

The Salmonella effector protein GtgE functions as a cysteine protease to cleave a subset of the Rab-family GTPases and to prevent delivery of antimicrobial agents to the Salmonella-containing vacuole.
Keywords: cysteine proteases, GtgE, Rab GTPase, Salmonella Typhi, Salmonella Typhimurium
Abstract
GtgE is an effector protein from Salmonella Typhimurium that modulates trafficking of the Salmonella-containing vacuole. It exerts its function by cleaving the Rab-family GTPases Rab29, Rab32 and Rab38, thereby preventing the delivery of antimicrobial factors to the bacteria-containing vacuole. Here, the crystal structure of GtgE at 1.65 Å resolution is presented, and structure-based mutagenesis and in vivo infection assays are used to identify its catalytic triad. A panel of cysteine protease inhibitors were examined and it was determined that N-ethylmaleimide, antipain and chymostatin inhibit GtgE activity in vitro. These findings provide the basis for the development of novel therapeutic strategies to combat Salmonella infections.
1. Introduction
Salmonella enterica continues to be a significant public health concern, with 93.8 million cases of non-typhoidal salmonellosis and 21 million cases of typhoid fever estimated worldwide each year (Majowicz et al., 2010 ▶; Crump & Mintz, 2010 ▶). There are thousands of S. enterica serovars, which can infect a multitude of hosts with varying outcomes. Some serovars, such as Salmonella Typhimurium (S. Typhimurium), can infect a broad range of vertebrate species, causing self-limiting gastroenteritis, or ‘food poisoning’. Others, such as Salmonella Typhi (S. Typhi), are only able to infect humans, causing a life-threatening systemic disease known as typhoid fever. Recent studies have begun to provide the first mechanistic explanations of the diversity in clinical presentation and host specificity exhibited by these different serovars.
Recent reports indicate that the ability of S. Typhi to cause typhoid fever is linked to the acquisition of a set of genes that encode typhoid toxin and that are unique to this, and related, serovars (Spanò et al., 2008 ▶, 2011 ▶; Song et al., 2013 ▶). In contrast, the inability of S. Typhi to explore other niches is, at least in part, owing to the absence of the type III secreted protein effector GtgE (Spanò & Galán, 2012 ▶). This effector protein, which is widely distributed among broad host-range salmonellae, proteolytically targets a very specific set of highly related Rab-family GTPases: Rab29, Rab32 and Rab38 (Spanò & Galán, 2012 ▶; Spanò et al., 2011 ▶; Schwartz et al., 2007 ▶). Rab32 and Rab38 function in combination with the BLOC-1, BLOC-2 and BLOC-3 proteins to deliver enzymes to lysosome-related organelles and presumably antimicrobial factors to the Salmonella-containing vacuole (SCV), resulting in pathogen death (Bultema et al., 2012 ▶; Raposo & Marks, 2007 ▶; Spanò & Galán, 2012 ▶). By targeting these GTPases, GtgE allows survival of the pathogen by preventing the delivery of antimicrobial factors to the SCV (Spanò & Galán, 2012 ▶). GtgE expression in S. Typhi enables this human-adapted serovar to survive within non-permissive host cells (Spanò & Galán, 2012 ▶).
These findings imply a crucial role for GtgE in expanding the host repertoire of Salmonella, prompting characterization of the proteolytic mechanism of GtgE to aid in the understanding of the biological activity of GtgE and to potentially assist in the development of GtgE inhibitors. Here, we identify GtgE as a cysteine protease with a catalytic triad of Cys45-His151-Asp169 using X-ray crystallography and biochemical analysis, and characterize an initial set of cysteine protease inhibitors that may prove useful for further inhibitor design.
2. Materials and methods
2.1. Protein expression and purification
S. Typhimurium LT2 GtgE1–228 and GtgE79–214 and human Rab381–211 were cloned into a modified pCDF-Duet-1 vector (Novagen) between the SalI and NotI restriction sites. All GtgE1–228 point mutants were generated by PCR mutagenesis. The protein was expressed in Escherichia coli BL21(DE3) cells with 0.75 mM IPTG for 16 h at 18°C. Selenomethionine-substituted GtgE79–214 was expressed in E. coli 834 cells in selenomethionine-supplemented minimal medium with 0.75 mM IPTG at 18°C for 12 h. The harvested cells were pelleted, resuspended in 50 mM Tris–HCl pH 8.0, 300 mM NaCl, 5 mM imidazole and lysed via homogenization. The cleared lysate was run over Ni–NTA resin (Qiagen) and the protein was eluted in 50 mM Tris–HCl pH 8.0, 300 mM NaCl, 500 mM imidazole. The protein was dialyzed against 25 mM Tris–HCl pH 8.0, 250 mM NaCl, 5 mM DTT and the N-terminal histidine tag was cleaved off with 6×His-rhinovirus 3C protease. The material was passed over Ni–NTA to remove the histidine tag and 3C protease. The final purification step was size-exclusion chromatography using a 120 ml Superdex 75 column (GE Healthcare). Native and selenomethionine-substituted GtgE79–214 was purified into 25 mM HEPES pH 8.0, 250 mM NaCl, 5 mM DTT. GtgE1–228 constructs of the wild type and catalytic mutants were purified into 25 mM Tris–HCl pH 8.0, 250 mM NaCl, 5 mM DTT. All Rab381–211 buffers contained 5 mM MgCl2 and final purification was performed on a 120 ml Superdex 200 column (GE Healthcare) into 25 mM Tris–HCl pH 8.0, 250 mM NaCl, 5 mM MgCl2, 5 mM DTT. The purified protein was stored at −80°C.
