

Abstract
Pathogenesis of type 1 diabetes (T1D) is mediated by effector T cells and CD4 Th1 and Th17 T cells have important roles in this process. While effector function of Th1 cells is well established, due to their inherent plasticity Th17 cells have been more controversial. Th17 cells contribute to pathogenicity, but several studies indicate that Th17 cells transfer disease through conversion to Th1 cells in vivo. CD4 T cells are attracted to islets by β-cell antigens which include insulin and the two new autoantigens, chromogranin A and islet amyloid polypeptide, all proteins of the secretory granule. Peptides of insulin and ChgA bind to the NOD class II molecule in an unconventional manner and since autoantigenic peptides may typically bind to MHC with low affinity, it is postulated that post-translational modifications of β-cell peptides could contribute to the interaction between peptides, MHC, and the autoreactive TCR.
Introduction
Interest in how T cells become pathogenic and mediate the autoimmune events that lead to islet destruction in type 1 diabetes (T1D) has spanned three decades. It was apparent by the mid-80's that both CD4 and CD8 T cells contributed to the disease process and the subsequent isolation of diabetogenic T cell lines and clones indicated that at least under some circumstances, either CD4 or CD8 T cell clones could induce the disease process in diabetes-prone rodents [1]. T cell receptor transgenic (TCR-Tg) mice followed, the first example being the BDC-2.5 TCR-Tg mouse [2], bearing the TCR from the diabetogenic CD4 T cell clone BDC-2.5 and widely used to investigate both pathogenic and regulatory events in NOD autoimmune diabetes [1]. In recent years, the paradigm of Th1/Th2 balance has shifted due to the increasing body of information on other CD4 subsets, in particular regulatory T cells (Tregs) and Th17 T cells. A much pursued goal has been the identification of the autoantigens that drive pathogenic T cells in T1D, not only because of the need to better understand etiology and the breakdown of tolerance, but also due to the growing interest in antigen-specific therapies. Insulin has been the dominant beta cell autoantigen, but the recent discovery of other beta cell secretory granule proteins as autoantigens leads to new avenues of investigation. In this review, we will highlight some of the latest developments in our knowledge of pathogenic CD4 T cells and the autoantigens that activate them.
CD4 Th1 Effector T cells
CD4 Th1 T cells have traditionally been regarded as playing a key role in the pathogenesis of T1D. Isolation and characterization of Th1 T cell clones from NOD mice and investigations of how pathogenic cells are activated and regulated using TCR-Tg mice have provided the basis of much of our understanding of how Th1 T cells contribute to autoimmune diabetes [1], but more importantly, the relevance of Th1 cells to T1D in humans has been confirmed by many studies on CD4 T cells isolated from human patients. Although recent attention has been more focused on various treatments to prevent the destructive activity of Th1 T cells, there continues to be interest in how Th1 T cells function, how they encounter antigen, and how they are triggered. By understanding these events in detail, new therapeutic approaches can be developed.
For example, the importance of costimulation in Th1 activation and function has been highlighted through studies of the CD40-CD154 signaling pathway, as well as CD28 and CTLA4 [3–6], but only recently has it been appreciated that CD40 operates in Th1 cells. CD40 was first noted to be elevated on T cells in autoimmune strains [7] and its presence on NOD-derived diabetogenic CD4 T cell clones suggested a functional role in the inflammatory response [8]. The observation that CD40 has a costimulatory function in Th1 cells [9,10] and that CD40+ (but not CD40-) CD4 T cells contained the diabetogenic population in adoptive transfers of disease from NOD mice [11,12], further supported a functional role. More recent work has shown that signaling between T cells occurs through CD40 and CD154 co-expressed on pathogenic CD4 T cells (Fig. 1), and that impaired CD40 signaling in NOD T cells through retroviral expression of a dominant-negative form of CD40 abrogates their ability to mediate T1D (Baker and Haskins, unpublished). Thus there appear to be intrinsic mechanisms in Th1 cells that contribute to their pathogenic properties, prolonging and enhancing their inflammatory activity and perhaps thereby promoting a more favorable environment for Th1 cells than for Tregs.
