the longest to be solved to high resolution using X-ray crystallography, provides clues to decipher the molecular determinants of protein-fiber assembly, stability and flexibility.
Figure 1.

In-frame extension of tail needle gp26. (a) Amino-acid sequence of the engineered fiber gp26-2M that contains two tandemly repeated MiCRUs (highlighted in blue and pink). Trimerization heptad repeats are numbered from 1 to 22 and residues in each heptad are designated a–b–c–d–e–f–g according to standard convention (Lupas, 1996 ▶). (b) Ribbon diagram of the gp26 MiCRU (residues 84–139 of PDB entry 3c9i).
2. Methods
2.1. Protein expression, purification and crystallization
An expression plasmid encoding gp26-2M was constructed as described previously (Bhardwaj et al., 2009 ▶). Recombinant gp26-2M fused to an N-terminal maltose-binding protein (MBP) was expressed in Escherichia coli. The MBP tag was cleaved with PreScission Protease and the resulting gp26-2M was separated from MBP using anion-exchange chromatography. Purified gp26-2M was subjected to size-exclusion chromatography on a Superdex S200 16/60 (GE Healthcare) column equilibrated in 20 mM Tris–HCl, 175 mM NaCl. Fractions containing gp26-2M protein were pooled and concentrated by ultrafiltration to 10 mg ml−1. Purified gp26-2M was screened for crystallization using the hanging-drop vapor-diffusion method at 298 K using crystallization kits from Hampton Research (California, USA). Protein droplets were prepared by mixing 3 µl of 10 mg ml−1 protein solution in 20 mM Tris buffer pH 8, 175 mM NaCl with 3 µl reservoir solution and equilibrating against 600 µl reservoir solution. Several conditions under which crystals appeared were further optimized by varying the concentrations of protein and salts at different pH values. Large diffraction-quality elongated rod-shaped crystals of dimensions ∼500 × 200 µm grew in 15–18 d using reservoir solution consisting of 0.1 M dibasic potassium phosphate, 0.1 M sodium citrate pH 4.5, 20%(w/v) polyethylene glycol (PEG) 8000 at 290 K. Prior to data collection, crystals were cryoprotected by quick passage through a solution consisting of mother-liquor solution supplemented with 27% ethylene glycol.
2.2. Data collection, structure determination and refinement
Crystals were screened on beamlines X6A and X29 at the Brookhaven National Synchrotron Light Source (NSLS), Upton, New York, USA and beamline F1 at the Cornell High Energy Synchrotron Source (CHESS), Ithaca, New York, USA under a constant stream of liquid nitrogen maintained at 100 K. The best diffraction data were collected at the F1 station on an ADSC Q270 CCD detector using an X-ray wavelength of 0.92 Å. Diffraction data were processed and scaled with the HKL-2000 suite (Otwinowski & Minor, 1997 ▶; Table 1 ▶).
Table 1. Crystallographic data-collection and refinement statistics.
Values in parentheses are for the outer resolution shell.
| Data-collection statistics | |
| Wavelength (Å) | 0.92 |
| Space group | P1 |
| Unit-cell parameters (Å, °) | a = 38.7, b = 147.9, c = 151.0, α = 87.9, β = 90.0, γ = 89.9 |
| Resolution range (Å) | 30–2.7 (2.8–2.7) |
| Wilson B factor (Å2) | 44.6 |
| Total observations | 187108 |
| Unique observations | 84119 |
| Completeness (%) | 92.0 (78.8) |
| R merge † (%) | 10.7 (26.1) |
| 〈I〉/〈σ(I)〉 | 13.7 (3.9) |
| Refinement statistics | |
| No. of reflections (15–2.7 Å) | 83626 |
| R work/R free ‡ (%) | 22.9/26.8 |
| No. of copies in asymmetric unit | 4 |
| No. of water molecules | 764 |
| Average B factors of model (Å2) | |
| Fiber | |
| A | 62.0 |
| B | 66.6 |
| C | 68.8 |
| D | 68.9 |
| Waters | 36.5 |
| Ions | 52.9 |
| R.m.s.d. from ideal bond lengths (Å) | 0.005 |
| R.m.s.d. from ideal bond angles (°) | 0.991 |
| Ramachandran plot (%) | |
| Core | 94.7 |
| Allowed | 4.9 |
| Generously allowed | 0.4 |
| Disallowed | 0.0 |
R
merge = 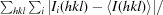
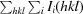 , where I
i(hkl) and 〈I(hkl)〉 are the ith and the mean measurement of the intensity of reflection hkl.
, where I
i(hkl) and 〈I(hkl)〉 are the ith and the mean measurement of the intensity of reflection hkl.
The R free value was calculated using 2136 reflections selected in thin resolution shells.
Acentric reflections were subjected to the H-test (Yeates, 1988 ▶), which gave a mean |H| of ∼0.083 (where 0.50 corresponds to untwinned and 0.0 corresponds to 50% twinned) and a mean H 2 of ∼0.016 (where 0.33 corresponds to untwinned and 0.0 corresponds to 50% twinned), indicative of twinning. Data-quality analysis using the phenix.xtriage routine from the PHENIX software suite v.1.8.2 (Adams et al., 2010 ▶) revealed the presence of pseudo-merohedral twinning with twin law (h, −k, −l) and an estimated twin fraction equal to 0.393. The structure was solved by molecular replacement with Phaser (McCoy, 2007 ▶) using three fragments of P22 gp26 (PDB entry 3c9i; Olia et al., 2009 ▶) as a search model corresponding to residues 1–84, 85–140 and 141–233. Four gp26-2M fibers were located in the asymmetric unit, which results in an estimated solvent content of 42.5%. The model was subjected to iterative cycles of positional refinement and isotropic B-factor refinement using 62 TLS groups in phenix.refine (Afonine et al., 2012 ▶) as well as manual building using Coot (Emsley & Cowtan, 2004 ▶). All steps of crystallographic refinement were carried out using a twin target function and twin law (h, −k, −l). The final refined twin fraction output by PHENIX was 0.48. The final refined model has an R work and an R free of 22.9 and 26.8%, respectively (Table 1 ▶). R free was calculated using 2136 reflections (2.55%) selected in thin resolution shells. MolProbity (Chen et al., 2010 ▶) evaluation of the Ramachandran plot gave 96.31% in the favored region, 3.63% in the allowed region and 0.06% outliers. Coordinates were deposited in the PDB with accession code 4lin.