2.2. Limited proteolysis of GtgE43–214
90 µg GtgE43–214 was treated with subtilisin protease (Sigma–Aldrich) at a range of 0.55–10.92 µg with 5 mM CaCl2 for 20 min at 4°C (Fig. 1 ▶ a). The reaction was terminated by the addition of 10 mM PMSF and SDS loading buffer. The cleavage products were separated by 15% SDS–PAGE, transferred to a PVDF membrane (Millipore) and stained with SYPRO Ruby protein stain (Sigma–Aldrich). The major protein bands were cut from the membrane and sent to the Columbia University Protein Core Facility (New York, USA) for N-terminal Edman degradation sequencing.
Figure 1.

(a) GtgE43–214 was subjected to limited proteolysis by subtilisin protease (Sigma–Aldrich) as described. Edman sequencing indicated that the two main cleavage products were N-terminally truncated to residue 79. C-terminal residue limits were estimated based on the molecular weight of the cleavage products as observed by SDS–PAGE. The crystallized construct is shown in red. Molecular-mass markers (left lane) are labelled in kDa. (b) Native and selenomethionine-derivative crystals of GtgE79–214 grown in 0.2 M Li2SO4, 1.75 M ammonium sulfate, 0.1 M Tris pH 7.0 at 4°C. (c) 2F o − F c model-phased electron-density map contoured at 2σ shown in blue with the final refined model shown.
2.3. Crystallization and structure determination
Native GtgE79–214 was crystallized in a hanging-drop format at a concentration of 10 mg ml−1 at 4°C in 0.2 M Li2SO4, 1.75 M ammonium sulfate, 0.1 M Tris pH 7.0 (Fig. 1 ▶ b). Selenomethionine-substituted GtgE79–214 was crystallized by two rounds of seeding with native crystals at 10 mg ml−1 concentration at 4°C in 0.2 M Li2SO4, 1.75 M ammonium sulfate, 0.1 M Tris pH 7.0 (Fig. 1 ▶ b). The crystals were cryoprotected in 0.3 M LiSO4, 2.5 M ammonium sulfate, 0.1 M Tris pH 7.0 and were flash-cooled in liquid nitrogen for data collection. X-ray data were collected on beamline X29 at Brookhaven National Synchrotron Light Source (NSLS). Native and selenomethionine-substituted GtgE79–214 diffraction data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). Phasing and initial model building of the selenomethione-substituted protein was performed using AutoSol in PHENIX (Adams et al., 2010 ▶; Terwilliger et al., 2009 ▶; McCoy et al., 2007 ▶; Grosse-Kunstleve & Adams, 2003 ▶). Automated building produced a model with 133 residues, and the initial model was subsequently used with ARP/wARP to build a model using the higher resolution native data (Langer et al., 2008 ▶). Refinement of this model was carried out with REFMAC5 (Winn et al., 2011 ▶; Murshudov et al., 2011 ▶) and manual model building was performed in Coot (Emsley et al., 2010 ▶). TLS refinement (Winn et al., 2003 ▶; Painter & Merritt, 2006 ▶) was used in the last stages to generate a final model spanning residues 80–213 with an R and an R free of 19.3 and 23.0%, respectively (Table 1 ▶). No electron density was observed for residues 79, 145, 146, 171, 193–199 and 214, so these residues were not modeled into the final structure. 98% of the residues fall into the most favored region of the Ramachandran plot, with no outliers. Figures were generated using CCP4mg (McNicholas et al., 2011 ▶).
Table 1. Crystallographic data-collection and structure-determination statistics.