Figure 1.

Pathogenic CD4 T cells in the islet infiltrate. Th1 and Th17 T cells are the primary CD4 effector T cells mediating islet inflammation in T1D. Th17 cells are more “plastic” and can be converted into cells with a dual phenotype (Th1/Th17) co-expressing Th1 and Th17 cytokines, or into Th1 cells. Th17 cells have also been reported to be a distinct pathogenic subset. Upon activation by antigen presented to Th1 cells by IAg7 on dendritic cells (DC), Th1 cells co-express both CD154 and CD40 which permits interaction with other Th1 cells through these molecules, further promoting activation and pathogenic function of Th1 cells. Cytokines (IFNγ, TNFα) produced by Th1 cells can act directly on islet β-cells, but can also activate macrophages (Mϕ) to produce inflammatory cytokines such as TNFα and IL-1β. Chemokines (chemoattractant cytokines) produced by both Th1 and Th17 cells are thought to be involved in the recruitment of other immune effectors into the site, including CD8 T cells, NK cells and macrophages. Thus the inflammatory environment is initiated and perpetuated, ultimately resulting in β-cell destruction.
Strategies to block CD4 Th1 T cells can provide insight into the pathogenic mechanisms employed by this subset and novel vaccination protocols have suggested potential therapeutic targets. For example, the prevention of diabetes in NOD mice by mature dendritic cells was found to work through affecting the migration of Th1 T cells and attracting CD4 T cells with a Th2 phenotype [13]. Administration of a galectin-9 plasmid, a negative regulator that works through the T cell Ig mucin 3, expressed on terminally differentiated Th1 T cells, was another treatment found to downregulate the Th1 T cell response in NOD mice and protect against disease [14]. An interesting new study has examined the role of B7x in T1D. Absence of islet expression of this inhibitory member of the B7/CD28 family resulted in expansion of both Th1 and Th17 cells in the pancreas after transfer of BDC-2.5 TCR-Tg T cells, whereas over-expression of B7x inhibited disease due to inhibition of IFNγ production rather than expansion of Tregs [15*]. Another study using BDC-2.5 TCR-Tg T cells in transfers suggests that activated effector Th1 cells are involved in their own feedback regulation by stimulating expansion of Tregs [16*]. These new studies are just some of those illustrating how Th1 effector function can be elucidated through efforts to prevent pathogenic CD4 T cell activity. A comprehensive list of other immunotherapies being investigated in T1D has been recently evaluated, with particular focus on targeting CD4 T cells through antigen-specific tolerance induction [17*], a topic we return to below.
CD4 Th17 T cells
The exact role of Th17 cells in autoimmune diabetes remains somewhat controversial. On the one hand there are data suggesting that Th17 cells play a pathogenic role in diabetes onset and that tolerogenic strategies that impede this population inhibit disease. For example, Jain et al reported that pathogenicity of IL-17-producing cells in NOD mice could be suppressed by antigen (GAD2) inserted into an immunoglobulin molecule, resulting in disease inhibition if Ig-GAD2 was administered during, but not prior to insulitis [18]. In another report where intervention was successful in pre-diabetic NOD mice, it was found that treatment with anti-IL17 antibodies or with the IL-17 inhibitory family member, IL-25, led to reduction of disease [19]. A recent study in diabetes-prone BB rats indicates that increased numbers of both Th1 and Th17 cells shift the balance toward effector T cells, leading to disease versus protection [20]. Transfers of OVA-specific Th17 T cells into RIP-mOVA mice suggested that Th17 T cells may be needed for stimulation of CD8 CTL effectors [21]. Studies with RelBlo dendritic cells indicated that tolerizing DC therapy works in young (4 wk) prediabetic NOD mice, but not in older (14-wk old) NOD animals where the increased production of IL-1β impairs Treg function and promotes Th17 differentiation [22]. These examples all suggest that Th17 T cells can act as pathogenic effectors, although at least in some instances via effects on other cells.