2.3. Structure analysis
Structural superimpositions were performed using the secondary-structure matching algorithm in Coot (Emsley & Cowtan, 2004 ▶; Krissinel & Henrick, 2004 ▶). SOCKET (Walshaw & Woolfson, 2001 ▶) was used to determine coiled-coil regions and to assign heptad positions using a packing cutoff of 7.0 Å. Detailed coiled-coil geometry analysis was performed using TWISTER (Strelkov & Burkhard, 2002 ▶). Interhelical distances were calculated using interhlx (K. Yap, University of Toronto). Interface surface areas were analyzed using the PISA server (Krissinel & Henrick, 2007 ▶). Ribbon diagrams and electron-density representations were prepared using PyMOL (DeLano, 2002 ▶).
2.4. Sedimentation analysis
Sedimentation-velocity (SV) and sedimentation-equilibrium (SE) analyses were carried out with a ProteomeLab XL-I analytical ultracentrifuge (Beckman Coulter, Palo Alto, California, USA) using an eight-hole An-50 Ti rotor and a two-sector centerpiece for velocity runs and a six-sector centerpiece for equilibrium runs. Prior to centrifuge runs, the gp26-2M samples were extensively dialyzed against 0.02 M Tris–HCl pH 8.0, 0.1 M NaCl buffer at 4°C. The partial specific volume of gp26-2M (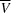 ), solvent density and relative viscosity values (0.7281 ml g−1, 1.00293 g ml−1 and 0.0010309 Pa s, respectively) along with the molecular mass were calculated using SEDNTERP v.1.09 (Laue et al., 1992 ▶; John Philo, Thousand Oaks, California, USA and RASMB; http://bitcwiki.sr.unh.edu). For SV runs, a two-sector 1.2 cm Epon centerpiece was loaded with 400 µl 70 µM (2.2 mg ml−1) gp26-2M (Table 2 ▶) and 420 µl dialysis buffer in the reference chamber. The runs were performed at 35 000 rev min−1 at a constant temperature of 10°C. Over ∼16 h, until complete sample sedimentation, absorbance values were collected at a fixed wavelength of 275 nm. The resulting data were fitted using a continuous sedimentation coefficient [c(s)] distribution model in SEDFIT (Schuck, 2000 ▶) and an estimated molecular mass was obtained. Similarly, for SE analysis, 0.3 cm six-sector Epon centerpieces were loaded with 100 µl gp26-2M sample at three different concentrations, 16 µM (0.5 mg ml−1), 35 µM (1.1 mg ml−1) and 57 µM (1.8 mg ml−1) (Table 2 ▶), and 120 µl dialysis buffer as a reference in the parallel opposite sector. SE scans were collected by spinning samples at four different velocities of 8000, 12 000, 18 000 and 24 000 rev min−1 until equilibrium was attained (∼12 h). For molecular-weight analysis, we used the ‘species analysis’ model available in SEDPHAT with RI noise baseline correction (Schuck, 2005 ▶). Analysis was performed for each protein concentration separately and the molecular masses were determined from the average obtained from the analyses of the three protein concentrations.
), solvent density and relative viscosity values (0.7281 ml g−1, 1.00293 g ml−1 and 0.0010309 Pa s, respectively) along with the molecular mass were calculated using SEDNTERP v.1.09 (Laue et al., 1992 ▶; John Philo, Thousand Oaks, California, USA and RASMB; http://bitcwiki.sr.unh.edu). For SV runs, a two-sector 1.2 cm Epon centerpiece was loaded with 400 µl 70 µM (2.2 mg ml−1) gp26-2M (Table 2 ▶) and 420 µl dialysis buffer in the reference chamber. The runs were performed at 35 000 rev min−1 at a constant temperature of 10°C. Over ∼16 h, until complete sample sedimentation, absorbance values were collected at a fixed wavelength of 275 nm. The resulting data were fitted using a continuous sedimentation coefficient [c(s)] distribution model in SEDFIT (Schuck, 2000 ▶) and an estimated molecular mass was obtained. Similarly, for SE analysis, 0.3 cm six-sector Epon centerpieces were loaded with 100 µl gp26-2M sample at three different concentrations, 16 µM (0.5 mg ml−1), 35 µM (1.1 mg ml−1) and 57 µM (1.8 mg ml−1) (Table 2 ▶), and 120 µl dialysis buffer as a reference in the parallel opposite sector. SE scans were collected by spinning samples at four different velocities of 8000, 12 000, 18 000 and 24 000 rev min−1 until equilibrium was attained (∼12 h). For molecular-weight analysis, we used the ‘species analysis’ model available in SEDPHAT with RI noise baseline correction (Schuck, 2005 ▶). Analysis was performed for each protein concentration separately and the molecular masses were determined from the average obtained from the analyses of the three protein concentrations.
Table 2. Summary of biophysical parameters used to study gp26-2M by AUC.
| Method of analysis† | Sample concentration (µM) | An-50 Ti rotor speed(s) (rev min−1) | Model used for data analysis | Calculated molecular mass (kDa) |
|---|---|---|---|---|
| SV | 16 | 35000 | Continuous distribution Lamm equation | 110.0 |
| SV | 70 | 35000 | Continuous distribution Lamm equation | 110.1 |
| SE | 16 | 8000, 12000, 18000, 24000 | Species analysis | 96.3 |
| SE | 35 | 8000, 12000, 18000, 24000 | Species analysis | 97.8 |
| SE | 57 | 8000, 12000, 18000, 24000 | Species analysis | 99.3 |
SV, sedimentation velocity; SE, sedimentation equilibrium.