Values in parentheses are for the highest resolution shell.
| Native | Selenomethionine-substituted | |
|---|---|---|
| Data collection | ||
| Space group | P41212 | P41212 |
| Unit-cell parameters (, ) | a = b = 56.18, c = 125.12, = = = 90 | a = b = 56.13, c = 125.03, = = = 90 |
| Wavelength () | 1.0750 | 0.9790 |
| Resolution () | 50.001.65 (1.711.65) | 50.002.38 (2.472.38) |
| No. of reflections | 420901 | 425153 |
| No. of unique reflections | 23155 (2428) | 15090 (1550) |
| R merge † | 0.072 (0.894) | 0.162 (0.705) |
| Mean I/(I) | 30.13 (3.2) | 16.45 (4.67) |
| Completeness (%) | 92.6 (100.0) | 99.0 (100.0) |
| Multiplicity | 11.2 (11.7) | 15.3 (15.5) |
| Refinement | ||
| Resolution () | 19.631.65 (1.6931.65) | |
| No. of reflections | 21891 (1778) | |
| R factor‡ | 0.193 (0.258) | |
| R free ‡ | 0.230 (0.250) | |
| No. of atoms | ||
| Total | 1129 | |
| Macromolecules | 957 | |
| Ligands | 5 | |
| Waters | 167 | |
| No. of protein residues | 124 | |
| R.m.s.d., bonds () | 0.017 | |
| R.m.s.d., angles () | 1.75 | |
| Average B factor (2) | 30.40 | |
| Ramachandran favored (%) | 98 | |
| Ramachandran outliers (%) | 0 | |
As defined and calculated by HKL-2000.
As defined and calculated by REFMAC5. The R free test-set size was 5%.
2.4. Gel-based activity assay
An activity assay was performed using Rab38 and full-length GtgE purified as detailed above. GtgE and Rab38 were mixed in a 1:4 molar ratio in the presence of 10 mM CaCl2 and 10 mM MgCl2. End-point assays were performed at 4°C for 30 min and time-point assays were sampled at 4°C as described in Fig. 4(c). Reactions were visualized by 15% SDS–PAGE and Coomassie staining. Time-point assays were run in triplicate and quantification of the GtgE and Rab38 band intensity was performed with ImageJ (Schneider et al., 2012 ▶). Uncleaved and cleaved Rab38 band intensities were first normalized against the intensity of the loading control GtgE. The normalized Rab38 cleavage-product intensities were then expressed relative to the total Rab38 in the reaction and these cleavage-product percentage values were summed to convey the product formation in terms of the total Rab38 in the reaction.
2.5. Analysis of GtgE catalytic mutants in vivo
CFP-Rab29 and CFP-Rab38 were expressed in COS-1 cells through viral transduction using an LZRS-based retroviral vector. Pseudotyped virus was produced by co-transfecting 4 µg pLZRS-CFP-Rab29 or pLZRS-CFP-Rab38, 4 µg pVSVG and 4 µg pGag/Pol plasmids in a 10 cm dish of HEK-293 cells using 30 µl Fugene 6 Transfection Reagent (Roche). Cell-culture supernatants were collected 48 h after transfection and were used at a dilution of 1:5 to transduce COS-1 cells. 40 h after transduction, the cells were infected with S. enterica serovar Typhi (S. Typhi) strain ISP2825 (Galán & Curtiss, 1991 ▶) bearing an empty low-copy vector or the same vector expressing wild-type GtgE or GtgE catalytic mutants, all full-length, under the control of the Salmonella rpsM constitutive promoter. To visualize bacteria in the imaging experiment, S. Typhi was also transformed with a vector expressing the fluorescent protein mCherry (pSB4010; Spanò et al., 2011 ▶). The cells were either lysed for Western blot analysis or imaged 2.5 h after infection. Live-cell imaging was performed at 37°C in a temperature-, humidity- and CO2-controlled live chamber (Pathology Devices) using a 60× oil objective (numerical aperture 1.4) of an Improvision spinning-disk confocal microscope equipped with a Nikon TE2000 microscope. For Western blot analysis cells were lysed in Laemmli SDS buffer, boiled and run on a 12% SDS–PAGE. Western blotting was performed with a rabbit anti-GFP antibody (Invitrogen, A-6455) using ECL-based detection.
2.6. Inhibition assay
N-Ethylmaleimide, antipain and chymostatin were solubilized in ethanol, water and DMSO, respectively. In a reaction volume of 15 µl, 5.75 µM full-length GtgE-WT was incubated at room temperature for 15 min with 10 mM MgCl2, 10 mM CaCl2 and one of the following additives: water (positive control), 2.5% ethanol/25% DMSO (delivery controls) and inhibitor [0.25 mM N-ethylmaleimide (Sigma–Aldrich), 5 mM antipain (Sigma–Aldrich) and 2.5 mM chymostatin (Santa Cruz Biotechnology)]. For the leupeptin assay, leupeptin (Sigma–Aldrich) was reconstituted in water and used at the following concentrations: 10 µM, 50 µM, 100 µM, 500 µM, 1 mM, 2 mM and 5 mM. 5.75 µM full-length GtgE-C45A was also incubated at room temperature with water as a negative control. 25.3 µM Rab38 was then added and the reaction was allowed to proceed at 4°C for 30 min. The reaction was ended by the addition of SDS running buffer and boiling. The results were visualized via 15% SDS–PAGE and Coomassie staining.