Th17/Th1 conversion
On the other hand, groups using transfer systems have shown that while purified polarized Th17 cells do indeed have the ability to transfer diabetes, this arises through in vivo conversion of these cells to Th1-like cells. The concept of plasticity in CD4 Th cell phenotypes – nicely outlined in a recent review [23] – has been discussed at length in recent years, but the circumstances under which cells undergo conversion are not well understood. Studies with the BDC-2.5 TCR-Tg mouse have helped to establish that T helper cell flexibility is characteristic of the autoimmune disease process and its regulation. BDC-2.5 TCR-Tg T cells can readily be activated to be Th1 effectors, converted to Tregs in the presence of TGFβ, and as recently demonstrated, can also display a Th17 phenotype. Two groups showed that disease could be transferred to recipient NOD.scid mice by BDC-2.5 TCR-Tg T cells polarized in vitro to a Th17 phenotype [24,25]. However, analysis of the transferred cells indicated that T cells that were Th17 at the time of transfer were converted into Th1 T cells in vivo. Lexberg et al reported that Th17 cells isolated ex vivo could not be converted into Th1 T cells by IL-12 [26], although later work by this group indicated that following culture with both IFNγ and IL-12, ex vivo Th17 cells could be converted to a Th1/Th17 phenotype, co-expressing IFNγ and IL-17, as well as the transcription factors, T-bet and RORγt [27**]. A new study by Bending et al has revealed that while the Il12rb2 gene is expressed in Th17 T cells generated in vitro, little expression is seen in Th17 cells isolated ex vivo; Il12rb2 expression can however be increased in ex vivo Th17 T cells following in vitro stimulation, thereby allowing conversion to a Th1 phenotype in the appropriate cytokine milieu [28**]. This study may point toward an important in vivo mechanism of Th17 cell differentiation and provide in part an explanation for differing observations with regard to Th17 plasticity. There is also evidence for conversion of Th17 to Th1 T cells in human autoimmune disease, at least in the instance of childhood autoimmune arthritis [29].
Autoantigens for CD4 T cells
There is a good body of data suggesting that the presence of autoreactive T cells in the pancreas is a reflection of the presence of antigen [30,31*]. Various reviews have provided a comprehensive list of the autoantigen targets of B cells and T cells in both humans and the NOD mouse [32–34]. We will discuss here only secretory granule proteins, insulin and two newly discovered antigens for CD4 T cells, chromogranin A (ChgA) and islet amyloid polypeptide (IAPP). These proteins are all proteolytically processed within the secretory granules and are thereby truncated into small peptide fragments such as insulin C peptide and in the case of ChgA, WE14.
Insulin has been the preeminent beta cell autoantigen, first as the target of autoantibodies in patients and rodents [35], and then following the first report of insulin-reactive T cell lines from pre-diabetic islets of NOD mice [36]; subsequently, insulin peptide specific T cells were found in the PLN of diabetic patients [37]. Recent studies have extended our understanding of interactions between insulin and MHC and the complexities of the T cell response to insulin and its peptides. For example, the ability of insulin B:9-23 to bind to IAg7 in two different registers has been described previously [38,39], and recently it was postulated that a third binding register exists in which disease-relevant peptides bind with low affinity, allowing their escape from thymic deletion; thus pathogenic CD4 T cells could target epitopes with unfavorable MHC binding motifs [40**]. The use of insulin in antigen-induced tolerance regimes has also been intensely investigated. One promising avenue for induction of antigen-specific tolerance involves administration of antigenic peptides coupled to cells with the chemical crosslinker, ethylene carbodiimide (ECDI), an approach thought to involve both the activation of Tregs and PD-L1/PD-1 mediated anergy. This approach is already in clinical trials for multiple sclerosis and a second trial is planned with insulin-coupled PBLs for T1D [17*].