3. Results and discussion
3.1. In-frame extension of the gp26 helical core by one MiCRU yields a trimeric fiber
In this paper, we sought to determine the atomic structure of the gp26-2M tail needle that contains two tandemly repeated MiCRUs (Fig. 1 ▶ a). At first, we investigated the quaternary structure of this engineered fiber in solution to determine whether in-frame insertion of an MiCRU alters the trimeric oligomeric state of gp26. To this end, purified gp26-2M was subjected to analytical ultracentrifugation (AUC) analysis working under sedimentation-velocity (SV) mode. Sedimentation data (in a concentration range between 16 and 70 µM) were fitted to a distribution of Lamm equation solutions to determine the diffusion-free sedimentation-coefficient distribution [c(s)] (Table 2 ▶, Fig. 2 ▶ a). At all concentrations tested, the gp26-2M sedimentation boundary exhibited a monophasic sigmoidal behavior indicative of a single major component in solution. The sedimentation coefficient distribution c(s) was then converted into a molar mass distribution c(M), suggesting a molecular mass of ∼110 kDa, which is higher than the theoretical mass expected for a trimeric fiber (3 × 31.7 = 95.1 kDa) but slightly smaller than that for a tetramer (4 × 31.7 = 126.8 kDa). Since SV can be shape-biased (Cole et al., 2008 ▶), especially for very elongated molecules, we also analyzed gp26-2M by sedimentation-equilibrium (SE) analysis. SE data obtained at three concentrations (16 µM (0.5 mg ml−1), 35 µM (1.1 mg ml−1) and 57 µM (1.8 mg ml−1) at four different rotor speeds were analyzed globally using the ‘species analysis’ model in SEDPHAT (Table 2 ▶). The resultant fit suggested a molecular mass of 97.8 ± 1.5 kDa (with very low residuals of <0.1%), remarkably close to the expected size of a trimer (molecular weight of ∼95.1 kDa; Figs. 2 ▶ b–2 ▶ d). Thus, SV and SE analyses demonstrated that in-frame extension of a MiCRU inside the gp26 helical core results in a homogeneous fiber that, like gp26, exists as a trimer in solution.
Figure 2.

Analysis of the oligomeric state of gp26-2M by AUC. (a) Sedimentation-velocity profiles of gp26-2M dissolved in 0.02 M Tris–HCl pH 8.0, 0.1 M NaCl buffer at 10°C. The top panel shows the raw absorbance at 275 nm plotted as a function of radial position. Data at intervals of 1 h are shown as dots for sedimentation at 35 000 rev min−1. The middle panel shows the residuals between the fitted curve and the raw data. The bottom panel shows that the fitted distribution of the apparent sedimentation coefficient (s*) calculated for gp26-2M is 3.76 S, which corresponds to an estimated molecular mass of ∼110 kDa. (b–d) Sedimentation-equilibrium profiles of gp26-2M at 16 µM (b), 35 µM (c) and 57 µM (d) measured at four different rotor speeds of 8000, 12 000, 18 000 and 24 000 rev min−1. Distributions were analyzed as part of a global fitting to the absorbance data at multiple loading concentrations (top panels). Solid lines are the global best fit to distributions, resulting in a molecular mass of 97.8 ± 1.5 kDa consistent with a trimer with very low residuals (<0.1%) (bottom panels).
3.2. Crystallization and structure determination of a 320 Å long fiber
We crystallized gp26-2M in the presence of a high concentration of PEG 8000 at pH 4.5. As was observed for wild-type gp26 (gp26-wt) needles, the crystals grew as elongated rods, mainly in clusters (Cingolani et al., 2006 ▶). In diffraction experiments, most of the gp26-2M crystals displayed fiber-like diffraction patterns, similar to those observed for crystalline fibers of A-DNA (Arnott & Hukins, 1972 ▶), characterized by anisotropic diffraction and smearing of diffraction spots along layer-lines. A few crystals gave discrete diffraction maxima indicative of a three-dimensional lattice; cryo-annealing proved to be essential to improve both the diffraction quality and the resolution limit (Cingolani et al., 2006 ▶). Although the best crystals diffracted beyond ∼2 Å resolution, the diffraction was anisotropic, limiting the resolution of complete data to 2.7 Å. Crystallographic analysis revealed that the gp26-2M crystals belonged to a triclinic space group with four fibers in the asymmetric unit and a total solvent content of 42.5% (Table 1 ▶). Diffraction data were phased by molecular replacement using the P22 gp26 structure (PDB entry 3c9i; Olia et al., 2009 ▶) as a search model. To perform the molecular replacement it was essential to divide up the search model into three trimeric fragments spanning residues 1–84, 85–140 and 141–233, each comprising about one third of the total molecule. Exhaustive molecular-replacement searches at 6 Å resolution identified four entire gp26-2M fibers in the asymmetric unit adopting two significantly distinct conformations. The four fibers are arranged as two dimers related by a pseudo-twofold symmetry axis parallel to the a axis. Crystallographic refinement without imposing fourfold NCS restraints and modeling four calcium ions, eight chloride ions and 764 water molecules lowered the R work and R free of the final model to 22.9 and 26.8%, respectively, calculated using all data between 15 and 2.7 Å resolution (Table 1 ▶). A representative section of the final electron density of gp26-2M is shown in Fig. 3 ▶(a) and a ribbon diagram of the final model is illustrated in Fig. 3 ▶(b). The ∼95 kDa trimeric fiber spans approximately 320 Å but measures only 20–25 Å in width at the N-terminal tip and 30–35 Å at the C-terminal tip (Fig. 3 ▶ b). The helical core is continuous between residues 28 and 195 to give a total length of 250 Å. These 168 residues form an uninterrupted trimeric bundle of helices characterized by the absence of any stutters or stammers (Brown et al., 1996 ▶). Each protomer in gp26-2M presents a progressive left-handed helical twist that turns the structure by >600° over the length of the helical core (Fig. 3 ▶ c). Hydrophobic residues are directed towards the center of the trimeric helical bundle, and although individual protomers lack a hydrophobic core, the tightly packed trimeric interface buries a total surface area of ∼35 350 Å2, which is comparable to the total occupied molecular surface area (∼40 660 Å2). Accordingly, the ratio of buried surface area (at the trimer interface) to total solvent-accessible surface area is exceptionally high in gp26-2M compared with most soluble proteins (0.87 versus <0.4; Janin et al., 1988 ▶; Lins et al., 2003 ▶).