3. Results and discussion
3.1. GtgE is a cysteine protease
The structure of GtgE (residues 80–213) was solved to 1.65 Å resolution using selenomethionine-substituted protein crystals, and the final model was refined to R and R free values of 19.3 and 23.0% against a native data set (§2, Table 1 ▶ and Fig. 1 ▶). The final structure is comprised of a six-stranded β-sheet that is sandwiched between three helices on one side of the sheet and one helix on the other (Fig. 2 ▶). The β-strands are arranged in an antiparallel topology as depicted in Fig. 2 ▶(b). Attempts to crystallize GtgE proved challenging, and multiple rounds of limited proteolysis were performed in order to arrive at a construct that was amenable to crystallization. Only after introducing an N-terminal truncation were well diffracting crystals obtained. The difficulties in crystallizing a larger construct of GtgE were likely to be the consequence of an extended, surface-exposed loop immediately N-terminal to residue 80. This region is highly susceptible to limited proteolysis (Fig. 1 ▶ a) and is substantially truncated in homologous structures. It is likely that this loop region inhibits the ordered protein packing necessary for crystal formation.
Figure 2.

(a) The overall fold of GtgE residues 80–213. Helices are shown in red, β-strands are shown in blue and areas without observable electron density are represented by dashed lines. His151 and Asp169 are depicted as sticks in green. (b) Topology diagram of GtgE residues 80–213 generated in PDBSum (Laskowski, 2009 ▶). (c) The full sequence of GtgE with the secondary-structure elements displayed for the crystallized residues 80–213. Gray regions of the sequence indicate residues that are not present in the crystallized sequence. There are five helices, which are numbered and shown in red; six β-strands are depicted in blue and labeled by their sheets; and the active residues Cys45, His151 and Asp169 are indicated by a red box. β-Turns are represented by β and β-hairpins by  . Originally generated in PDBSum (Laskowski, 2009 ▶).
. Originally generated in PDBSum (Laskowski, 2009 ▶).
Our structure reveals significant similarity to the cysteine protease superfamily, specifically to the clan CA families C1, C2 and C39 (as identified by DaliLite v.3; Holm & Rosenström, 2010 ▶). Members of the cysteine protease superfamily participate in a diverse array of biological pathways. The conserved active-site triad is remarkably adaptable to seemingly disparate chemical reactions, functioning within enzymes as different as proteases, acetyltransferases, transglutamases, deamidases and deubiqutinases. The C1 family members primarily serve as proteolytic enzymes in the lysosomal pathway, while the C2 family members, the calcium-dependent calpains, function in a variety of cellular processes such as signal transduction, apoptosis and cytoskeletal remodeling (Rawlings et al., 2012 ▶). The C39 endopeptidases are bacterial proteins responsible for the maturation of bacteriocin, a secreted bacterial antibiotic protein (Dirix et al., 2004 ▶). The catalytic activity of these enzymes is driven by a nucleophilic cysteine thiol and an electron-accepting histidine and often requires a third residue for the proper orientation of this cysteine–histidine pair. The superfamily members share a conserved overall fold, but differ in the placement of catalytic triad residues within their canonical active site.
Interestingly, a sizeable portion of characterized bacterial T3SS effector proteins belong to the cysteine protease superfamily, utilizing this cysteine-powered catalytic core to manipulate the cellular processes of the host in a variety of ways. For example, E. coli-encoded Cif functions as a deamidase, using a Cys-His-Gln catalytic core to deamidate a critical glutamine in the ubiquitin-like NEDD8 protein (Hsu et al., 2008 ▶; Cui et al., 2010 ▶). Through binding to and deamidating NEDD8, Cif effectively prevents the E3 ligase activity of neddylated CRL complexes, resulting in cell-cycle arrest, the formation of stress fibers and host apoptosis (Cui et al., 2010 ▶; Merlet et al., 2009 ▶; Rabut & Peter, 2008 ▶; Saha & Deshaies, 2008 ▶; Crow et al., 2012 ▶; Taieb et al., 2011 ▶; Jubelin et al., 2010 ▶; Yao et al., 2012 ▶). Another effector, Yersinia pestis YopJ, employs its cysteine-dependent triad, His-Glu-Cys, to acetylate serine and threonine residues on MAPK kinases and the IκB kinase complex, inhibiting both MAPK signaling and activation of the NFκB pathway, respectively (Mukherjee et al., 2007 ▶; Ding et al., 1996 ▶). The catalytic activity of YopJ ultimately leads to the inhibition of innate and adaptive immunity responses and induction of cell death (Mukherjee et al., 2007 ▶; Paquette et al., 2012 ▶; Orth, 2002 ▶; Viboud & Bliska, 2005 ▶). In addition, S. Typhimurium produces a deubiquitinase, SseL, which functions by using a His-Asn-Cys triad to remove Lys63-linked ubiquitin chains from SCV-associated aggregates that are targeted for autophagic degradation (Mesquita et al., 2012 ▶; Rytkönen et al., 2007 ▶). In this manner, SseL decreases the autophagic flux within the host, consequently contributing to down-modulation of NF-κB-dependent cytokine production and macrophage-delayed cytotoxicity (Mesquita et al., 2012 ▶; Rytkönen et al., 2007 ▶; Figueira & Holden, 2012 ▶; Le Negrate et al., 2008 ▶).