Chromogranin A (ChgA) and islet amyloid polypeptide (IAPP) are two newly discovered autoantigens for CD4 T cells in T1D. Using a biochemical purification strategy combined with mass spectrometric analysis of chromatographic fractions antigenic for diabetogenic T cell clones, we demonstrated that these proteins are the source of antigenic ligands for diabetogenic CD4 T cell clones [41**,42*]. Initial studies identified at least one peptide ligand from each protein, WE14 from ChgA and KS20 from IAPP. The IAPP sequence KS20 strongly stimulates a T cell clone BDC-5.2.9 [42*], but although WE14 from ChgA can stimulate at least three T cell clones including BDC-2.5, the natural sequence, a proteolytic cleavage product of ChgA, is only antigenic at high concentrations of peptide [41**]. Of note, another group has identified a peptide, ChgA 29-42, from vasostatin-1 which itself is generated from ChgA; ChgA 29-42 was shown to be a better binder of IAg7 and could stimulate T cells from the BDC-2.5 TCR-Tg mouse [43], findings that appear to challenge the status of WE14 as the ligand for BDC-2.5. Subsequent studies, however, have demonstrated that the antigenicity of the WE14 peptide for BDC-2.5 is dramatically altered if the peptide is treated with transglutaminase, an enzyme known to be involved in various post-translational modification processes. The details of how this reaction compares to events taking place within inflamed islets are not yet known, but our in vitro data, coupled with the fact that the natural form of the antigen in beta-cell extracts is far more potent than unmodified WE14, would suggest that an altered form of this peptide is the ligand for BDC-2.5. The enzymatically converted form of WE14 also elicits responses from BDC-2.5 TCR-Tg T cells and from primary CD4 T cells of NOD mice (Delong, Haskins et al, unpublished results). Furthermore, preliminary studies on T cells from human T1D patients indicate similar responsiveness to WE14, with a significant difference in responses from patients versus controls to the converted peptide (Gottlieb, Haskins et al, unpublished results).
Post-translational modification is a well-established mechanism for the generation of autoantigens in several autoimmune diseases; notable examples include citrullination in rheumatoid arthritis and multiple sclerosis, deamidation in celiac disease, and phosphorylation in systemic lupus erythromatosis [44–47]. Another example is the iodination of thyroglobulin which only occurs in the thyroid gland, resulting in iodinated epitopes shown to be pathologically relevant in autoimmune thyroid disease [48,49]. Evidence of post-translational modification in T1D comes from studies on CD4 T cells from a human patient in which it was found that the stimulating epitope was a peptide from the insulin A chain, A1-13, containing oxidized adjacent cysteines resulting in a vicinal disulfide bond [50]. With this exception, however, there has been little other investigation into post-translational modification as the basis for autoantigenicity in T1D. The inflammatory conditions within islet beta cells could lead to a variety of enzymatic and/or spontaneous non-enzymatic post-translational modifications of secretory granule proteins. In addition, secretory granule peptides are prone to aggregation, an event that can readily occur in a dysregulated cell environment, perhaps making them more attractive targets for antigen-presenting cells. We hypothesize that during the autoimmune progression of disease in islets, neo-antigens such as an altered WE14 peptide ligand are generated due to post-translational modification (Fig. 2). In addition to disulfide formation, these reactions could include deamidation, citrullination, acetylation and others (Fig. 3).
Figure 2.

Hypothetical generation of β-cell autoantigens through post-translational modification. Proteins such as insulin (Ins), chromogranin A (ChgA), and islet amyloid polypeptide (IAPP) normally undergo proteolytic cleavage in the secretory granules to yield functional peptides. If the environment of the β-cell is undergoing stress generated by inflammatory events, these peptides can undergo aggregation and/or other modifications. Although post-translational modifications (PTM) are a normal part of protein processing, under conditions of cellular stress, dysfunctional PTM can occur. Increases in intracellular levels of calcium can lead to the activation of enzymes (e.g., transglutaminase) that catalyze PTM. Spontaneous (non-enzymatic) PTM (e.g., oxidation or glycation) are also increased. PTM can result in potentially harmful changes, such as altered amino acids that could result in recognition of a peptide as “non-self” and/or in altered processing within APC. Aggregated forms of peptides may be more readily taken up by antigen-presenting cells (APC) for internal processing to neo-epitopes. Post-translationally modified peptides might also be bound with higher affinity by the class II IAg7 molecule on the APC whereas unmodified self-peptides are thought to be poor binders.