Figure 3.

Crystal structure of a 320 Å long gp26-2M engineered fiber. (a) Representative 2F o − F c electron-density map of gp26-2M contoured at 1.5σ above background. The density is overlaid on residues Thr223–Arg228 of the final model, which are shown as sticks. (b) Ribbon diagram of gp26-2M determined crystallographically at 2.7 Å resolution. The engineered fiber is shown in light gray with MiCRU-I and MiCRU-II colored blue and pink, respectively. Magnified views of the gp26-2M N- and C-termini are shown in the top panels. (c) Ribbon diagram of a representative protomer of gp26-2M.
3.3. Structural determinants of gp26-2M stability
The α-helical structure of gp26-2M displays all the characteristics of canonical left-handed, parallel and in-register coiled coils. Analysis of the gp26-2M structure with the SOCKET server (using the default distance cutoff of 7.0 Å) indicates that between residues 42 and 196 and 261 and 281 most positions a and d of the heptad repeat are occupied by hydrophobic residues (Fig. 1 ▶ a) that form ‘knobs’ packed into ‘holes’ generated between side chains of neighboring helices. Positions e and g are usually charged residues (Fig. 1 ▶ a). In total, gp26-2M contains 22 consecutive heptad repeats that stabilize the fiber structure by generating a spine of inter-chain hydrophobic interactions mainly mediated by amino acids at positions a and d. This continuous ‘knobs-into-holes’ arrangement causes each of the three protomers to spiral around one another to generate a left-handed supercoil (Fig. 4 ▶ a, top panel). Analysis of buried residues within the coiled-coil regions of gp26-2M reveals that position a of heptads 5–22 (corresponding to residues 70–195) is exclusively populated by the β-branched amino acids Leu, Ile and Val (e.g. Leu70, Ile77, Val84, Ile91, Ile98, Val105, Ile112, Val119, Val126, Ile133, Val140, Ile147, Ile154, Val161, Ile168, Val175, Val182 and Ile189), whereas position d is often occupied by polar residues such as histidine and asparagine in addition to β-branched hydrophobic amino acids and alanines (e.g. His73, Leu80, His87, Asn94, Leu101, Ala108, Leu115, Leu122, Ala129, Leu136, His143, Asn150, Leu157, Ala164, Leu171, Leu178, Ala185 and Leu192) (Figs. 1 ▶ a and 4 ▶ a, top panel). Interestingly, asparagine in the d position also occurs in a number of trimeric autotransporters that are known to bind anions such as chloride (Hartmann et al., 2009 ▶).
Figure 4.

Determinants for the structural stability of the gp26-2M fiber. (a) Middle: a cartoon (tube) representation of gp26-2M displaying buried ions and water molecules inside the helical core. Top: a magnified view of the gp26-2M helical core with the aliphatic side chains of β-branched chain hydrophobic residues shown as green spheres. Bottom: magnified view of the gp26-2M helical core showing charged residues involved in interchain salt bridges (O and N atoms are shown as red and blue spheres, respectively). (b) Magnified view of the calcium (Ca) ion, chloride ions (Cl1 and Cl2) and water molecules (W1, W2 and W3) buried inside gp26-2M cavities showing interacting residues.
In addition to its hydrophobic intersubunit interactions, gp26-2M presents a network of surface interhelical salt bridges that latch the three helices together (Fig. 4 ▶ a, bottom panel). Most of these salt bridges are originated by polar residues located at the e and g positions of the 22 trimerization heptads (Fig. 1 ▶ a). Notably, each MiCRU contains the trimerization motif R-hxxh-E, which was first identified by Kammerer et al. (2005 ▶) in another context, where arginine and glutamic acid occupy positions g and e, respectively, h is a hydrophobic residue (Ile, Leu, Val, Met) and x can be any amino acid. This motif has been shown to enhance structural stability and control the topology of coiled coils in a number of parallel trimeric coiled coils (Kammerer et al., 2005 ▶). This trimerization motif is repeated twice per MiCRU and therefore four times in gp26-2M (between residues 104–109, 125–130, 160–165 and 181–186) with amino-acid sequence RVTTAE.
Finally, the gp26-2M helical core contains three ions and three water molecules trapped inside polar cavities (Fig. 4 ▶ a). These cavities are formed at the intersection of helical chains, at points in the helical core of increased separation between chains and local coiled-coil unwinding. The gp26-2M helical core contains a calcium ion (Ca) bound to the side chains of Asn63 and Gln66 and a chloride ion (Cl1) in the MiCRU-I region that interacts with the side chains of Asn94 protruding from each of the three protomers (Fig. 4 ▶ b); both ions were previously identified in gp26-wt (Olia et al., 2009 ▶). Similar to Cl1 in the MiCRU-I region, an additional chloride ion (Cl2) is also located in the MiCRU-II region interacting with the side chains of Asn150 of all three protomers. Three well ordered waters (W1, W2 and W3) were also identified to interact with the side chains of His73, His87 and His143; two waters (W1 and W2) have been previously observed in gp26-wt (Olia et al., 2009 ▶; Fig. 4 ▶ b). Because of these buried ions and water molecules, the helical distance (defined as the distance between the midpoint of helices from adjacent protomers) in regions occupied by ions increases to 15 Å, compared with an average value of ∼10 Å elsewhere in the gp26-2M coiled coil (calculated using interhlx; K. Yap, University of Toronto). Interruptions of the tight hydrophobic core at cavities occupied by buried ions has been shown to favor coiled-coil structural stability by providing the correct ‘register’ (Olia et al., 2009 ▶; Guardado-Calvo et al., 2009 ▶). Likewise, a central chloride ion coordinated by asparagine residues seems to be a common feature among parallel trimeric coiled coils of viral fusion proteins and adhesins (Guardado-Calvo et al., 2009 ▶; Olia et al., 2009 ▶; Hartmann et al., 2009 ▶).