GtgE is the newest addition to this growing list of bacterial effectors that function using an active-site cysteine. Produced by S. Typhimurium, GtgE cleaves its Rab GTPase substrates, Rab29, Rab32 and Rab38, preventing the delivery of antimicrobial agents to the SCV and thereby subverting one facet of the host’s defense mechanism. In this manner, GtgE contributes to the ability of broad-host strains of Salmonella to maintain a diverse repertoire of host species.
3.2. The catalytic triad of GtgE
The overall fold of GtgE and the placement of its active-site histidine (His151) align best with its cysteine protease clan CA homologs (Fig. 3 ▶). His151 was first identified as a critical catalytic residue by Spanò and coworkers through mutagenesis (Spanò et al., 2011 ▶). This work confirms the importance of this residue in its active-site positioning (Fig. 4 ▶ a). Our crystal structure did not contain a cysteine in the canonical position, nor did it contain any cysteines in proximity to the other active-site residues. In addition, the cysteine-containing helix present in members of this superfamily was not present in the active site. Since the crystal structure lacks the first 78 residues of GtgE, we hypothesized that the active-site cysteine must be located in the missing N-terminal domain. To confirm this hypothesis, we made point mutations (Cys to Ala) of each of the eight cysteine residues in GtgE and tested the ability of the enzyme to cleave Rab38. Cys45 was the only mutation that resulted in a loss of activity (Fig. 4 ▶ b). Although Cys45 is missing from our structure, we predict, based on the homologous enzymes, that it resides at the beginning of a helix formed by the N-terminal residues not included in the crystallized construct. The third triad member in GtgE is Asp169, which was also determined by point mutation (Asp to Ala) and activity analysis (Fig. 4 ▶ a).
Figure 3.

Alignment of GtgE80–213 (green) with a C39 family structural homolog, the peptidase domain of ComA (PDB entry 3k8u, purple; Ishii et al., 2010 ▶). The active residues of GtgE80–213, His151 and Asp169 (labeled in bold), align with the active histidine and aspartic acid residues of the peptidase domain of ComA. The active cysteine is missing in the GtgE80–213 structure owing to N-terminal truncation, but is predicted to reside at the beginning of an α-helix, homologous to the active cysteine of ComA depicted here. The active residues of the peptidase domain of ComA, Cys17, His97 and Asp112, are labeled in bold italics.
Figure 4.

(a) Mutations (C45A, H151A and D169A) of the catalytic triad of full-length GtgE greatly reduce the enzyme activity in vitro. GtgE runs slightly larger than Rab38, and the row of bands corresponding to GtgE is indicated with an asterisk to the right of the panel. (b) The Cys-to-Ala mutation activity profile for GtgE. Each cysteine mutant was tested for cleavage activity against Rab38 as described. Of all eight cysteine mutants, only Cys45Ala shows a loss of function, indicating that it is the active cysteine of GtgE. The top row of bands corresponds to GtgE and is indicated with an asterisk to the right of the panel. (c) The catalytic triad residues (Cys45, His151 and Asp169) were mutated to alanines and their ability to cleave Rab38 is charted as a percentage of the total Rab38 cleaved over time in minutes. The standard error of the mean is indicated with black bars for each time point.
To quantitate the effect of these catalytic mutations on GtgE activity, we monitored cleavage of Rab38 as a percentage of total Rab38 cleaved over time (Fig. 4 ▶ c). Under the reaction conditions used in this study, wild-type GtgE cleaved 36% of Rab38 over the course of 30 min. It is unclear why more Rab38 was not cleaved, although the in vitro conditions may poorly recapitulate those within the cellular context. In addition, it is not clear how active GtgE must be, and for how long, in order to achieve a biologically significant effect. Alanine mutations to the catalytic core, Cys45 and His151, significantly hindered the activity of GtgE, reducing the activity of the enzyme by 94 and 89%, respectively (Fig. 4 ▶ c). An alanine mutation of the third triad member in GtgE, Asp169, led to a 72% decrease in activity (Fig. 4 ▶ c), which is reflective of the important, though supplementary, role of Asp159 in the reaction. Taken together, these data indicate that Cys45, His151 and Asp169 are key components of the active site of GtgE.