Figure 3.
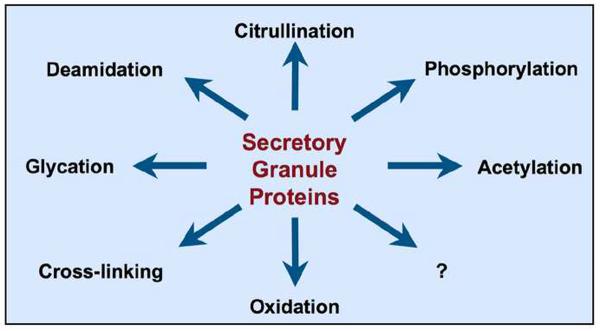
Potential post-translational modifications in the islet β-cell. A variety of post-translational modifications could take place in secretory granule proteins within the islet β-cell under the inflammatory conditions that prevail during progression of T1D.
Insulin and IAPP are proteins originating in beta cells, but ChgA is found in secretory granules of other tissues besides the pancreatic islet and may therefore be less obvious as a candidate autoantigen. One hypothesis for the autoantigenic activity of certain ligands in T1D is that these peptides bind to the predisposing MHC class II in an unusual manner, a mechanism that has precedent in other autoimmune disorders such as EAE [51,52], and is exemplified by the ability of insulin B:9-23 to bind to IAg7 in various binding registers [39,40]. The WE14 peptide from ChgA is also a weak binder to IAg7, filling only part of the class II groove [41**]. If weak binding to the class II molecule is a characteristic of autoantigenic peptides, some additional mechanisms, such as post-translational modification, may be necessary in order to increase peptide affinity for MHC and/or TCR. For example, the conversion of WE14 by transglutaminase could simulate an intracellular process in which peptide multimers are formed or the peptide is covalently bound to IAg7. It is possible that post-translational modification could be relevant to new findings by Mohan et al indicating that at least some diabetogenic insulin-reactive T cells respond to insulin peptide generated only in the secretory granules [53*], and a second paper describing effective vaccination of NOD mice with an insulin mimotope but not natural insulin sequences [54].
Conclusions
The immune system has evolved strategies to ensure that it has the capability to respond to infectious agents, but does not respond to self-antigens. Among the central and peripheral tolerogenic mechanisms involved is deletion orchestrated by the expression of transcription factors such as Aire that enable the expression of peripheral self-antigens, both in the thymus and in secondary lymphoid organs [55]. However, the antigens expressed at these sites will not undergo the same post-translational modifications that occur in the peripheral organs themselves. Proteolytic processing of self-antigens clearly occurs in the pancreatic beta cell and in autoimmune diabetes, there are ample opportunities for post-translational modifications to take place. Th1 and Th17 T cells are key participants in the autoreactive response although the factors governing initiation of this response remain unclear. Topics for future study include the further clarification of when Th1 and Th17 T cells contribute to disease, post-translational modification as a general mechanism in β-cells for generation of antigenic epitopes for autoreactive T cells, and identification of more peptides (or altered peptides) for use in targeted induction of tolerance.
Highlights
CD4 Th1 and Th17 T cells are primary mediators of autoimmune diabetes
Pathogenicity of Th17 cells may result from conversion to Th1 cells in vivo
Chromogranin A and islet amyloid polypeptide are new autoantigens for CD4 T cells
β-cell peptides bind to MHC class II unconventionally and with low affinity
Antigenic epitopes in T1D may be generated through post-translational modification
Acknowledgements
We thank Rocky Baker, Thomas Delong, and Jing He for helpful comments and suggestions, and to Brenda Bradley for assistance with the figures.