3.4. Structural evidence for fiber conformational flexibility
The triclinic unit cell of the gp26-2M crystal structure contains four trimers packed as two antiparallel dimers of fibers (referred to as fibers A and C and fibers B and D; Fig. 5 ▶ a). We performed secondary-structure matching superimposition analysis to identify putative differences among the four fibers and found that, although not perfectly identical, fibers A and B are superimposable, as are fibers C and D, with an overall root-mean-square deviation (r.m.s.d.) of only ∼0.7 Å (Fig. 5 ▶ b). In contrast, r.m.s.d. values greater than 4.0 Å were observed when fibers A or B are compared with fibers C or D, and this is mainly owing to large differences at the N-termini, which have swung away in C and D compared with A and B (Fig. 5 ▶ c). Thus, there are two structurally distinct conformations of gp26-2M trapped in the crystallographic asymmetric unit; these are exemplified by fibers A and C. To understand the contribution of individual coiled-coil residues to the conformational flexibility of gp26-2M, we carried out a comparative analysis of coiled-coil parameters using TWISTER (Strelkov & Burkhard, 2002 ▶). Firstly, we compared the coiled-coil characteristics of the gp26-2M fibers observed in the gp26-2M asymmetric unit using gp26-wt (PDB entry 3c9i) as a reference (Table 3 ▶). We found that the Crick angles (which define the position of each residue relative to the coiled-coil axis) for the a and d positions match reasonably well among all fibers. Also, local helical parameters such as the number of residues per turn, the rise per residue along the coiled-coil axis and the α-helical radius were comparable in all fibers. In contrast, significant deviations were observed in the local coiled-coil radius, the pitch, the phase per residue and the radius of curvature along the α-helical axis (Table 3 ▶). Consistent with structural superimposition (Figs. 5 ▶ b and 5 ▶ c), the coiled-coil parameters matched well between fibers A and B and between fibers C and D, but significant differences were observed between fibers A and B and fibers C and D. gp26-2M molecules A and B have much tighter coiled-coil packing when compared with molecules C and D; approximately five fewer residues (∼93 versus 98 residues) are sufficient for fibers A and B to make a complete superhelical turn, which results in a tighter pitch compared with fibers C and D (∼134 versus ∼141 Å). Similarly, tighter packing allows fibers A and B to revolve by up to ∼600° over as few as 154 residues, ∼28° greater than fibers C and D (which revolve by ∼572° over 154 residues). This results in a shorter overall radius of curvature for fibers A and B as opposed to fibers C and D (∼66.7 versus ∼71.1 Å) and a much tighter packing of coiled-coil residues. We extended our analysis to the MiCRU regions (MiCRU-I and MiCRU-II) of gp26-2M fibers and gp26-wt. Consistent with the results obtained from the analysis of fibers, MiCRU-I and MiCRU-II of both fibers C and D seemed to have a slightly relaxed coiled-coil pitch compared with fibers A and B (average of ∼141 versus 133 Å). Thus, fibers A and B are more rigid and tightly packed compared with fibers C and D, underlying differences in how the residues in the MiCRU region interact, thus causing flexibility.
Figure 5.

Determinants of conformational flexibility. (a) The arrangement of four trimers packed in the triclinic unit cell (shown in yellow) of the gp26-2M crystal structure (referred to as fibers A, B, C and D). Structural superimposition of (b) fiber A versus fiber B, fiber C versus fiber D and (c) fiber A versus fiber C. The N-terminus of fiber A or B sways away by ∼28.7° with respect to fiber C or D. (d, e) Structural superimposition among individual protomers of fibers A and C. The arrow indicates the region of maximum displacement at the N- and C-termini within individual protomers.
Table 3. Relative comparison of coiled-coil parameters for gp26-2M fibers and gp26-wt.
| gp26-2M fibers | gp26-wt (PDB entry 3c9i) | ||||
|---|---|---|---|---|---|
| Fiber | A | B | C | D | Molecule A |
| Coiled-coil parameters | |||||
| Residues | 42–195 | 42–195 | 42–195 | 42–195 | 42–139 |
| Coiled-coil radius (Å) | 6.15 ± 0.44 | 6.15 ± 0.45 | 6.15 ± 0.41 | 6.19 ± 0.45 | 6.13 ± 0.47 |
| Residues/superhelical turn | 93.6 | 93 | 98 | 98 | 95 |
| Coiled-coil pitch (Å) | 134.3 ± 19.9 | 133.5 ± 21.1 | 141.2 ± 19.3 | 140.5 ± 21.9 | 138.0 ± 16.9 |
| Coiled-coil phase (°)/No. of residues | −598.91/154 | −601.52/154 | −571.95/154 | −573.83/154 | −370.65/98 |
| α-Helical parameters | |||||
| Residues per turn | 3.63 ± 0.09 | 3.64 ± 0.10 | 3.63 ± 0.12 | 3.63 ± 0.12 | 3.64 ± 0.09 |
| Rise per residue (Å) | 1.51 ± 0.05 | 1.51 ± 0.05 | 1.51 ± 0.05 | 1.51 ± 0.05 | 1.51 ± 0.04 |
| α-Helix radius (Å) | 2.28 ± 0.06 | 2.28 ± 0.06 | 2.28 ± 0.07 | 2.28 ± 0.07 | 2.30 ± 0.05 |
| Radius of curvature (Å) | 66.34 | 67.15 | 71.51 | 70.72 | 80.52 |
| Crick angles | |||||
| Position a (°) | 18.33 ± 2.6 | 17.36 ± 3.11 | 17.72 ± 2.74 | 18.20 ± 2.81 | 17.39 ± 3.01 |
| Position d (°) | −31.99 ± 4.23 | −32.56 ± 4.83 | −32.52 ± 4.15 | −32.60 ± 4.16 | −34.49 ± 3.31 |
To assess how individual gp26-2M protomers contribute to the flexibility of these fibers, we superimposed individual protomers within each fiber. This revealed that fiber A protomers differ mainly at their N-termini, with a maximum displacement of ∼21 Å at residue 28 (Fig. 5 ▶ d). In contrast, fiber C protomers present structural differences throughout the entire length of the fiber, with increased deviations at both the N- and C-termini (Fig. 5 ▶ e). The maximum displacement is observed at residue 255, corresponding to ∼18 Å. Overall, all six promoters of fibers A and C are non-identical, with r.m.s.d. values of between 1.7 and 5.0 Å; likewise, small yet significant differences are observed between fiber B and D protomers, which explains why the observed crystal form belongs to space group P1 (with four fibers in the asymmetric unit) as opposed to P21 with two fibers per asymmetric unit related by a twofold screw axis. In conclusion, the gp26-2M crystallographic asymmetric unit contains six structurally distinct conformers of the gp26-2M protomer assembled to form two trimeric fibers.