3.3. Catalytic mutants in vivo
GtgE is able to cleave Rab29, Rab32 and Rab38 and prevent their recruitment to the SCV (Spanò & Galán, 2012 ▶; Spanò et al., 2011 ▶). To test whether the residues of the catalytic triad are necessary for the activity of GtgE in vivo, we infected COS-1 cells with S. Typhi expressing either wild-type GtgE or GtgE with a catalytic triad point mutation (C45A, H151A or D169A) and analyzed the effect of GtgE mutant expression on Rab29 and Rab38 and their subcellular localization (Fig. 5 ▶). Cells infected with wild-type S. Typhi show intact Rab29 and Rab38 (Fig. 5 ▶ a) as well as recruitment of these Rab GTPases to the surface of the SCV (Fig. 5 ▶ b). In contrast, Rab29 and Rab38 are cleaved in cells infected with S. Typhi expressing wild-type GtgE (Fig. 5 ▶ a) and are not detected on the SCV (Fig. 5 ▶ b). Importantly, when cells are infected with S. Typhi expressing the catalytic triad point mutants of GtgE they show intact Rab29 and Rab38 (Fig. 5 ▶ a), and these two Rab GTPases are promptly recruited to the surface of the SCV (Fig. 5 ▶ b). These data indicate that mutations of the GtgE catalytic triad residues are sufficient to render GtgE inactive in vivo.
Figure 5.

COS-1 cells expressing CFP fusions of Rab29 and Rab38 were left uninfected or were infected with wild-type S. Typhi (wt) or S. Typhi expressing full-length wild-type GtgE (pGtgE) or catalytic triad point mutations of GtgE (pGtgEH151A, pGtgEC45A and pGtgED169A). Two and a half hours after infection, Rab29 and Rab38 were analyzed by Western blotting (a) or fluorescence microscopy (b). Nonspecific bands associated with the Rab38 experiments are indicated by a red asterisk.
3.4. GtgE inhibition
The inhibition of GtgE may serve as a means to alter the host specificity of broad-host salmonellae. Equipped with an understanding of the function of GtgE at the mechanistic level, we were able to identify three small molecules from a panel of cysteine protease inhibitors that are capable of inhibiting GtgE function in vitro: N-ethylmaleimide (NEM), antipain and chymostatin (Fig. 6 ▶ a). NEM covalently modifies cysteine residues, making it the least specific, although the most potent, of the three inhibitors, showing inhibition at a 1:43 molar ratio of GtgE to NEM.
Figure 6.
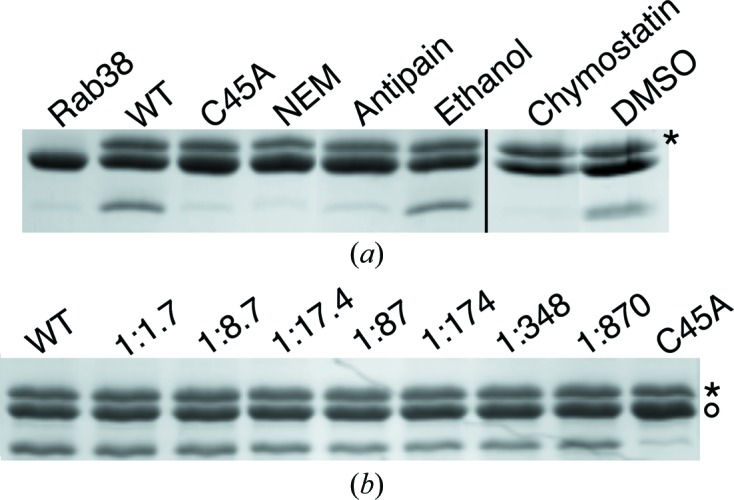
(a) Full-length GtgE is inhibited by N-ethylmaleimide (NEM), antipain and chymostatin. The reactions with chymostatin and its delivery control, DMSO, were performed separately using identical conditions to those of the reactions with NEM and antipain; thus, this is indicated with a black line. (b) Leupeptin does not inhibit GtgE at the highest concentration that could be tested given the solubility of leupeptin. At a molar ratio of 1:870 GtgE:leupeptin, leupeptin did not inhibit the ability of GtgE to cleave Rab38. The activity of GtgE-Cys45Ala is shown as a control. An asterisk to the right of each panel indicates the row of bands corresponding to GtgE; in (b) a circle denotes the row of bands corresponding to full-length Rab38.