The authors acknowledge financial support from the National Institutes of Health, the Juvenile Diabetes Foundation, and the American Diabetes Association (KH); and from the Wellcome Trust, the UK Medical Research Council, and the European Union (AC).
References
- 1.Haskins K. Pathogenic T-cell clones in autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol. 2005;87:123–162. doi: 10.1016/S0065-2776(05)87004-X. [DOI] [PubMed] [Google Scholar]
- 2.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 5.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 3.Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol. 2009;21:265–272. doi: 10.1016/j.smim.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009;21:293–300. doi: 10.1016/j.smim.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev. 2011;241:180–205. doi: 10.1111/j.1600-065X.2011.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner DH, Jr., Newell E, Sanderson RJ, Freed JH, Newell MK. Increased expression of CD40 on thymocytes and peripheral T cells in autoimmunity: a mechanism for acquiring changes in the peripheral T cell receptor repertoire. Int J Mol Med. 1999;4:231–242. doi: 10.3892/ijmm.4.3.231. [DOI] [PubMed] [Google Scholar]
- 8.Wagner DH, Jr., Vaitaitis G, Sanderson R, Poulin M, Dobbs C, Haskins K. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc Natl Acad Sci U S A. 2002;99:3782–3787. doi: 10.1073/pnas.052247099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munroe ME, Bishop GA. A Costimulatory Function for T Cell CD40. J Immunol. 2007;178:671–682. doi: 10.4049/jimmunol.178.2.671. [DOI] [PubMed] [Google Scholar]
- 10.Baker RL, Wagner DH, Jr., Haskins K. CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J Autoimmun. 2008;31:385–392. doi: 10.1016/j.jaut.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Waid DM, Vaitaitis GM, Wagner DH., Jr. Peripheral CD4loCD40+ auto-aggressive T cell expansion during insulin-dependent diabetes mellitus. Eur J Immunol. 2004;34:1488–1497. doi: 10.1002/eji.200324703. [DOI] [PubMed] [Google Scholar]
- 12.Munroe ME. Functional roles for T cell CD40 in infection and autoimmune disease: the role of CD40 in lymphocyte homeostasis. Semin Immunol. 2009;21:283–288. doi: 10.1016/j.smim.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Morel PA, Srinivas M, Turner MS, Fuschiotti P, Munshi R, Bahar I, Feili-Hariri M, Ahrens ET. Gene expression analysis of dendritic cells that prevent diabetes in NOD mice: analysis of chemokines and costimulatory molecules. J Leukoc Biol. 2011 doi: 10.1189/jlb.0311126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou FC, Shieh SJ, Sytwu HK. Attenuation of Th1 response through galectin-9 and T-cell Ig mucin 3 interaction inhibits autoimmune diabetes in NOD mice. Eur J Immunol. 2009;39:2403–2411. doi: 10.1002/eji.200839177. [DOI] [PubMed] [Google Scholar]
- 15*.Wei J, Loke P, Zang X, Allison JP. Tissue-specific expression of B7x protects from CD4 T cell-mediated autoimmunity. J Exp Med. 2011;208:1683–1694. doi: 10.1084/jem.20100639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, Martin GH, Elhage R, Derian N, Carpentier W, et al. Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J Clin Invest. 2010;120:4558–4568. doi: 10.1172/JCI42945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Luo X, Herold KC, Miller SD. Immunotherapy of type 1 diabetes: where are we and where should we be going? Immunity. 2010;32:488–499. doi: 10.1016/j.immuni.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain R, Tartar DM, Gregg RK, Divekar RD, Bell JJ, Lee HH, Yu P, Ellis JS, Hoeman CM, Franklin CL, et al. Innocuous IFNgamma induced by adjuvant-free antigen restores normoglycemia in NOD mice through inhibition of IL-17 production. J Exp Med. 2008;205:207–218. doi: 10.1084/jem.20071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, Shapiro AM. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. 2009;58:1302–1311. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van den Brandt J, Fischer HJ, Walter L, Hunig T, Kloting I, Reichardt HM. Type 1 diabetes in BioBreeding rats is critically linked to an imbalance between Th17 and regulatory T cells and an altered TCR repertoire. J Immunol. 2010;185:2285–2294. doi: 10.4049/jimmunol.1000462. [DOI] [PubMed] [Google Scholar]