4. Discussion
Crystallization of protein fibers has proven to be more challenging than that of globular proteins. A query of the PDB for protein structures containing a helical length of over 100 Å yielded only 44 results, most of which encompass extended monomeric regions of globular proteins and engineered dimeric proteins fused to the GCN4 motif. An interesting example is the triple coiled-coil region of adhesin protein UspA1 from the pathogenic bacterium Moraxella catarrhalis (PDB entry 2qih; Conners et al., 2008 ▶). Residues 527–665 of UspA1 form a left-handed trimeric coiled-coil structure of approximately 200 Å in length (Fig. 6 ▶ c), very similar to gp26-2M coiled-coil residues 42–195 but ∼25 Å shorter in length and similar in width (∼20 Å) (Fig. 6 ▶ b). Similarly, E. coli immunoglobulin-binding domain protein (EIBD) fused to GCN4 adaptors (PDB entry 2xzr) forms a 160 Å long trimeric coiled-coil structure (Leo et al., 2011 ▶; Fig. 6 ▶ d). Despite the similar structure, the coiled-coil pitch of these two structures is >150 Å, slightly more relaxed than in gp26-2M (∼138 Å). Among non-trimeric coiled-coil structures, a 230 Å long dimeric cytoplasmic domain of a bacterial chemoreceptor from Thermotoga maritima (PDB entry 2ch7) is the longest structure of a helical fiber formed by a tight tetrameric coiled coil (Park et al., 2006 ▶; Fig. 6 ▶ e). Similarly, a 7 Å resolution crystal structure of a 400 Å long coiled-coil tropomyosin is the longest structure to be determined for a dimeric coiled coil (PDB entry 1c1g; Whitby & Phillips, 2000 ▶; Fig. 6 ▶ f), although the detailed chemistry of intrasubunit packaging is not known owing to the limited resolution. Interestingly, the helical core of gp26-2M described in this paper contains a 225 Å long uninterrupted coiled-coil structure that to our knowledge is the longest segment of any triple coiled-coil protein for which a high-resolution structure has been determined (Fig. 6 ▶). Among all of these fibers, gp26-2M is the only example of a crystallized fiber in which the N- and C-terminal ends are knotted by flanking domains. This may contribute to increased fiber stability and promote crystallization by the association of the non-helical domain flanking the coiled-coil core.
Figure 6.

Crystal structures of helical coiled-coil fibers longer than 100 Å. (a–d) The crystal structures of trimeric gp26-wt, gp26-2M, UspA1 (residues 527–665) and E. coli immunoglobulin-binding protein (EIBD) residues 391–438 fused to GCN4 adaptors (PDB entries 3c9i, 4lin, 2qih and 2xzr). (e) A 2.5 Å resolution crystal structure of the dimeric (tetrameric coiled coil) cytoplasmic domain of a bacterial chemoreceptor from T. maritima (PDB entry 2ch7). (f) A 7.0 Å resolution structure of tropomyosin (PDB entry 1c1g). The relative length of continuous helical regions is indicated.
Why are protein fibers recalcitrant to form three-dimensional crystals? A possible explanation is that the intrinsic conformational flexibility of fibers prevents stabilization into an ordered three-dimensional lattice. As suggested in this study, the triclinic crystal form of gp26-2M contains two distinct trimeric fibers (Fig. 5 ▶ c) which show as many as six drastically different conformations of the same protomer (Fig. 5 ▶ e). Accordingly, our attempt to crystallize an even longer engineered fiber containing three MiCRUs (gp26-3M; Bhardwaj et al., 2009 ▶) were unsuccessful, despite this fiber being biochemically well behaved, extremely stable (melting temperature of >85°C) and perfectly monodisperse in solution, like gp26-2M. It is possible that gp26-2M represents the upper limit of crystallizability for the tail needle gp26 and that above ∼320 Å the number of structural conformers in solution decreases the concentration to below that required for nucleation, preventing crystallization.
5. Biological implications
Surface-exposed fibers emanating from a viral capsid or projecting from a bacteriophage tail (Bhardwaj et al., 2013 ▶) represent the first part of a virion to sense the outside environment. For instance, the P22 tail spike interacts with Salmonella lipopolysaccharide chains and mediates phage adhesion to the host surface (Casjens & Molineux, 2012 ▶), which promotes the ejection of the tail needle gp26 inside the host (Israel, 1976 ▶, 1978 ▶). Owing to the tremendous rate at which these events occur in nature, tail-fiber genes evolve faster than other phage genes (Veesler & Cambillau, 2011 ▶) and genetic exchange of fiber genes can occur via horizontal gene transfer among phages crossing host phylogenetic boundaries (Hendrix et al., 1999 ▶).