Antipain and chymostatin are both microbial-derived small peptide inhibitors which contain a C-terminal aldehyde that inhibits cysteine proteases by forming a hemiacetal adduct with the active thiol (Umezawa, 1982 ▶). Antipain is bulky in structure (Arg-Val-Arg-Phe) and broadly targets cysteine and serine proteases. Chymostatin is composed of two Phe residues, capreomycidine, a large residue unique to microbes, and a variable hydrophobic residue. Exhibiting greater specificity than antipain, chymostatin inhibits papain, chymotrypsin and cathepsins A/B/C/H/L. Chymostatin was a twofold more potent inhibitor of GtgE than was antipain, inhibiting GtgE at a molar ratio of 1:435 GtgE to chymostatin compared with a molar ratio of 1:870 GtgE to antipain (Fig. 6 ▶ a). Conversely, leupeptin, another microbial-derived inhibitor comprised of residues with relatively small side chains (Leu-Leu-Arg), does not inhibit GtgE at the highest tested molar ratio (1:870 GtgE to leupeptin; Fig. 6 ▶ b).
GtgE cleaves between a Gly and Val located in the Switch I region of Rab29/32/38 (Spanò et al., 2011 ▶). This region is highly conserved among these three Rab GTPases, consisting predominantly of hydrophobic residues and several residues with bulkier side chains, such as aspartic acid and phenylalanine. We theorize that chymostatin and antipain are more suited to inhibit GtgE because they both contain aromatic residues, whereas leupeptin does not; therefore, these inhibitors are better able to mimic the Rab29/32/38 cleavage site and are best suited to provide the foundation for more targeted inhibitor design.
In conclusion, through X-ray crystallography and biochemical assays we were able to characterize the catalytic triad of GtgE and provide novel insight into the cysteine-dependent catalytic function of this S. Typhimurium-encoded effector protease. The information gleaned from this work can be used to design GtgE-specific inhibitors that may aid in the attenuation of broad-host strains of Salmonella.
Supplementary Material
PDB reference: GtgE, 4mi7
Acknowledgments
This work was funded in part by NIH ARRA Award R01AI52182 to CES and NIAID Grants R01AI055472 and R01AI079022 to JEG. We thank D. Oren at Rockefeller University and W. Yu at Brookhaven beamline X29 for access to and assistance with crystallographic equipment. We also thank M. Holt and N. Shah for valuable insight. ACK performed protein purification, crystallization and structure determination, in vitro catalytic mutant assays and inhibition assays. SS performed in vivo catalytic mutant assays. All authors contributed to manuscript preparation. The authors declare that they have no conflict of interest.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Bultema, J. J., Ambrosio, A. L., Burek, C. L. & Di Pietro, S. M. (2012). J. Biol. Chem. 287, 19550–19563. [DOI] [PMC free article] [PubMed]
- Crow, A., Hughes, R. K., Taieb, F., Oswald, E. & Banfield, M. J. (2012). Proc. Natl Acad. Sci. USA, 109, E1830–E1838. [DOI] [PMC free article] [PubMed]
- Crump, J. A. & Mintz, E. D. (2010). Clin. Infect. Dis. 50, 241–246. [DOI] [PMC free article] [PubMed]
- Cui, J., Yao, Q., Li, S., Ding, X., Lu, Q., Mao, H., Liu, L., Zheng, N., Chen, S. & Shao, F. (2010). Science, 329, 1215–1218. [DOI] [PMC free article] [PubMed]
- Ding, J., McGrath, W. J., Sweet, R. M. & Mangel, W. F. (1996). EMBO J. 15, 1778–1783. [PMC free article] [PubMed]
- Dirix, G., Monsieurs, P., Dombrecht, B., Daniels, R., Marchal, K., Vanderleyden, J. & Michiels, J. (2004). Peptides, 25, 1425–1440. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Figueira, R. & Holden, D. W. (2012). Microbiology, 158, 1147–1161. [DOI] [PubMed]
- Galán, J. E. & Curtiss, R. III (1991). Infect. Immun. 59, 2901–2908. [DOI] [PMC free article] [PubMed]
- Grosse-Kunstleve, R. W. & Adams, P. D. (2003). Acta Cryst. D59, 1966–1973. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Hsu, Y., Jubelin, G., Taieb, F., Nougayrède, J.-P., Oswald, E. & Stebbins, C. E. (2008). J. Mol. Biol. 384, 465–477. [DOI] [PMC free article] [PubMed]