- 21.Ankathatti Munegowda M, Deng Y, Chibbar R, Xu Q, Freywald A, Mulligan SJ, van Drunen Littel-van den Hurk S, Sun D, Xiong S, Xiang J. A Distinct Role of CD4(+) Th17- and Th17-Stimulated CD8(+) CTL in the Pathogenesis of Type 1 Diabetes and Experimental Autoimmune Encephalomyelitis. J Clin Immunol. 2011 doi: 10.1007/s10875-011-9549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertin-Maghit S, Pang D, O'Sullivan B, Best S, Duggan E, Paul S, Thomas H, Kay TW, Harrison LC, Steptoe R, et al. Interleukin-1beta produced in response to islet autoantigen presentation differentiates T-helper 17 cells at the expense of regulatory T-cells: Implications for the timing of tolerizing immunotherapy. Diabetes. 2011;60:248–257. doi: 10.2337/db10-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009;39:216–224. doi: 10.1002/eji.200838475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bending D, De la Pena H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, Cooke A. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009;119:565–572. doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lexberg MH, Taubner A, Forster A, Albrecht I, Richter A, Kamradt T, Radbruch A, Chang HD. Th memory for interleukin-17 expression is stable in vivo. Eur J Immunol. 2008;38:2654–2664. doi: 10.1002/eji.200838541. [DOI] [PubMed] [Google Scholar]
- 27**.Lexberg MH, Taubner A, Albrecht I, Lepenies I, Richter A, Kamradt T, Radbruch A, Chang HD. IFN-gamma and IL-12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. Eur J Immunol. 2010;40:3017–3027. doi: 10.1002/eji.201040539. [DOI] [PubMed] [Google Scholar]
- 28**.Bending D, Newland S, Krejci A, Phillips JM, Bray S, Cooke A. Epigenetic changes at Il12rb2 and Tbx21 in relation to plasticity behavior of Th17 cells. J Immunol. 2011;186:3373–3382. doi: 10.4049/jimmunol.1003216. [DOI] [PubMed] [Google Scholar]
- 29.Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, Evans JG, Cimaz R, Bajaj-Elliott M, Wedderburn LR. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A. 2010;107:14751–14756. doi: 10.1073/pnas.1003852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lennon GP, Bettini M, Burton AR, Vincent E, Arnold PY, Santamaria P, Vignali DA. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity. 2009;31:643–653. doi: 10.1016/j.immuni.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci U S A. 2011;108:1561–1566. doi: 10.1073/pnas.1018973108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lieberman SM, DiLorenzo TP. A comprehensive guide to antibody and T-cell responses in type 1 diabetes. Tissue Antigens. 2003;62:359–377. doi: 10.1034/j.1399-0039.2003.00152.x. [DOI] [PubMed] [Google Scholar]
- 33.Babad J, Geliebter A, DiLorenzo TP. T-cell autoantigens in the non-obese diabetic mouse model of autoimmune diabetes. Immunology. 2010;131:459–465. doi: 10.1111/j.1365-2567.2010.03362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moser A, Hsu HT, van Endert P. Beta cell antigens in type 1 diabetes: triggers in pathogenesis and therapeutic targets. F1000 Biol Rep. 2010;2:75. doi: 10.3410/B2-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castano L, Eisenbarth GS. Type-I diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu Rev Immunol. 1990;8:647–679. doi: 10.1146/annurev.iy.08.040190.003243. [DOI] [PubMed] [Google Scholar]
- 36.Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. 1995;25:1056–1062. doi: 10.1002/eji.1830250430. [DOI] [PubMed] [Google Scholar]