There are several examples in virology whereby the length and flexibility of a surface-exposed fiber directly affect the host specificity and virus infectivity. In adenovirus, natural differences in the length of the virion-exposed fiber have important biological consequences. Adenovirus (Ad) fiber is a homotrimeric molecule extending from each of the 12 vertices of the icosahedral capsid. The fiber N-terminus attaches to the capsid and is followed by a central shaft domain of variable length and a C-terminal knob containing a receptor-binding site (Nicklin et al., 2005 ▶). The fiber shaft is formed by a triple β-spiral fold (van Raaij et al., 1999 ▶) composed of 6–23 repeats depending on the Ad serotype. The length of the Ad shaft determines the binding affinity to the CAR receptor and hence the infectivity, with shorter shafts usually leading to reduced CAR binding and infectivity (Shayakhmetov & Lieber, 2000 ▶). Cryo-electron microscopic (EM) studies suggested that longer fibers are more flexible and therefore less visible in cryo-EM reconstructions compared with short fibers, and thus both the length and flexibility of the Ad fiber shaft play a central role in receptor interaction (Chiu et al., 2001 ▶). Similarly, we have identified P22-like phages (and prophages) that encode longer or shorter tail needles than P22 gp26 owing to insertions and/or deletions in the α-helical coiled-coil core (Bhardwaj et al., 2009 ▶). For instance, phages HS1 and Eco82-1 have five more trimerization heptads than P22-gp26 (19 versus 14 heptads) and are only three heptads shorter than gp26-2M (∼22 heptads) described in this paper. How does the length of a tail needle helical core affect infectivity and host specificity? We recently determined that the domain immediately downstream of the gp26 helical core does not confer host specificity, but substitutions at this position affect the kinetics of P22 genome ejection in Salmonella (Leavitt et al., 2013 ▶). Likewise, chimeras of P22 carrying a shorter tail needle (lacking 2–3 heptads) are considerably less infectious than wild-type phages under laboratory conditions and slower at ejecting DNA in vitro (Leavitt & Casjens, 2013 ▶). We are currently testing how mutations that extend the gp26 helical core affect the rate of P22 genome delivery and phage infectivity.
In summary, randomly occurring mutations and horizontal gene transfer are likely to be responsible for extending and/or shortening surface-exposed viral fibers. In-frame insertion of trimerization heptads, or of a region containing multiple heptads, results in modular extension of surface-exposed fibers, as observed for tail needles of the gp26 superfamily. This may lead to an increase in structural stability (Bhardwaj et al., 2009 ▶) and conformational flexibility, as shown in this paper, and confer new biological properties such as the ability to explore a large volume in the search for a cell or to bind to a specific receptor. The high-resolution crystal structure of the engineered fiber gp26-2M presented in this work enhances our understanding of coiled-coil heptad repeats and provides a framework to decipher the structural determinants of protein-fiber stability and flexibility.
Supplementary Material
PDB reference: gp26-2M, 4lin
Acknowledgments
We are grateful to the staff at National Synchrotron Light Source beamlines X6A and X29 as well as the staff at MacCHESS for beamtime and beamline assistance. This work was supported by National Institutes of Health Grant 1R01GM100888-01A1 to GC. The research in this publication includes work carried out at the Kimmel Cancer Center X-ray Crystallography and Molecular Interactions Facility, which is supported in part by NCI Cancer Center Support Grant P30 CA56036.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Arnott, S. & Hukins, D. W. (1972). Biochem. Biophys. Res. Commun. 47, 1504–1509. [DOI] [PubMed]
- Berget, P. B. & Poteete, A. R. (1980). J. Virol. 34, 234–243. [DOI] [PMC free article] [PubMed]
- Bhardwaj, A., Molineux, I. J., Casjens, S. R. & Cingolani, G. (2011). J. Biol. Chem. 286, 30867–30877. [DOI] [PMC free article] [PubMed]
- Bhardwaj, A., Olia, A. S. & Cingolani, G. (2013). Curr. Opin. Struct. Biol. 10.1016/j.sbi.2013.10.005. [DOI] [PMC free article] [PubMed]
- Bhardwaj, A., Olia, A. S., Walker-Kopp, N. & Cingolani, G. (2007). J. Mol. Biol. 371, 374–387. [DOI] [PubMed]
- Bhardwaj, A., Walker-Kopp, N., Casjens, S. R. & Cingolani, G. (2009). J. Mol. Biol. 391, 227–245. [DOI] [PMC free article] [PubMed]
- Bhardwaj, A., Walker-Kopp, N., Wilkens, S. & Cingolani, G. (2008). Protein Sci. 17, 1475–1485. [DOI] [PMC free article] [PubMed]
- Botstein, D., Waddell, C. H. & King, J. (1973). J. Mol. Biol. 80, 669–695. [DOI] [PubMed]
- Brown, J. H., Cohen, C. & Parry, D. A. (1996). Proteins, 26, 134–145. [DOI] [PubMed]
- Casjens, S. R. & Molineux, I. J. (2012). Adv. Exp. Med. Biol. 726, 143–179. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Chiu, C. Y., Wu, E., Brown, S. L., Von Seggern, D. J., Nemerow, G. R. & Stewart, P. L. (2001). J. Virol. 75, 5375–5380. [DOI] [PMC free article] [PubMed]