- Ishii, S., Yano, T., Ebihara, A., Okamoto, A., Manzoku, M. & Hayashi, H. (2010). J. Biol. Chem. 285, 10777–10785 [DOI] [PMC free article] [PubMed]
- Jubelin, G., Taieb, F., Duda, D. M., Hsu, Y., Samba-Louaka, A., Nobe, R., Penary, M., Watrin, C., Nougayrède, J.-P., Schulman, B. A., Stebbins, C. E. & Oswald, E. (2010). PLoS Pathog. 6, e1001128. [DOI] [PMC free article] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. (2009). Nucleic Acids Res. 37, D355–D359. [DOI] [PMC free article] [PubMed]
- Le Negrate, G., Faustin, B., Welsh, K., Loeffler, M., Krajewska, M., Hasegawa, P., Mukherjee, S., Orth, K., Krajewski, S., Godzik, A., Guiney, D. G. & Reed, J. C. (2008). J. Immunol. 180, 5045–5056. [DOI] [PubMed]
- Majowicz, S. E., Musto, J., Scallan, E., Angulo, F. J., Kirk, M., O’Brien, S. J., Jones, T. F., Fazil, A. & Hoekstra, R. M. (2010). Clin. Infect. Dis. 50, 882–889. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Merlet, J., Burger, J., Gomes, J. E. & Pintard, L. (2009). Cell. Mol. Life Sci. 66, 1924–1938. [DOI] [PMC free article] [PubMed]
- Mesquita, F. S., Thomas, M., Sachse, M., Santos, A. J., Figueira, R. & Holden, D. W. (2012). PLoS Pathog. 8, e1002743. [DOI] [PMC free article] [PubMed]
- Mukherjee, S., Hao, Y.-H. & Orth, K. (2007). Trends Biochem. Sci. 32, 210–216. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Orth, K. (2002). Curr. Opin. Microbiol. 5, 38–43. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). J. Appl. Cryst. 39, 109–111.
- Paquette, N., Conlon, J., Sweet, C., Rus, F., Wilson, L., Pereira, A., Rosadini, C. V., Goutagny, N., Weber, A. N., Lane, W. S., Shaffer, S. A., Maniatis, S., Fitzgerald, K. A., Stuart, L. & Silverman, N. (2012). Proc. Natl Acad. Sci. USA, 109, 12710–12715. [DOI] [PMC free article] [PubMed]
- Rabut, G. & Peter, M. (2008). EMBO Rep. 9, 969–976. [DOI] [PMC free article] [PubMed]
- Raposo, G. & Marks, M. S. (2007). Nature Rev. Mol. Cell Biol. 8, 786–797. [DOI] [PMC free article] [PubMed]
- Rawlings, N. D., Barrett, A. J. & Bateman, A. (2012). Nucleic Acids Res. 40, D343–D350. [DOI] [PMC free article] [PubMed]
- Rytkönen, A., Poh, J., Garmendia, J., Boyle, C., Thompson, A., Liu, M., Freemont, P., Hinton, J. C. & Holden, D. W. (2007). Proc. Natl Acad. Sci. USA, 104, 3502–3507. [DOI] [PMC free article] [PubMed]
- Saha, A. & Deshaies, R. J. (2008). Mol. Cell, 32, 21–31. [DOI] [PMC free article] [PubMed]
- Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. (2012). Nature Methods, 9, 671–675. [DOI] [PMC free article] [PubMed]
- Schwartz, S. L., Cao, C., Pylypenko, O., Rak, A. & Wandinger-Ness, A. (2007). J. Cell Sci. 120, 3905–3910. [DOI] [PubMed]
- Song, J., Gao, X. & Galán, J. E. (2013). Nature (London), 499, 350–354. [DOI] [PMC free article] [PubMed]
- Spanò, S. & Galán, J. E. (2012). Science, 338, 960–963. [DOI] [PMC free article] [PubMed]
- Spanò, S., Liu, X. & Galán, J. E. (2011). Proc. Natl Acad. Sci. USA, 108, 18418–18423. [DOI] [PMC free article] [PubMed]
- Spanò, S., Ugalde, J. E. & Galán, J. E. (2008). Cell Host Microbe, 3, 30–38. [DOI] [PubMed]
- Taieb, F., Nougayrède, J. P. & Oswald, E. (2011). Toxins, 3, 356–368. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Adams, P. D., Read, R. J., McCoy, A. J., Moriarty, N. W., Grosse-Kunstleve, R. W., Afonine, P. V., Zwart, P. H. & Hung, L.-W. (2009). Acta Cryst. D65, 582–601. [DOI] [PMC free article] [PubMed]
- Umezawa, H. (1982). Annu. Rev. Microbiol. 36, 75–99. [DOI] [PubMed]
- Viboud, G. I. & Bliska, J. B. (2005). Annu. Rev. Microbiol. 59, 69–89. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Methods Enzymol. 374, 300–321. [DOI] [PubMed]
- Yao, Q., Cui, J., Wang, J., Li, T., Wan, X., Luo, T., Gong, Y.-N., Xu, Y., Huang, N. & Shao, F. (2012). Proc. Natl Acad. Sci. USA, 109, 20395–20400. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: GtgE, 4mi7