- 37.Kent SC, Chen Y, Bregoli L, Clemmings SM, Kenyon NS, Ricordi C, Hering BJ, Hafler DA. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435:224–228. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 38.Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol. 2007;178:6051–6057. doi: 10.4049/jimmunol.178.10.6051. [DOI] [PubMed] [Google Scholar]
- 39.Suri A, Levisetti MG, Unanue ER. Do the peptide-binding properties of diabetogenic class II molecules explain autoreactivity? Curr Opin Immunol. 2008;20:105–110. doi: 10.1016/j.coi.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A. 2010;107:10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42*.Delong T, Baker RL, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Barbour G, Bradley B, Haskins K. Islet Amyloid Polypeptide Is a Target Antigen for Diabetogenic CD4+ T Cells. Diabetes. 2011;60 doi: 10.2337/db11-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikoopour E, Sandrock C, Huszarik K, Krougly O, Lee-Chan E, Masteller EL, Bluestone JA, Singh B. Cutting edge: vasostatin-1-derived peptide ChgA29-42 is an antigenic epitope of diabetogenic BDC2.5 T cells in nonobese diabetic mice. J Immunol. 2011;186:3831–3835. doi: 10.4049/jimmunol.1003617. [DOI] [PubMed] [Google Scholar]
- 44.Anderton SM. Post-translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol. 2004;16:753–758. doi: 10.1016/j.coi.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Wu CT, Gershwin ME, Davis PA. What makes an autoantigen an autoantigen? Ann N Y Acad Sci. 2005;1050:134–145. doi: 10.1196/annals.1313.015. [DOI] [PubMed] [Google Scholar]
- 46.Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Ann N Y Acad Sci. 2005;1050:1–9. doi: 10.1196/annals.1313.001. [DOI] [PubMed] [Google Scholar]
- 47.Petersen J, Purcell AW, Rossjohn J. Post-translationally modified T cell epitopes: immune recognition and immunotherapy. J Mol Med (Berl) 2009;87:1045–1051. doi: 10.1007/s00109-009-0526-4. [DOI] [PubMed] [Google Scholar]
- 48.Champion BR, Page KR, Parish N, Rayner DC, Dawe K, Biswas-Hughes G, Cooke A, Geysen M, Roitt IM. Identification of a thyroxine-containing self-epitope of thyroglobulin which triggers thyroid autoreactive T cells. J Exp Med. 1991;174:363–370. doi: 10.1084/jem.174.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rasooly L, Rose NR, Saboori AM, Ladenson PW, Burek CL. Iodine is essential for human T cell recognition of human thyroglobulin. Autoimmunity. 1998;27:213–219. doi: 10.3109/08916939808993833. [DOI] [PubMed] [Google Scholar]
- 50.Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, Kay TW, Rossjohn J, Falk BA, Nepom GT, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med. 2005;202:1191–1197. doi: 10.1084/jem.20051251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He XL, Radu C, Sidney J, Sette A, Ward ES, Garcia KC. Structural snapshot of aberrant antigen presentation linked to autoimmunity: the immunodominant epitope of MBP complexed with I-Au. Immunity. 2002;17:83–94. doi: 10.1016/s1074-7613(02)00340-0. [DOI] [PubMed] [Google Scholar]
- 52.Bankovich AJ, Girvin AT, Moesta AK, Garcia KC. Peptide register shifting within the MHC groove: theory becomes reality. Mol Immunol. 2004;40:1033–1039. doi: 10.1016/j.molimm.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 53*.Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11:350–354. doi: 10.1038/ni.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med. 2011;208:1501–1510. doi: 10.1084/jem.20110574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardner JM, Devoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, Johannes KP, Su MA, Chang HY, Krummel MF, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science. 2008;321:843–847. doi: 10.1126/science.1159407. [DOI] [PMC free article] [PubMed] [Google Scholar]