- Cingolani, G., Andrews, D. & Casjens, S. (2006). Acta Cryst. F62, 477–482. [DOI] [PMC free article] [PubMed]
- Cole, J. L., Lary, J. W., Moody, T. P. & Laue, T. M. (2008). Methods Cell Biol. 84, 143–179. [DOI] [PMC free article] [PubMed]
- Conley, M. P. & Wood, W. B. (1975). Proc. Natl Acad. Sci. USA, 72, 3701–3705. [DOI] [PMC free article] [PubMed]
- Conners, R., Hill, D. J., Borodina, E., Agnew, C., Daniell, S. J., Burton, N. M., Sessions, R. B., Clarke, A. R., Catto, L. E., Lammie, D., Wess, T., Brady, R. L. & Virji, M. (2008). EMBO J. 27, 1779–1789. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Guardado-Calvo, P., Fox, G. C., Llamas-Saiz, A. L. & van Raaij, M. J. (2009). J. Gen. Virol. 90, 672–677. [DOI] [PubMed]
- Hartmann, M. D., Ridderbusch, O., Zeth, K., Albrecht, R., Testa, O., Woolfson, D. N., Sauer, G., Dunin-Horkawicz, S., Lupas, A. N. & Alvarez, B. H. (2009). Proc. Natl Acad. Sci. USA, 106, 16950–16955. [DOI] [PMC free article] [PubMed]
- Hendrix, R. W., Smith, M. C., Burns, R. N., Ford, M. E. & Hatfull, G. F. (1999). Proc. Natl Acad. Sci. USA, 96, 2192–2197. [DOI] [PMC free article] [PubMed]
- Hyman, P., Valluzzi, R. & Goldberg, E. (2002). Proc. Natl Acad. Sci. USA, 99, 8488–8493. [DOI] [PMC free article] [PubMed]
- Israel, V. (1976). J. Virol. 18, 361–364. [DOI] [PMC free article] [PubMed]
- Israel, V. (1978). J. Gen. Virol. 40, 669–673. [DOI] [PubMed]
- Janin, J., Miller, S. & Chothia, C. (1988). J. Mol. Biol. 204, 155–164. [DOI] [PubMed]
- Kammerer, R. A., Kostrewa, D., Progias, P., Honnappa, S., Avila, D., Lustig, A., Winkler, F. K., Pieters, J. & Steinmetz, M. O. (2005). Proc. Natl Acad. Sci. USA, 102, 13891–13896. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lander, G. C., Tang, L., Casjens, S. R., Gilcrease, E. B., Prevelige, P., Poliakov, A., Potter, C. S., Carragher, B. & Johnson, J. E. (2006). Science, 312, 1791–1795. [DOI] [PubMed]
- Laue, T. M., Shah, B. D., Ridgeway, T. M. & Pelletier, S. L. (1992). Analytical Ultracentrifugation in Biochemistry and Polymer Science, edited by S. E. Harding, A. J. Rowe & J. C. Horton, pp. 90–125. Cambridge: Royal Society of Chemistry.
- Leavitt, J. C. & Casjens, S. R. (2013). Personal communication.
- Leavitt, J. C., Gogokhia, L., Gilcrease, E. B., Bhardwaj, A., Cingolani, G. & Casjens, S. R. (2013). PLoS One, 8, e70936. [DOI] [PMC free article] [PubMed]
- Leo, J. C., Lyskowski, A., Hattula, K., Hartmann, M. D., Schwarz, H., Butcher, S. J., Linke, D., Lupas, A. N. & Goldman, A. (2011). Structure, 19, 1021–1030. [DOI] [PubMed]
- Lins, L., Thomas, A. & Brasseur, R. (2003). Protein Sci. 12, 1406–1417. [DOI] [PMC free article] [PubMed]
- Lupas, A. (1996). Methods Enzymol. 266, 513–525. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- Mitraki, A. & van Raaij, M. J. (2005). Methods Mol. Biol. 300, 125–140. [DOI] [PubMed]
- Nicklin, S. A., Wu, E., Nemerow, G. R. & Baker, A. H. (2005). Mol. Ther. 12, 384–393. [DOI] [PubMed]
- Olia, A. S., Al-Bassam, J., Winn-Stapley, D. A., Joss, L., Casjens, S. R. & Cingolani, G. (2006). J. Mol. Biol. 363, 558–576. [DOI] [PubMed]
- Olia, A. S., Casjens, S. & Cingolani, G. (2007). Nature Struct. Mol. Biol. 14, 1221–1226. [DOI] [PubMed]
- Olia, A. S., Casjens, S. & Cingolani, G. (2009). Protein Sci. 18, 537–548. [DOI] [PMC free article] [PubMed]
- Olia, A. S., Prevelige, P. E. Jr, Johnson, J. E. & Cingolani, G. (2011). Nature Struct. Mol. Biol. 18, 597–603. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Park, S.-Y., Borbat, P. P., Gonzalez-Bonet, G., Bhatnagar, J., Pollard, A. M., Freed, J. H., Bilwes, A. M. & Crane, B. R. (2006). Nature Struct. Mol. Biol. 13, 400–407. [DOI] [PubMed]
- Raaij, M. J. van, Mitraki, A., Lavigne, G. & Cusack, S. (1999). Nature (London), 401, 935–938. [DOI] [PubMed]
- Schuck, P. (2000). Biophys. J. 78, 1606–1619. [DOI] [PMC free article] [PubMed]
- Schuck, P. (2005). Modern Analytical Ultracentrifugation: Techniques and Methods, edited by D. J. Scott, S. E. Harding & A. J. Rowe, pp. 26–60. Cambridge: The Royal Society of Chemistry.
- Shayakhmetov, D. M. & Lieber, A. (2000). J. Virol. 74, 10274–10286. [DOI] [PMC free article] [PubMed]
- Strauss, H. & King, J. (1984). J. Mol. Biol. 172, 523–543. [DOI] [PubMed]
- Strelkov, S. V. & Burkhard, P. (2002). J. Struct. Biol. 137, 54–64. [DOI] [PubMed]
- Tang, J., Lander, G. C., Olia, A. S., Olia, A., Li, R., Casjens, S., Prevelige, P. Jr, Cingolani, G., Baker, T. S. & Johnson, J. E. (2011). Structure, 19, 496–502. [DOI] [PMC free article] [PubMed]
- Veesler, D. & Cambillau, C. (2011). Microbiol. Mol. Biol. Rev. 75, 423–433. [DOI] [PMC free article] [PubMed]
- Walshaw, J. & Woolfson, D. N. (2001). J. Mol. Biol. 307, 1427–1450. [DOI] [PubMed]
- Whitby, F. G. & Phillips, G. N. (2000). Proteins, 38, 49–59. [PubMed]
- Yeates, T. O. (1988). Acta Cryst. A44, 142–144. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: gp26-2M, 4lin